
Abstract
Background
In Bacillus subtilis, two major transcriptional factors, GlnR and TnrA, are involved in a sophisticated network of adaptive responses to nitrogen availability. GlnR was reported to repress the transcription of the glnRA, tnrA and ureABC operons under conditions of excess nitrogen. As GlnR and TnrA regulators share the same DNA binding motifs, a genome-wide mapping of in vivo GlnR-binding sites was still needed to clearly define the set of GlnR/TnrA motifs directly bound by GlnR.
Methods
We used chromatin immunoprecipitation coupled with hybridization to DNA tiling arrays (ChIP-on-chip) to identify the GlnR DNA-binding sites, in vivo, at the genome scale.
Results
We provide evidence that GlnR binds reproducibly to 61 regions on the chromosome. Among those, 20 regions overlap the previously defined in vivo TnrA-binding sites. In combination with real-time in vivo transcriptional profiling using firefly luciferase, we identified the alsT gene as a new member of the GlnR regulon. Additionally, we characterized the GlnR secondary regulon, which is composed of promoter regions harboring a GlnR/TnrA box and bound by GlnR in vivo. However, the growth conditions revealing a GlnR-dependent regulation for this second category of genes are still unknown.
Conclusions
Our findings show an extended overlap between the GlnR and TnrA in vivo binding sites. This could allow efficient and fine tuning of gene expression in response to nitrogen availability. GlnR appears to be part of complex transcriptional regulatory networks, which involves interactions between different regulatory proteins.
Similar content being viewed by others


Background
The response of the Gram-positive bacterium Bacillus subtilis to nitrogen availability is an example of a highly sophisticated system to detect nitrogen levels and transmit this signal to effect intracellular enzyme activity and gene regulation. In this bacterium, ammonium assimilation occurs via the glutamine synthetase-glutamate synthase (GS-GOGAT) pathway to generate glutamate, the precursor for amino acids and nucleotides biosynthesis [1]. Glutamine is the B. subtilis preferred nitrogen source followed by arginine and ammonium [2, 3].
Two transcription factors, TnrA and GlnR, and one enzyme, the GS, play a major role in the B. subtilis nitrogen regulatory network [4,5,6]. TnrA and GlnR both control the expression of nitrogen-regulated genes with partial overlap of their respective regulon. They are active under different nutritional conditions. Under nitrogen-limited conditions of growth, TnrA acts on the transcription of a large regulon comprising at least 35 transcriptional units [7,8,9,10,11,12,13]. In particular, TnrA exerts an activating effect on the transcription of its own gene tnrA [4, 14] and represses that of glnRA and gltAB operons encoding GS and GOGAT, respectively [6, 15, 16]. On the contrary, in an excess of nitrogen, GlnR acts as a repressor of tnrA, glnRA and ureABC expression [4, 5, 17,18,19].
Glutamine acts as the metabolic signal for nitrogen availability. When glutamine is in excess it binds to and feedback inhibits GS by forming the complex FBI-GS that in turn directly interacts and sequesters TnrA, thus inhibiting its DNA-binding function [12, 20]. FBI-GS activates GlnR through a chaperoning interaction, which results in transcriptional repression of the tnrA and glnRA genes [5, 21,22,23].
TnrA binding sites have been defined as 17-bp inverted repeat sequences with the consensus TGTNANATTTTNTNACA [8, 13]. Indeed, GlnR and TnrA bind in vitro the same site upstream of the tnrA and the glnRA operon, albeit with different specificity [19]. It is proposed that the differences in GlnR and TnrA motifs appeared limited but large enough to bring about some specificity in their binding profile [24].
Despite knowledge of GlnR-regulated genes, a global identification of the TnrA/GlnR motifs directly bound by GlnR was still missing. Here, we used chromatin immunoprecipitation of GlnR-DNA complexes coupled with hybridization of DNA to tiled oligonucleotides arrays (ChIP-on-chip) to identify the GlnR DNA-binding sites in vivo, at the genome scale. We showed that GlnR binds efficiently 61 regions on the chromosome and overlaps partially the previously defined TnrA primary regulon [8]. Analysis with real-time in vivo transcriptional profiling allowed to show that GlnR represses expression of the TnrA-dependent alsT gene. Additionally, we characterized the GlnR secondary regulon, which is composed of promoter regions harboring a GlnR/TnrA box and bound by GlnR in vivo.
Methods
Bacterial strains and growth conditions
The B. subtilis strains used in this work are listed in Table 1. Luria–Bertani (LB) medium was used to cultivate E. coli and B. subtilis. B. subtilis cells were also grown in a modified Spizizen minimal medium containing 62 mM K2HPO4, 44 mM KH2PO4, 17 mM trisodium citrate, 11 mM K2SO4, 0.6% glycerol, 1 mM MgSO4, 1 mM CaCl2, 100 µM FeCl3 citrate, 112 µM ZnCl2, 5 µM MnCl2, 2.5 µM CuCl2, and 0.3% glutamate or 0.3% glutamine. When necessary, ampicillin, erythromycin, chloramphenicol, and spectinomycin were added at 100, 8, 5 and 100 µg ml−1, respectively. To obtain solid media, 20 g Agar noble l-1 (Difco) were added to the liquid media. To transform E. coli or B. subtilis cells, standard procedures were used as described in [25, 26].
DNA manipulations
DNA manipulations and cloning procedures were performed as described elsewhere [25]. DNA polymerase, restriction enzymes, and phage T4 DNA ligase were used according to the manufacturer’s instructions (Biolabs).
Construction of a glnR::glnR-spa and perR::perR-spa strains
A B. subtilis strain was constructed to express a C-terminal SPA-tagged GlnR protein (hereafter GlnRSPA). A translational fusion between the glnR coding sequence and the sequential peptide affinity (SPA) tag sequence was integrated in the chromosome as described in [27, 28]. The pMUTIN-SPALIC vector (described by Doherty et al. [29]) was used to construct a pMUTIN-SPALIC derivative containing C-terminal SPA-tagged glnR gene. After transformation of wild-type BSB1 strain with this plasmid and selection for erythromycin-resistance, the strain Bs005 was obtained in which the expression of glnR-spa is under the control of the native glnR promoter, and the resulting GlnRSPA is the only source of GlnR. The same strategy was used to construct the Bs013 strain expressing the PerRSPA protein.
Construction of ΔglnR deletion
The glnR mutant BSB21 was constructed by homologous replacement of the glnR coding sequence with the spectinomycin-resistance gene spc using a joining PCR technique [30]. Integration of the spc cassette at the glnR locus and deletion of the glnR gene were confirmed by DNA sequencing.
Construction of luciferase promoter fusion strains
We used the strategy described previously in [8] by using the pUC18 cm-luc plasmid and the assembly Gibson’s procedure [31]. The primers used for PCR are indicated in Additional file 1: Table S2.
Luciferase assay
We measured the luciferase activity as already described in details in [8] using a PerkinElmer Envision 2104 Multilabel Reader. Relative luminescence unit (RLU) and OD600 were measured at 5 min intervals.
Genome-wide determination of the GlnR-binding sites by ChIP-on-chip
To measure the chromosome-wide DNA-binding profiles of GlnR, chromatin immunoprecipitation assays were performed as described previously [32]. The strain Bas005 was grown at 37 °C until an OD600 of 0.6 in minimal medium containing glutamine supplemented with 0.5 mM IPTG and 1 µg erythromycin ml−1. After cells treatment with formaldehyde, cellular DNA was extracted and sonicated. To purify the DNA regions specifically cross-linked to GlnRSPA an antibody against the FLAG was used. The immuno-precipitated DNA (IP) and the control whole cell DNA extract (WCE) were labeled with Cy3 and Cy5, respectively, and co-hybridized to the B. subtilis Roche-NimbleGen tiled microarrays [33].
Peak sequence extraction and analysis
To detect possible GlnR-binding sites from the chips, signal peaks were extracted, then the IP/WCE ratios (log2) were corrected and each peak was assigned a ChipScore as described in details in [34] and [35]. This score is based on the distribution of the peak height values and estimates for each peak its relative distance from the median. Only the regions associated with a peak scoring ≥4.0 in at least the two replicates were considered as putative GlnR-binding sites.
SPA-tag pull-down experiments
The strains expressing the SPA fusions were grown to exponential phase in LB medium and the cells were recovered by centrifugation. Cells were frozen in liquid nitrogen. For tandem affinity purifications, cell pellets were resuspended with 5 ml of 10 mM Tris–Cl pH 8.0, 150 mM NaCl, 1 mg lysozyme ml−1, and 5 U Benzonase ml−1 (Novagen). Wild-type cells, which did not harbor a SPA fusion, were used as a control (no-SPA containing strain). GlnRSPA, PerRSPA and No-SPA containing protein complexes were isolated and analyzed as described in [36].
Results
C-terminally SPA-tagged GlnR is a functional regulator
The B. subtilis glnR locus was modified to express the GlnR protein fused at its C-terminus with the SPA tag (GlnRSPA). In the resulting glnR::glnR-spa strain, the expression of the gene encoding the GlnRSPA protein is under the control of its native transcriptional signals (see Methods section). To check the activity of the GlnRSPA fusion protein, expression of the tnrA gene was compared in wild-type and glnR::glnR-spa strains. The expression of tnrA is known to be inhibited by GlnR [19]. The tnrA promoter region was fused with the luc reporter gene and introduced at the native tnrA locus in wild-type, glnR::glnR-spa and glnR::spc strains (Table 1). Light emission, which results from the activity of the luc-encoded firefly luciferase, was recorded every 5 min during growth in minimal medium with glutamine as sole nitrogen source. Expression of the tnrA promoter was repressed in the wild-type and glnR::glnR-spa strains whereas it was increased by a twofold factor in ΔglnR cells during the exponential growth phase (Fig. 1). We noticed that the transcription rate increased with time. This may be due to glutamine consumption from the medium in the used conditions. This entailed a decrease of GlnR repressive effect and an increase of TnrA activating effect on tnrA expression during the growth. Thus, GlnRSPA was able to repress tnrA expression as GlnRWT. We concluded that the GlnRSPA fusion protein was functional for transcriptional regulation.

Expression of tnrA under the control of GlnRWT and GlnRSPA. Promoter activity (RLU/OD) of a PtnrA′-luc transcriptional fusion with the luc reporter gene is indicated: purple line, wild-type; red line, ΔglnR cells; green line, glnR::glnR-spa cells. Strains were grown in minimal medium supplemented with glutamine as the sole nitrogen source. Growth (OD600nm) was monitored every 5 min: black lines, wild type; grey lines, ΔglnR; blue lines, glnR::glnR-spa. For each strain, one representative curve, out of three independent replicates realized, is shown
Genome-wide mapping of GlnR binding sites
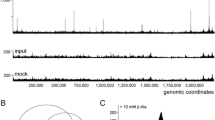
To identify GlnR-binding targets in B. subtilis genome, we carried out ChIP-on-chip experiments. The glnR::glnR-spa strain was grown in minimal medium with glutamine as the nitrogen source to exponential phase. After cross-linking, GlnR-bound DNA was immunoprecipitated using a FLAG specific antibody. Significantly GlnR-enriched DNA regions were identified as explained in the Methods section. Overall 61 enriched DNA regions were identified from the ChIP-on-chip signals (Additional file 2: Table S1). We retrieved GlnR-binding sites for the 3 well-characterized GlnR regulated promoters, glnR, tnrA and ureA (Fig. 2). In addition, 41 GlnR binding sites were detected less than 300 base-pairs upstream of a start codon. This suggests a GlnR-dependent expression of the nearest genes and therefore the existence of new candidates in the GlnR regulon. Finally, 17 peaks were located within intragenic regions more than 30 base-pairs downstream of a start codon (Fig. 2) (Additional file 2: Table S1). The location of these sites was intriguing since no GlnR intragenic binding sites have been described so far. It is possible that GlnR could bind to these intragenic sites to mediate repression by a roadblock mechanism, as described for the B. subtilis CcpA and CodY regulators [37, 38].

Analysis pipeline of the GlnR-binding sites detected by ChIP-on-chip. Several promoter regions associated to GlnR-binding sites are proposed to be classified in the two groups: GlnR primary (in red) and secondary (in blue) regulons
GlnR-binding sites overlap the TnrA regulon
The GlnR and TnrA regulators are known to bind to DNA sites (GlnR/TnrA sites) that have similar pattern. Therefore, we compared the set of the newly identified GlnR-binding sites with the previously defined TnrA primary regulon [8, 13]. Fifteen of the GlnR-bound regions are located in TnrA-dependent promoter regions (Fig. 2) (Table 2). As one region is involved in the regulation of two divergent promoters (nasA and nasB) in total we recovered 16 well-characterized TnrA regulated promoters. In addition, 5 GlnR-binding sites overlapped the TnrA secondary regulon whose members are bound by TnrA in vivo but are not differentially regulated in a ΔtnrA strain [8]. These sites are located upstream of braB and codV translational start sites as well as in the encoding region of ykoH, ypqP and yobI (Additional file 2: Table S1).
We further performed in silico analyses to investigate the presence of GlnR/TnrA boxes within the 38 newly identified inter- and intragenic GlnR-binding sites, which did not harbor a previously predicted GlnR/TnrA box. We used the MEME standard bioinformatic method [39] to identify common motifs among genomic regions representing 150 bp centered at each GlnR-binding site. We did not impose a constraint that the motif must be an inverted repeat sequence on the search. This yielded 16-nt sequences present in 3 GlnR-binding sites and matching the previously reported 17-nt TnrA box consensus with at least 10 identical nucleotides (Fig. 3) [8]. These potential GlnR/TnrA motifs are located in the promoter region of bceA, yjcN and yraH genes.

Identification of a 16-nt consensus sequence similar to the GlnR/TnrA motif in 3 in vivo GlnR binding sites. The 3 identified sequences are aligned with the previously reported GlnR/TnrA box
Half of the GlnR-binding sites detected by ChIP-on-chip did not display a significant match to the GlnR/TnrA box consensus. Using MEME, we were unable to identify a common DNA sequence motif among GlnR targets that lack a canonical GlnR/TnrA box motif. These suggest that GlnR recognizes degenerated GlnR/TnrA motif sequences, or that other factors are required for GlnR binding at these sites.
In vivo GlnR-binding correlates with transcriptional regulation of the alsT gene
We then tested the correlation between in vivo GlnR-binding and GlnR-dependent expression of the closer genes. Expression of 9 candidate genes containing a GlnR/TnrA box motif in their promoter region and covering the different groups that are illustrated on Fig. 2 was tested. We choose alsT, amtB, pucI, pucR, braB, codV, bceA, yjcN and yraH (Table 2). For this purpose, we used transcriptional fusions between the promoter regions and the luciferase gene in wild type and ΔglnR cells. Luciferase activity was recorded during exponential growth in minimal medium with glutamine as sole nitrogen source. In these conditions, transcription rate from PalsT was fourfold increased in a glnR mutant compared to wild-type (Fig. 4). As a control, expression of alsT was not altered in ΔglnR cells in the presence of glutamate as sole nitrogen source. In the glutamate-containing medium, alsT expression appeared repressed by both TnrA and GlnR in wild-type cells since alsT was derepressed in a tnrA mutant (Fig. 4) [13]. These results validated the GlnR-dependent regulation of the alsT gene. No difference in luciferase activity was observed for the 8 other gene fusions between wild type and ΔglnR strains in the conditions used (data not shown).

Expression of alsT under the dual control of GlnR and TnrA. Strains were grown in minimal medium supplemented with glutamate or glutamine as the sole nitrogen source. Growth (OD600nm) was monitored every 5 min: black lines, wild type; grey lines, ΔglnR; blue lines, ΔtnrA. a Promoter activity (RLU/OD) of a PalsT′-luc transcriptional fusion with the luc reporter gene is indicated: purple line, wild-type; red line, ΔglnR cells; green line, ΔtnrA cells. Strains were grown in the presence of glutamine. For each strain, one representative curve, out of three independent replicates realized, is shown. b Promoter activity (RLU/OD) of a PalsT′-luc transcriptional fusion with the luc reporter gene is indicated: purple line, wild-type; red line, ΔglnR cells; green line, ΔtnrA cells. Strains were grown in the presence of glutamate. For each strain, one representative curve, out of three independent replicates realized, is shown
GlnRSPA is associated to the glutamate synthase and to TnrA in vivo
To provide insight putative interactions of GlnR with other transcriptional factors in vivo, we sought to identify GlnRSPA binding partners. The strain expressing the glnR-spa fusion was grown in the nitrogen-rich LB medium in exponential phase. GlnR-associated proteins were purified and identified by mass spectrometry. Strains expressing no SPA-tagged protein and a SPA fusion to PerR, a non-related protein of B. subtilis, were used as negative controls [40, 41]. The TnrA and GltA proteins were specifically and reproducibly detected in the GlnRSPA pull-down complexes (Table 3) based on the protein abundance index (PAI, established according to [42]. Therefore the GlnRSPA protein is found in complex with the glutamate synthase and the TnrA regulator.
Discussion
Using the ChIP-on-chip methodology, we have identified 61 enriched DNA-regions in the B. subtilis chromosome that are reproducibly bound by the GlnR regulator in abundant nitrogen growth conditions. As we recovered the known GlnR regulon, the whole GlnR binding sites identified by ChIP-on-chip could be considered as relevant. Our analyses revealed that a large overlap exists between the location of GlnR-binding sites and genes whose expression is regulated by TnrA. Fifteen GlnR-binding regions belong to the previously defined TnrA primary regulon (Fig. 1) [8]. Real-time in vivo transcriptional profiling enabled us to validate the repression of the alsT gene by GlnR in excess-nitrogen conditions (Fig. 3). Hence, alsT is submitted to a dual regulation by GlnR and TnrA, depending on the nutritional conditions. These data allow to define the GlnR primary regulon which is now composed of 4 transcription units (glnRA, ureABC, tnrA and alsT) fulfilling three criteria: (1) GlnR binding in ChIP-on-chip experiments; (2) the presence of a GlnR/TnrA box; (3) GlnR-dependent expression regulation.
Remarkably, 5 GlnR-binding sites are associated to regions reported to belong to the TnrA secondary regulon whose members are bound by TnrA in vivo but for which the conditions of a potential TnrA-dependent regulation are still unknown [8]. In addition, 3 GlnR-bound DNA regions correlates with the presence of in silico predicted GlnR/TnrA motifs (Fig. 3). Under conditions that maximize GlnR activity, expression of braB, codV, bceA, yjcN and yraH was similar in wild-type and glnR mutant cells. However, regulation of these genes is known to be driven by other transcription factors (Additional file 2: Table S1). Therefore the existence of complex regulatory networks could mask GlnR activity.
Altogether, the ChIP-on-chip approach allowed us to define a GlnR secondary regulon, which is composed of 23 genomic regions fulfilling two criteria: (1) in vivo GlnR binding in ChIP-on-Chip experiments; (2) the presence of a GlnR/TnrA motif. We propose that GlnR might play a regulatory role in specific unknown conditions. The composition of the secondary regulon cannot be clearly delimited and is opened to permutations with the primary regulon depending on the discovery of yet unknown conditions involving GlnR-dependent regulation. We assume that expression of some genes could respond to specific growth conditions leading to intermediate levels of GlnR activity, as exemplified by the regulation of braB by the CodY regulator [43]. Moreover, we observed that GlnR belong to a protein complex in vivo with the glutamate synthase GltA. The potential role of a direct interaction between GlnR and GltA in the control of transcriptional regulation deciphers further investigations.
Finally, we reported a set of 35 GlnR binding DNA sites, which did not harbor a canonical GlnR-binding motif, suggesting that GlnR recognizes degenerated GlnR box sequences or that other factors are required for GlnR binding at these sites. It was previously shown that a GlnR protein truncated in the C-terminal domain repressed more tightly the expression of its target genes than the wild-type GlnR [44]. Deletions in the C-terminal region of GlnR [44] or TnrA [45,46,47] abolished their interaction with GS. Thus, we cannot exclude that addition of a SPA tag in the C-terminal part of GlnR might have changed its binding affinity to DNA as well as the interaction specificity with GS and the regulatory control. This could also explain that GS was not detected as protein partner in the GlnRSPA pull-down complexes.
The binding characterization of GlnR to DNA regions without evident GlnR-binding motif would be an important improvement to understand the role of GlnR and require further studies. In vitro assays could be performed to study the direct interaction between the native GlnR protein and the DNA regions that do not have a GlnR-binding motif. However, the binding of GlnR to these sites might require other unknown regulatory factors or specific conditions. It will be necessary to develop in vivo approaches to study the binding of GlnR to the newly identified targets and the consequences on the regulation of the nearest genes. Moreover, the surprising interaction detected between GnR and GltA deserve further investigations.
Conclusions
In the light of our results, we propose that binding of GlnR and TnrA to the same DNA binding sites may allow fine control over gene expression in response to various nitrogen levels. GlnR appears to be a part of complex transcriptional regulatory networks, which involves interactions between different regulatory proteins. In vivo, GlnR is found in complex with the GltA and TnrA proteins. Further investigations are required to define the exact role of the GlnR regulator in the control of the newly identified in vivo binding sites.
Abbreviations
- ChIP-on-chip:
-
chromatin immunoprecipitation coupled with hybridization to tiled oligonucleotides arrays
- GS:
-
glutamine synthetase
- bp:
-
base pair
- SPA tag:
-
sequential peptide affinity tag
- OD:
-
optical density
- RLU:
-
relative luminescence unit
- PCR:
-
polymerase chain reaction
References
Dean DR, Aronson AI. Selection of Bacillus subtilis mutants impaired in ammonia assimilation. J Bacteriol. 1980;141(2):985–8.
Fisher SH, Debarbouille M. Nitrogen soource utilization and its regulation. In: Sonenshein AL, Hoch JA, Losick JA, editors. Bacillus subtilis and its closest relatives: frome genes to cells. Washington DC: ASM Press; 2002. p. 181–91.
Hu P, Leighton T, Ishkhanova G, Kustu S. Sensing of nitrogen limitation by Bacillus subtilis: comparison to enteric bacteria. J Bacteriol. 1999;181(16):5042–50.
Fisher SH. Regulation of nitrogen metabolism in Bacillus subtilis: vive la difference! Mol Microbiol. 1999;32(2):223–32.
Schreier HJ, Brown SW, Hirschi KD, Nomellini JF, Sonenshein AL. Regulation of Bacillus subtilis glutamine synthetase gene expression by the product of the glnR gene. J Mol Biol. 1989;210(1):51–63.
Wray LV Jr, Ferson AE, Rohrer K, Fisher SH. TnrA, a transcription factor required for global nitrogen regulation in Bacillus subtilis. Proc Natl Acad Sci USA. 1996;93(17):8841–5.
Brandenburg JL, Wray LV Jr, Beier L, Jarmer H, Saxild HH, Fisher SH. Roles of PucR, GlnR, and TnrA in regulating expression of the Bacillus subtilis ure P3 promoter. J Bacteriol. 2002;184(21):6060–4.
Mirouze N, Bidnenko E, Noirot P, Auger S. Genome-wide mapping of TnrA-binding sites provides new insights into the TnrA regulon in Bacillus subtilis. Microbiologyopen. 2015;4(3):423–35.
Nakano MM, Hoffmann T, Zhu Y, Jahn D. Nitrogen and oxygen regulation of Bacillus subtilis nasDEF encoding NADH-dependent nitrite reductase by TnrA and ResDE. J Bacteriol. 1998;180(20):5344–50.
Nakano MM, Yang F, Hardin P, Zuber P. Nitrogen regulation of nasA and the nasB operon, which encode genes required for nitrate assimilation in Bacillus subtilis. J Bacteriol. 1995;177(3):573–9.
Wray LV Jr, Atkinson MR, Fisher SH. The nitrogen-regulated Bacillus subtilis nrgAB operon encodes a membrane protein and a protein highly similar to the Escherichia coli glnB-encoded PII protein. J Bacteriol. 1994;176(1):108–14.
Wray LV Jr, Zalieckas JM, Fisher SH. Bacillus subtilis glutamine synthetase controls gene expression through a protein-protein interaction with transcription factor TnrA. Cell. 2001;107(4):427–35.
Yoshida K, Yamaguchi H, Kinehara M, Ohki YH, Nakaura Y, Fujita Y. Identification of additional TnrA-regulated genes of Bacillus subtilis associated with a TnrA box. Mol Microbiol. 2003;49(1):157–65.
Robichon D, Arnaud M, Gardan R, Pragai Z, O’Reilly M, Rapoport G, Debarbouille M. Expression of a new operon from Bacillus subtilis, ykzB-ykoL, under the control of the TnrA and PhoP-phoR global regulators. J Bacteriol. 2000;182(5):1226–31.
Belitsky BR, Sonenshein AL. Modulation of activity of Bacillus subtilis regulatory proteins GltC and TnrA by glutamate dehydrogenase. J Bacteriol. 2004;186(11):3399–407.
Belitsky BR, Wray LV Jr, Fisher SH, Bohannon DE, Sonenshein AL. Role of TnrA in nitrogen source-dependent repression of Bacillus subtilis glutamate synthase gene expression. J Bacteriol. 2000;182(21):5939–47.
Brown SW, Sonenshein AL. Autogenous regulation of the Bacillus subtilis glnRA operon. J Bacteriol. 1996;178(8):2450–4.
Wray LV Jr, Ferson AE, Fisher SH. Expression of the Bacillus subtilis ureABC operon is controlled by multiple regulatory factors including CodY, GlnR, TnrA, and Spo0H. J Bacteriol. 1997;179(17):5494–501.
Zalieckas JM, Wray LV Jr, Fisher SH. Cross-regulation of the Bacillus subtilis glnRA and tnrA genes provides evidence for DNA binding site discrimination by GlnR and TnrA. J Bacteriol. 2006;188(7):2578–85.
Hauf K, Kayumov A, Gloge F, Forchhammer K. The molecular basis of TnrA control by glutamine synthetase in Bacillus subtilis. J Biol Chem. 2015;291(7):3483–95.
Fisher SH, Wray LV Jr. Bacillus subtilis glutamine synthetase regulates its own synthesis by acting as a chaperone to stabilize GlnR-DNA complexes. Proc Natl Acad Sci USA. 2008;105(3):1014–9.
Nakano Y, Kimura K. Purification and characterization of a repressor for the Bacillus cereus glnRA operon. J Biochem. 1991;109(2):223–8.
Schumacher MA, Chinnam NB, Cuthbert B, Tonthat NK, Whitfill T. Structures of regulatory machinery reveal novel molecular mechanisms controlling B. subtilis nitrogen homeostasis. Genes Dev. 2015;29(4):451–64.
Groot Kormelink T, Koenders E, Hagemeijer Y, Overmars L, Siezen RJ, de Vos WM, Francke C. Comparative genome analysis of central nitrogen metabolism and its control by GlnR in the class Bacilli. BMC Genom. 2012;13:191.
Sambrook J, Fristch EF, Maniatis T, editors. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor Laboratory: Cold Spring Harbor; 1989.
Kunst F, Rapoport G. Salt stress is an environmental signal affecting degradative enzyme synthesis in Bacillus subtilis. J Bacteriol. 1995;177(9):2403–7.
Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433(7025):531–7.
Zeghouf M, Li J, Butland G, Borkowska A, Canadien V, Richards D, Beattie B, Emili A, Greenblatt JF. Sequential Peptide Affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J Proteome Res. 2004;3(3):463–8.
Doherty GP, Fogg MJ, Wilkinson AJ, Lewis PJ. Small subunits of RNA polymerase: localization, levels and implications for core enzyme composition. Microbiology. 2010;156(Pt 12):3532–43.
Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast. 1996;12(3):259–65.
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–5.
Nicolas P, Mader U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S, et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012;335(6072):1103–6.
Rasmussen S, Nielsen HB, Jarmer H. The transcriptionally active regions in the genome of Bacillus subtilis. Mol Microbiol. 2009;73:1043–57.
Reppas NB, Wade JT, Church GM, Struhl K. The transition between transcriptional initiation and elongation in E. coli is highly variable and often rate limiting. Mol Cell. 2006;24:747–57.
Buescher JM, Liebermeister W, Jules M, Uhr M, Muntel J, Botella E, Hessling B, Kleijn RJ, Le Chat L, Lecointe F, et al. Global network reorganization during dynamic adaptations of Bacillus subtilis metabolism. Science. 2012;335:1099–103.
Delumeau O, Lecointe F, Muntel J, Guillot A, Guedon E, Monnet V, Hecker M, Becher D, Polard P, Noirot P. The dynamic protein partnership of RNA polymerase in Bacillus subtilis. Proteomics. 2011;11(15):2992–3001.
Belitsky BR, Sonenshein AL. Roadblock repression of transcription by Bacillus subtilis CodY. J Mol Biol. 2011;411(4):729–43.
Choi SK, Saier MH Jr. Regulation of sigL expression by the catabolite control protein CcpA involves a roadblock mechanism in Bacillus subtilis: potential connection between carbon and nitrogen metabolism. J Bacteriol. 2005;187(19):6856–61.
Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34(Web Server issue):369–73.
Mirouze N, Ferret C, Yao Z, Chastanet A, Carballido-Lopez R. MreB-dependent inhibition of cell elongation during the escape from competence in Bacillus subtilis. PLoS Genet. 2015;11(6):e1005299.
Rueff AS, Chastanet A, Dominguez-Escobar J, Yao Z, Yates J, Prejean MV, Delumeau O, Noirot P, Wedlich-Soldner R, Filipe SR, et al. An early cytoplasmic step of peptidoglycan synthesis is associated to MreB in Bacillus subtilis. Mol Microbiol. 2014;91(2):348–62.
Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteom. 2005;4(9):1265–72.
Belitsky BR, Brinsmade SR, Sonenshein AL. Intermediate levels of Bacillus subtilis CodY activity are required for derepression of the branched-chain amino acid permease, BraB. PLoS Genet. 2015;11(10):e1005600.
Wray LV Jr, Fisher SH. Bacillus subtilis GlnR contains an autoinhibitory C-terminal domain required for the interaction with glutamine synthetase. Mol Microbiol. 2008;68(2):277–85.
Kayumov A, Heinrich A, Fedorova K, Ilinskaya O, Forchhammer K. Interaction of the general transcription factor TnrA with the PII-like protein GlnK and glutamine synthetase in Bacillus subtilis. FEBS J. 2011;278(10):1779–89.
Fedorova KP, Scharafutdinov IS, Turbina EY, Bogachev MI, Ilinskaja ON, Kayumov AR. The C-terminus of transcription factor TnrA from Bacillus subtilis controls DNA-binding domain activity but is not required for dimerization. Mol Biol. 2013;47(2):331–7.
Hauf K, Kayumov A, Gloge F, Forchhammer K. The molecular basis of TnrA control by glutamine synthetase in Bacillus subtilis. J Biol Chem. 2016;291(7):3483–95.
Authors’ contributions
SA conceived and designed the experiments and analyzed the data. SA wrote the manuscript. PR participated in the design of the experiments and in the analyses of the data. AA and OD performed the tandem affinity purification of PerR-SPA and GlnR-SPA. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to Dr. Mark Fogg for the gift of the vector pMUTIN-SPALIC and to Dr. Elena Bidnenko and Dr. Vania Rosas for the construct of the strain BSB21 (BaSySBio consortium). We thank Dr. Philippe Noirot for the financial support.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All supporting data are included in the main paper and in Additional files 1, 2.
Consent to publish
Not applicable.
Ethics approval and consent to participate
Not applicable. This report does not include animal or human data.
Funding
This work was supported by the EU-funded BaSysBio project LSHG-CT-2006-037469 and by the European Union, Marie Curie ITN AMBER, 317338.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
13104_2017_2703_MOESM2_ESM.xls
Additional file 2: Table S1. Mapping of GlnR DNA binding sites by ChIP-on-chip. ChIP-on-chip experiments were performed and data were analysed as previously described [32] using the method described by Reppas et al. [34]. GlnR was purified in two biological replicates for each condition of growth. This table lists all significantly enriched DNA regions by ChIP-on-chip experiment performed with a GlnRSPA expressing Bacillus subtilis strain.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Randazzo, P., Aucouturier, A., Delumeau, O. et al. Revisiting the in vivo GlnR-binding sites at the genome scale in Bacillus subtilis . BMC Res Notes 10, 422 (2017). https://doi.org/10.1186/s13104-017-2703-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-017-2703-9