
Abstract
Motivation
Since the advent of ColabFold, numerous software packages have been provided with Google Colaboratory-compatible ipynb files, allowing users to effortlessly test and reproduce results without the need for local installation or configuration. MEGADOCK, a protein–protein docking tool, is particularly well-suited for Google Colaboratory due to its lightweight computations and GPU acceleration capabilities. To increase accessibility and promote widespread use, it is crucial to provide a computing environment compatible with Google Colaboratory.
Results
In this study, we report the development of a Google Colaboratory environment for running our protein–protein docking software, MEGADOCK. We provide a comprehensive ipynb file, including the compilation of MEGADOCK with the FFTW library installation on Colaboratory, the introduction of related tools using PyPI/apt, and the execution and visualization of docking structures. This streamlined environment enables users to visualize docking structures with just one click. The code is available under a CC-BY NC 4.0 license from https://github.com/ohuelab/MEGADOCK-on-Colab.
Similar content being viewed by others


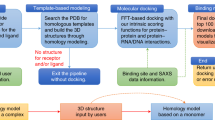
Introduction
Understanding protein–protein interactions is of paramount importance for elucidating complex biological phenomena and identifying potential drug targets. MEGADOCK [1, 2] is a protein docking software developed by our team to accurately predict protein–protein interactions. Designed to function on Linux-based systems, MEGADOCK supports GPU computing via CUDA and parallel computing on cluster machines. These capabilities render it particularly suitable for conducting comprehensive prediction validations, including one-to-many and many-to-many protein interactions [3]. In this paper, we introduce MEGADOCK-on-Colab, a tool that facilitates the execution of MEGADOCK on Google Colaboratory (hereinafter referred to as Colaboratory).
Colaboratory (https://colab.research.google.com) is a Jupyter Notebook hosting service provided by Google Research, enabling users to compose and execute Python code directly within their web browsers. This versatile service caters to a wide range of applications, including machine learning, data analysis, and education, while offering complimentary access to accelerators such as GPUs. Each computing environment is provisioned as a virtual machine (VM) with root privileges and is subsequently discarded after use, thereby permitting flexible installation of libraries and external tools. The implementation of AlphaFold2 [4] and ColabFold [5] via Colaboratory has enticed a growing number of life science researchers to utilize the platform. Notably, since the emergence of ColabFold, an increasing array of software packages have been accompanied by Colaboratory-compatible.ipynb files, enabling users to effortlessly test and reproduce results without necessitating local installation or configuration (e.g., small-molecule docking software DiffDock [6]).
In this study, we have successfully established a Colaboratory environment for executing MEGADOCK. We furnish a comprehensive ipynb file encompassing the compilation of MEGADOCK, inclusive of the installation of the FFTW library on Colaboratory, the incorporation of pertinent tools via PyPI/apt, and the execution and visualization of docking structures within the Colaboratory framework. This streamlined environment empowers users to visualize docking structures with a single click.
Implementation
The following is a step-by-step explanation of the MEGADOCK-on-Colab process flow and implementation method.
Preparation for input
In Colaboratory, user interfaces can be added to Python variables using annotations such as #@param{type:”string”}. For instance, by writing:
R_pdb_id = ”1CGI” #@param {type:”string”}
An input form for the variable R_pdb_id can be displayed with the pre-filled value of “1CGI”. Using this approach, we have configured the tool to accept two PDB IDs and chain names as input forms. Additionally, we incorporated input forms to receive the main arguments for MEGADOCK. The reason for preparing these inputs prior to tool installation is to ensure that the input forms are displayed as close to the top of the Jupyter Notebook as possible, making users aware of their presence.
Installation of MEGADOCK on Colaboratory VM
Next, we install the GPU version of MEGADOCK on the Colaboratory VM as follows. System commands can be used in Colaboratory by adding a ‘!’ prefix.
!git clone https://github.com/akiyamalab/MEGADOCK
!git clone https://github.com/NVIDIA/cuda-samples
!apt install -y libfftw3-dev libfftw3-single3
%cd ./MEGADOCK
!make -j 2 -f Makefile.colab
Since the operating system is based on Ubuntu, apt can be used (to determine the actual OS being used, run commands like !cat /etc/os-release). Note that the NVIDIA drivers and CUDA are pre-installed (to check the available GPU and CUDA version, execute !nvidia-smi).
Downloading PDB files
We download the specified PDB files from the Protein Data Bank using wget and use Biopython [7] to extract the specified chains. Biopython is available on PyPI and can be installed as follows:
!pip install biopython
Executing MEGADOCK
MEGADOCK can be executed with the following command:
!./megadock-gpu -R $MDPDBR -L $MDPDBL -t $MDt -N $MDN -o $MDOF
Please note that the arguments need to be passed from Python to the shell environment variables. For instance, we can set the values beforehand using something like:
os.environ[’MDPDBR’] = pdbr.
Visualizing predictions
NGLView [8] is a molecular viewer that operates within Jupyter Notebook. It is available through PyPI and can be installed with the following command:
!pip install nglview
To visualize the output.pdb file, use the following code:
from google.colab import output
output.enable_custom_widget_manager()
import nglview as nv
view = nv.show_structure_file(”output.pdb”)
view
Results
Figures 1 and 2 showcase example screenshots of MEGADOCK-on-Colab. Upon entering the requisite information, such as files and parameters, into the initial input form, users can automatically execute the entire series of processes by selecting the “Run all” option. As of April 2023, the free version of Colaboratory frequently assigns an NVIDIA Tesla T4 GPU to its virtual environments. Utilizing the Tesla T4 GPU, the MEGADOCK docking calculation for PDB 1CGI chain E (consisting of 245 residues) and 1CGI chain I (consisting of 56 residues) required approximately 5 s. The processing time for non-docking calculations, encompassing installation procedures, amounted to roughly 60 s. Additionally, although we have demonstrated how to execute the program using specified PDB IDs, it is also possible for users to upload their own PDB files and perform the docking calculations.

MEGADOCK-on-Colab input form

Visualization with NGLView in MEGADOCK-on-Colab
Discussion
In this study, we have delineated a method for constructing an execution environment on Colaboratory, utilizing MEGADOCK as a representative example.
Owing to its virtual nature, Colaboratory alleviates concerns regarding potential damage to the environment arising from installation failures, while consistently offering a nearly identical computing environment. However, it’s important to note that Colaboratory does not guarantee complete reproducibility. The libraries, OS version, GPU, CPU, and other factors are determined by the cloud and can vary, potentially affecting the results of compiled code. Thus, while Colaboratory facilitates reproducibility to some extent, it does not ensure it in all circumstances, enhancing the convenience of software utilization.
As evidenced in the present study, Colaboratory supports not only Python-based programs but also the compilation of diverse programming languages, execution of binary files, and implementation of system commands, effectively operating as a GPU-equipped server. In light of the ongoing development of an extensive array of GPU-compatible libraries, including those for deep learning, sharing reproducible execution environments on Colaboratory is progressively becoming a standard practice in software publication.
Limitations
While our study introduces significant advancements in protein–protein docking software, there are certain limitations that need to be acknowledged.
Firstly, the reproducibility aspect of Colaboratory is not absolute. Although Colaboratory offers a nearly identical computing environment, it does not guarantee complete reproducibility. Factors such as the libraries, OS version, GPU, and CPU are determined by the cloud and can vary, potentially affecting the results of compiled code. This inherent variability underscores the need for caution when interpreting results obtained solely from this platform.
Secondly, our current implementation does not delve deeply into the detailed analysis of protein–protein interaction surfaces. While we provide a general overview and visualization, intricate details, especially those crucial for understanding specific molecular interactions, are not extensively covered. This might limit the depth of analysis researchers can perform using our tool, especially when a nuanced understanding of interaction sites is required.
Lastly, the visualization capabilities of our tool, while robust, could benefit from the integration of more advanced molecular viewers. Tools like mol* [9] offer enhanced visualization features that can provide a more comprehensive and detailed view of molecular interactions. Incorporating such advanced viewers in future updates could significantly elevate the user experience and the depth of analysis possible.
In conclusion, while our tool offers a novel approach and several advantages, users should be aware of these limitations when utilizing it for their research. We aim to address these in future iterations, ensuring a more comprehensive and user-friendly experience.
Code availability
The code is available under a CC-BY NC 4.0 license from https://github.com/ohuelab/MEGADOCK-on-Colab.
References
Ohue M, Shimoda T, Suzuki S, et al. MEGADOCK 4.0: an ultra-high-performance protein–protein docking software for heterogeneous supercomputers. Bioinformatics. 2014;30(22):3281–3.
Ohue M, Matsuzaki Y, Uchikoga N, et al. MEGADOCK: an all-to-all protein–protein interaction prediction system using tertiary structure data. Protein Pept Lett. 2014;21(8):766–78.
Aoyama K, Watanabe H, Ohue M, Akiyama Y. Multiple HPC environments-aware container image configuration workflow for large-scale all-to-all protein–protein docking calculations. In: Proceedings of the SCFA2020, LNCS. 2020; 12082:23–39.
Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–9.
Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19(6):679–82.
Corso G, Stärk H, Jing B, Barzilay R, Jaakkola T. DiffDock: diffusion steps, twists, and turns for molecular docking. In: Proceedings of the ICLR2023. https://openreview.net/forum?id=kKF8_K-mBbS.
Cock PJ, Antao T, Chang JT, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25(11):1422–3.
Nguyen H, Case DA, Rose AS. NGLview-interactive molecular graphics for Jupyter notebooks. Bioinformatics. 2018;34(7):1241–2.
Sehnal D, Bittrich S, Deshpande M, et al. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021;49(W1):W431–7.
Funding
This work has been supported by the JST FOREST (JPMJFR216J) and JST ACT-X (JPMJAX20A3).
Author information
Authors and Affiliations
Contributions
MO conducted the entire study, implementation, data preparation, and writing of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
There are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ohue, M. MEGADOCK-on-Colab: an easy-to-use protein–protein docking tool on Google Colaboratory. BMC Res Notes 16, 229 (2023). https://doi.org/10.1186/s13104-023-06505-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-023-06505-w