
Abstract
Background
Transposable elements (TEs) are common and often present with high copy numbers in cellular genomes. Unlike in cellular organisms, TEs were previously thought to be either rare or absent in viruses. Almost all reported TEs display only one or two copies per viral genome. In addition, the discovery of pandoraviruses with genomes up to 2.5-Mb emphasizes the need for biologists to rethink the fundamental nature of the relationship between viruses and cellular life.
Results
Herein, we performed the first comprehensive analysis of miniature inverted-repeat transposable elements (MITEs) in the 5170 viral genomes for which sequences are currently available. Four hundred and fifty one copies of ten miniature inverted-repeat transposable elements (MITEs) were found and each MITE had reached relatively large copy numbers (some up to 90) in viruses. Eight MITEs belonging to two DNA superfamilies (hobo/Activator/Tam3 and Chapaev–Mirage–CACTA) were for the first time identified in viruses, further expanding the organismal range of these two superfamilies. TEs may play important roles in shaping the evolution of pandoravirus genomes, which were here found to be very rich in MITEs. We also show that putative autonomous partners of seven MITEs are present in the genomes of viral hosts, suggesting that viruses may borrow the transpositional machinery of their cellular hosts’ autonomous elements to spread MITEs and colonize their own genomes. The presence of seven similar MITEs in viral hosts, suggesting horizontal transfers (HTs) as the major mechanism for MITEs propagation.
Conclusions
Our discovery highlights that TEs contribute to shape genome evolution of pandoraviruses. We concluded that as for cellular organisms, TEs are part of the pandoraviruses’ diverse mobilome.
Similar content being viewed by others


Background
Transposable elements (TEs) are DNA fragments that can move from one place to another in their host genomes, often resulting in their own amplification in the process [1]. DNA transposons represent a specific group of TEs, and they can be classified as either “autonomous” or “non-autonomous” elements. Autonomous transposons can transpose by themselves, while non-autonomous elements require enzymes encoded in trans by autonomous elements to be mobile [2]. Miniature inverted–repeat transposable elements (MITEs) are non-autonomous elements typically showing high copy numbers and length homogeneity [3, 4]. MITEs are often flanked by terminal inverted repeats (TIRs) and produce a short target site duplication (TSD) upon integration. As a group, MITEs form several superfamilies classified according to sequence similarity between their TIRs or TSD and those of autonomous partners3. It has been shown that MITEs play important roles in eukaryotic evolution, including an increase in genome size, formation of new genes, and the regulation of gene expression [5,6,7,8].
The vertical inactivation theory predicts that TEs will not escape the final fate of an inevitable vertical extinction in their host genomes due to elimination or inactivation [9, 10]. However, two factors may promote the long-term persistence of TEs in organism genomes. First, TEs have the capacity to be introduced into a new genomic background via horizontal transfers (HTs) [11], and second, autonomous DNA transposons can generate numerous defective elements (or MITEs) that evade the host defense system, either because of their small in size (< 600-bp) and/or because they fail to trigger the host repression response [2]. MITEs have been found in numerous eukaryotes including representatives of plants, fungi, protozoans and metazoans [2,3,4,5, 8]. In contrast, only one MITE family with a substantial number of copies has been reported from the giant virus P. salinus [6]. To the best of our knowledge, the amount and impact of the canonical TEs (for example, typical Class I or Class II TEs) on the evolution of viruses remains largely unknown. All TEs reported so far in viral genomes, apart from one Tc1/mariner member recently discovered in the genome of Pandoravirus salinus [6], display very low copy numbers (usually one or two) [12,13,14,15,16,17,18,19]. This has been interpreted as suggesting that viruses simply act as vectors for horizontal transfer (HT) of specific TEs with no clear impact on viral genomes themselves. To investigate the impact of TEs on viral genome evolution, a large-scale systematic analysis was therefore required. For the present study, a comprehensive survey of MITEs was performed through a careful examination of all latest available viral genome sequences. We focused on MITEs because their small size may allow a long-term persistence in diverse lineages through evasion of the host defense system2 and because a substantial number of one MITE belonging to the Tc1/mariner superfamily has been found in the genome of a giant virus, P. salinus [6].
Results
Discovery and characterization of MITEs in viruses
To scan viral genomes for MITEs, we characterized MITEs do novo in the most recent publicly available database of viral genomes (5170 viruses) from viruSITE [20] as of May 2016 using MITE-hunter [21]. Ten candidate MITEs were retrieved and were classified into four superfamilies of DNA transposons (Additional file 1: Table S1). These excluded twelve elements whose TSD could not be determined (Additional file 2: Table S2). Boundaries of all the MITE candidates could be well-defined through alignment of multiple full-length copies along with their flanking sequences (Additional file 3: Figure S1a). These repeats shared all features reported for MITEs (3), including a small size (116–422-bp), TIRs, 2–8-bp TSDs, stable secondary structure, lack of coding capacity and high size homogeneity (Additional file 1: Table S1 and Additional file 3: Figure S1a,d). Submariner-NA or Submariner (see Methods for details on nomenclature) had previously been reported [6], but the remaining nine MITEs are reported here for the first time in viruses. Sequences of all MITEs and their autonomous elements identified in this study have been deposited in Repbase [22].
Our survey revealed that MITEs were present in the genomes of viruses belonging to the Ascoviridae, Polydnaviridae (PDV) and Pandoraviridae families (Fig. 1). The number of MITEs also displayed important differences among viral genomes. For example, only one MITE (CMC-NA_1) was found in the genome of Glypta fumiferanae ichnovirus (GfIV), whereas six MITEs (Submariner-NA, hAT-NA1, hAT-NA2, hAT-NA3, hAT-NA4, hAT-NA5, and hAT-NA6) were detected in the genomes of the three known pandoraviruses (Fig. 1). Nucleotide genetic distance between each MITE copy and the consensus sequence varied from 0.06 to 0.4 (Additional file 3: Figure S1e). Meanwhile, the distribution of pairwise divergence among MITEs usually overlap, suggesting that these MITEs might amplify at similar time in these viral genomes. Comparisons of copy numbers and contents of MITEs between PDV and the Pandoraviridae showed that MITEs had experienced the most successful amplification in Pandoravirus salinus, the virus with the largest genome characterized to date [23]. Here, MITEs generated up to 251 copies, contributing with about 37.4-kb of DNA and constituting almost 1.5% of the viral genome (Fig. 2). This proportion of MITEs is even higher than that assessed for many insect and plant genomes [5, 8].

The schematic representation of phylogenetic relationships of viruses. The presence of MITEs in viral genomes is shown using rectangles. Species abbreviations were shown in Additional file 1: Table S1

Copy number and content of ten MITEs in the viral genomes. a Comparison of copy numbers of ten MITEs in studied viruses. b Amount of nucleotide covered by each MITE in five viral genomes
To determine whether MITEs identified in viruses in this study have been experienced transposition, paralogous empty sites (i.e., not containing MITEs) were searched in viral genomes using the flanking sequences (50 or 100-bp on either side) of all full-length elements as queries. One paralogous empty site of CMC-NA_1 was found in the genome of GfIV, suggesting that CMC-NA_1 had experienced transposition activity in this virus (Additional file 3: Figure S1b). Alternatively, this result might be explained by host-to-virus transposition into only one of the paralogous sites. CMC-NA The six MITEs discovered in the genomes of the three known pandoraviruses (Fig. 1 and Additional file 1 Table S1) enabled investigation of their presence/absence polymorphism at viral orthologous genomic sites using synteny and collinearity analyses. The analyses showed that these MITEs were widely distributed in syntenic and collinear regions of pandoraviruses, and that 262 orthologous genomic sites were devoid of MITEs (i.e., orthologous empty sites) (Additional file 4: Figure S2). These results provided strong evidence that MITEs had been transpositionally active after divergence of these pandoraviruses, contributing to their genetic diversity. Our results also showed that some MITE copies shared at seven orthologous sites between different viruses. For hAT_NA1 and hAT_NA2, their copies were found at two and one orthologous sites shared by P. salinus and P. dulcis, respectively. For hAT_NA4, its copies were found at one orthologous site shared by P. salinus and P. inopinatum. For hAT_NA5, its copies were present at two orthologous sites shared by P. salinus and P. dulcis and also found at one orthologous site shared by P. salinus and P. inopinatum). This might imply that such MITEs have been conserved since the divergence of these viruses. Alternatively, it indicate that transposition might occur independently twice at the same locus in difference viruses.
Putative autonomous partners for MITE transposition
MITEs have no capacity of transposing and need to borrow the transpositional machinery from their master elements (2). Generally, the sequence similarity of MITEs and DNA transposon responsible for their transposition is restricted to TIRs. Therefore, we investigated potential autonomous elements of these MITEs in both viral and cellular host genomes (e.g. Acanthamoeba castellanii for pandoraviruses) using homology analysis. Autonomous DNA transposons potentially involved in the spread of eight MITEs were discovered in cellular hosts (or species related to their hosts) (Fig. 3 and Additional file 1: Table S1). We found that these autonomous elements had all the hallmarks of the corresponding superfamilies. For example, both hAT-5 and hATm-6 were flanked by 8-bp TSD and encoded an intact transposase with a conserved dimerization domain (PF05699) at the C-terminus, characteristic features of the hAT superfamily [24]. Interestingly, hAT-5 was identified in four amoebas (A. castellanii, A. polyphaga, A. pearcei and A. quina), three of which contained hAT-5 sharing 100% sequence identity at the nucleotide level, encoding a 620-amino-acid (aa) transposase (nucleotide positions: 279–2141) without disruption by premature stop codons. These data might suggest that hAT-5 is still active in the amoeba hosts of pandoraviruses. Phylogenetic analysis of hAT transposases showed that hAT-5 was a member of the Ac family (Additional file 5: Figure S3a). Although more than 74 hATm closely related to hATm-6 have been deposited in Repbase [22], knowledge about these transposons remains relatively limited. Our results suggested that hATm-6 might represent a fourth family of hAT transposons. First, the hATm family appears to be divergent from the other three reported hAT families (Ac, Buster and Tip) [24, 25] and forms a well supported clade, with bootstrap values of 100% (Additional file 5: Figure S3a). Second, we confirmed that 28 members of the hATm family had well-defined boundaries and that integration of these transposons generated 8 or 9-bp TSDs (Additional file 5: Figure S3b and Additional file 6: Table S3). Third, multiple alignments of their terminal sequences showed that hATm transposons were flanked by the conserved 5′-TAGGGTG and CACCCTA-3′ termini (Additional file 5: Figure S3c). We therefore propose to establish a new family within the hAT superfamily, designated hATm [22].

Multiple alignments of the 5′ and 3′ terminal sequences of hAT-NA1 to hAT-NA5 (a and b), hATm-NA6 (c), Submariner-NA (d), IS200/IS605_NA (e) and CMC-NA (f) with their corresponding potential autonomous elements. MITEs identified in viruses are shown using red color. Species abbreviations are shown in Additional file 1: Table S1. CfTEC was obtained from the previous report [26], and the rest of the CACTA transposons were retrieved from Repbase [20]. TIRs of hATm-6_RP and hATm-6_CV are 417 bp and 575 bp, respectively. Therefore, only 40 bp of both termini of these transposons are shown using arrows
The hAT-5 transposons identified in four amoebas had short TIRs (10 bp) that were identical to the terminal sequences of hAT-NA3 and hAT-NA5 (Fig. 3a and b, Additional file 1: Table S1). Similarly, we showed that 5 to 8 out of 10 nucleotides of hAT-5 TIRs were identical to the termini of hAT-NA1, hAT-NA2 and hAT-NA4 (Fig. 3b). This level of TIR similarity between MITEs and their autonomous partners is typical [26]. We also observed that the sequence identity between hAT-NA5 and hAT-5 was not limited to the TIRs but also encompassed parts of the internal sequences of hAT-5 (Fig. 3a). This result suggested that hAT-NA5 was an internal deletion derivative of hAT-5. In view of the high level of similarity reported here for TIRs and TSD, hAT-5 was likely responsible for the transposition of the five MITEs identified in pandoraviruses.
We also noticed similarity of termini (13/15 nucleotides) between hATm-NA6_CcBV and two autonomous elements from Cotesia vestalis and Rhodnius prolixus (Fig. 3c). Interestingly, autonomous DNA transposons sharing the same terminal sequences (5′-TAGGGTG and CACCCTA-3′) with that of hATm-NA6_CcBV were also present in the genomes of Cotesia congregata bracovirus (CcBV) (GenBank accession number MF689003) as well as in its insect host, C. congregata (Additional file 2: Table S2 and Additional file 5: Figure S3c). Analysis of its transposase showed that both autonomous elements had encoded a relatively recent function protein which suggests transposition activity occurred in the wasp genome impacting the virus.
Sun et al. [6] reported the presence of an autonomous partner of Submariner-NA_PS in the pandoravirus amoebal host A. castellanii. Here, we found that this autonomous element was also present in three other amoebas, A. lugdunensis, A. rhysodes and A. palestinensis (Additional file 1: Table S1). Our results showed that the autonomous partner discovered in A. rhysodes seemed to have a functional transposase, which encoded a 375-aa protein (nucleotide positions:339–1466) and is similar in size to other active transposases of Tc1/mariner transposons [27]. Moreover, the intact DDE signature, a central catalytic domain of Tc1/mariner transposases, was also identified in the A. rhysodes transposase sequence. All the above autonomous transposons included 29-bp TIRs that displayed high sequence identity with TIRs of Submariner-NA (Fig. 3d), suggesting that they might be involved in the transposition of Submariner-NA.
In the case of IS200/IS605_NA, its autonomous elements were found in two ascovirues and three viruses belonging to the family of Baculoviridae (Additional file 1: Table S1). IS200/IS605_NA are flanked by 10 bp TIRs, which are identical to those of its autonomous elements (Fig. 3d). However, these autonomous elements only encoded a protein of unknown function TnpB, and no transposase (TnpA) was found. Although no autonomous partner of CMC-NA_1 and CMC-NA_2 was found in the viral and cellular host genomes, sequence similarity was observed among TIRs of CMC-NA_1, CMC-NA_2 and the reported CACTA transposons (Fig. 3f). In particular, 12/15 nucleotides of TIRs of ENS1_Cis, a CACTA transposon described in the transparent tunicate Ciona savignyi [22, 28], was identical to the TIRs of CMC-NA_1_GfIV. In addition, both ENS1_Cis, CMC-NA_1 and CMC-NA_2 shared the invariable 5′-CAC and GTG-3′ termini and created a 2-bp TSD (Additional file 3: Figure S1a,b). Thus, our results suggests that CMC-NA_1 and CMC-NA_2 are two members of the CACTA transposons, which are affiliated to a larger “megafamily” known as CMC for Chapaev–Mirage–CACTA [29].
Horizontal transfers of MITEs between viruses and their hosts
To identify the possible HTs of MITEs between viruses and their cellular hosts or closely related species, each MITE found in viruses was used as a query to search against cellular genomes. In the case of CMC-NA_1, CMC-NA_2, Submariner-NA, hAT-NA3, hAT-NA4, hAT-NA5 and hATm-NA6, these searches yielded a number of significant hits (score ≥ 123, e-value ≤ 3 × 10− 11). CMC-NA and hATm-NA6 were distributed in two and three wasps, respectively (Additional file 1: Table S1). The remaining four MITEs were found in eight amoebas. The boundaries of these MITEs could be determined in at least one cellular genome, revealing that they created a TSD upon insertion similar to that of MITEs found in viruses (Additional file 3: Figure S1 and Additional file 7: Figure S4). Almost all sequences presented the characteristics of non-autonomous elements, namely short length and lack of coding capacity (Additional file 1: Table S1). The alignment revealed sequence identity from 63.7 to 96.9% across the whole length of seven MITEs between any two species (Fig. 4, Additional file 8: Table S4 and Additional file 9: Figure S5). This high sequence identity strongly suggests that they were acquired through HTs. To rule out the possibility that the latter conclusion is compromised because based on accidental DNA cross-contamination artifacts, we conducted two additional analyses. First, MITEs computationally identified in the viral genomes were confirmed using PCR amplification and sequencing of PCR products (GenBank accession numbers MF576508-MF576521). Second, we plotted the coverage of six MITEs plus 1000 bp before and after their genomic positions on the genomes of P. salinus and P. dulcis and found no significant drop in coverage (Additional file 10: Figure S6).

Next, we investigated the relationship between MITEs found in viral and host genomes. A phylogenetic tree of full-length copies of Submariner-NA elements indicated that pandoravirus elements formed a monophyletic clade (Fig. 5a), indicating that it was a single ancestral transfer, more likely from host to virus, followed by spread of the MITEs within viral genomes. We should also notice that this result can also be due to multiple host-to-virus HT from one or more hosts that have not been sequenced yet. In contrast, copies of the rest six MITEs from viruses intermingled with host elements in the phylogenies was indicative of possibly multiple HT events from a single ancestor TE into these species (Fig. 5b and Additional file 11: Figure S7). Encapsidated PDV genomes comprise multiple dsDNA circular segments (often referred to as “circles”), linear copies of which reside in the genomes of wasp hosts. The latter condition facilitates movement of TEs from wasp DNA to the integrated “proviral” PDV genome. This is apparently what happened in the case of the CMC-NA_1, CMC-NA_2 and hATm-NA6 found in the genomes of PDVs, as nearly identical copies of these TEs were observed in wasp DNA near proviral genome segments (Additional file 9: Figure S5b,c,g). Interestingly, no CMC-NA_1 were found (data not shown) in the genome of species closely related to GfIV, Apophua simplicipes ichnovirus (AsIV) [30], an observation that suggests GfIV acquired CMC-NA from the genome of G. fumiferanae as opposed to a viral ancestor of both GfIV and AsIV. With respect to CMC-NA_2 and hATm-NA6 from bracoviruses, they similarly appear to have originated from the genomes of their braconid host wasps, as we failed to detect them in nudiviruses, the viral progenitor of bracoviruses [31].

The phylogenetic relationship of two MITEs from viral and host genomes. a A phylogenetic tree of full-length copies of Submariner-NA elements from pandoraviruses and their host amoeba. b Phylogeny of full-length copies of hAT-NA5 elements from pandoraviruses and their host amoeba. Clade credibility values (> 70%) are shown at each node. Each intact copy from different species is indicated using distinguishable color lines. Because hAT-NA5 from A. castellanii and A. pearcei are identical, and they are displayed using fluorescent green lines
Discussion
Here, we reported on the successful invasion of virus genomes by additional nine MITEs, eight of which belonging to the CMC and hAT superfamilies were for the first time identified in viruses, further expanding the organismal scope of these two superfamilies. The hAT superfamily represents a very large and diverse group of DNA transposons widely distributed in the cellular world and the hAT transposons can also be domesticated to become important functional genes in eukaryotes [24, 25]. Here, our results clearly indicated that the hAT transposons were remarkable and reached high copy numbers in some DNA viruses (Fig. 2). This is in agreement with the capacity of hAT elements to transpose efficiently in eukaryotes [32]. A phylogenetic tree of hAT transposases and sequence analyses clearly supported the recognition of a fourth family (Additional file 5: Figure S3), which we here named hATm, present in a wide range of organisms.
There are six known families of giant viruses infecting Acanthamoeba species: Mimiviridae, Pandoraviridae, Marseilleviridae, Faustovirus, Pithoviridae, and Mollivirus [33], with genome sizes ≥ 500 kb. The genome of P. salinus, 2.5-Mb in size [23], is the largest genome of all sequenced viruses and is even larger than the genomes of some bacteria such as Tremblaya [34] and Rickettsia [35]. Therefore, the discovery of pandoraviruses with genomes up to 2.5-Mb emphasizes the need for biologists to rethink the fundamental nature of the relationship between viruses and cellular life. Because there are large differences in genome size – even among giant viruses – the factors driving the evolution of these genomes have been the subject of intense debate and research. Many studies showed that genome evolution of giant viruses was related to successive steps of gene duplication, gene transfer and amplification of other types of repeated sequences (e.g. IS607 from the bacteria and archaea) [23, 36,37,38]. MITEs were observed here to be most abundant in pandoraviruses, a condition that may have played a role in genome expansion of these viruses. This result is consistent with the earlier observations made on plants and insects [5, 8]. Interestingly, all hAT transposons identified in pandoraviruses have very similar termini (Fig. 3a), suggesting that they are derived from some close related progenitor elements. Generally, TEs reported in the viral genomes had only one or two copies. In contrast, we found that MITEs in every case have expanded within the ascovirus, PDV and pandoravirus genomes (Additional file 1: Table S1). In addition, we should note that other undetermined twelve repeat elements were found in other seven families of ds DNA viruses (Additional file 2: Table S2). Meanwhile, almost all these repeat elements have amplified in viral genomes. Together, our results may suggest that TEs have played important roles in the genome evolution of ds DNA viruses.
MITEs have been found both in ascovirus, PDVs and pandoraviruses, which may be related, in both cases, to low constraints on genome size, making it possible to accommodate insertions. Most viruses have a genome strictly limited in size, each single particle has to contain the whole set of genes required to support a new replication cycle and the size of genetic material incorporated strictly depends on physical capacity of the particles. This results in most virus genomes having high gene density. In such compact genomes, any insertion is likely to have a deleterious effect for the virus, which would result in its elimination by counter selection. By contrast, the capacity of giant viruses particles to incorporate DNA is high and the cumulative size of the packaged viral molecules of Polydnaviridae appears to be similar to that of “giant viruses” with over 800 kb for CcBV [39]. Moreover, PDVs are solely transmitted vertically as endogenous viruses of parasitic wasps, and there is no replication in infected cells. The genes involved in particle production reside permanently in the wasp genome and have been replaced in the DNAs packaged in the particles by virulence genes. These are introduced during wasp oviposition and are essential to alter parasitized host immune response and, more broadly, its physiology (metabolism, development), favouring wasp larvae development. Sequencing of encapsidated PDV genomes has shown that, resulting from these particular constraints, the coding density is unusual low for a virus, with about 70% of non coding sequence in the case of CcBV [40]. This may allow to maintenance of MITE insertions. Accordingly, several remnants of TEs have been previously identified in dsDNA circles packaged in the particles including retroelements Dong and DIRS and a large DNA transposon Maverick [12, 14]. PDV proviral segments from which circles are produced have been integrated for up to 100 million years in the wasp genomes [41] and, as such, they have been regularly the target of TE insertions including those of hATm-NA6 and other MITEs reported here.
Unlike most other viral hosts, amoebae are usually infected by a variety of microorganisms through amoebal attack [42]. Free-living amoeba have been described as “melting pots” for genetic exchanges because of the stable coexistence of different parasites in amoeba [37, 38, 43, 44]. This condition increases possibilities HTs between the host and its parasites and among the different parasites within their amoebal host. In the present study, HTs of four MITEs have taken place between pandoraviruses and their amoebal host. To the best of our knowledge, this is the first convinced documented example of MITEs from giant viruses and their hosts being involved in HTs. Meanwhile, three addition HT cases of MITEs, probably from wasps to PDVs, were found. PDVs represent a highly unconventional group of DNA viruses whose genomes have become integrated into the genome of their wasp hosts. This condition will be propitious to introduce MITEs from wasps to PDVs. The above results also provide evidence supporting that viruses could be served as vectors for transposon HTs. In contrast, the mechanisms of most HTTs have been lacking, although nearly three thousand cases of HTTs have been reported in organisms [45, 46].
Although the viral genomes, especially pandoraviruses, were found to be rich in MITEs, these TEs do not have the capacity to mediate their own transposition. From this observation, one question arises: how did MITEs amplify and colonize viral genomes. In this study, autonomous partners (Submariner, hAT-5 and hATm-6) of seven MITEs were detected in viral hosts. Sequence similarity between MITEs and these autonomous elements are largely limited to their termini or TIRs (Fig. 2), which are usually the only requirement for transposase recognition of DNA transposons during the process of transposition1. This result implies that Submariner, hAT-5 and hATm-6 may be responsible for transposition of the MITEs we found in DNA viruses. Because Submariner, hAT-5 and hATm-6 appear to be still active in viral hosts, the stage is set for MITEs found in viruses to be amplified again in the future.
Conclusions
Our discovery highlights that TEs contribute to shape genome evolution of pandoraviruses. We concluded that as for cellular organisms, TEs are part of the pandoraviruses’ diverse mobilome.
Methods
Identification and copy number determination of MITEs in viral genomes
We downloaded the latest publicly available genome assemblies of 5170 viruses from viruSITE [20] as of May 2016. MITEs from the above viral genomes were characterized de novo using MITE-hunter with default parameters [21]. Multiple sequence alignment (MSA) files generated by MITE-hunter were manually analyzed for boundaries and TIRs and TSD of potential TEs. Based on the characteristics of MITEs [3], TEs that meet the following criteria are considered as a false-positive: (1) Length of TEs longer than 800 bp; (2) TEs with no TIRs or TSD; (3) Compound TEs. The consensus sequences of candidate MITEs were reconstructed using DAMBE with default parameters [47], and were served as a custom repeat library to pass through a local version of RepeatMasker (http://www.repeatmasker.org) using the search engine RMBlast to calculate the MITE copy number. Fragments shorter than 80 bp were manually excluded. The secondary structure of MITEs was predicted using mFold [48]. Pairwise distance between each copy and the consensus sequence was calculated using MEGA 4 (Pairwise deletion, Maximum composite likelihood model) [49]. Paralogous empty sites of candidate MITEs were found using the method previously described [17].
Identification and characterization of candidate MITE-related sequences and putative autonomous partners in their potential host genomes
Based on similar TIRs and TSD with those of known DNA transposons [3], ten of candidate MITEs identified in viruses could be classified into the superfamilies. We noticed that these ten MITEs resided in viruses, which infect insects or amoebas. Therefore, we looked for these MITE-related sequences in the viral hosts. We used the consensus sequences or representatives of these ten MITEs as queries to BLASTn against the publicly available genome sequences of 227 insects and 15 amoebas as well as the National Center for Biotechnology Information (NCBI) Nucleotide collection database, and all obtained hits were manually checked. If MITEs found in one species were present in less than five full-length copies, which make us difficult to reconstruct reliable consensus sequences, the best match of the queries was used to calculate its copy number and construct multiple alignments together with MITEs from other species. To test for HTs, MITEs were considered to be present in one viral host if one transposon showed ≥ 60% similarity to the consensus sequences at the nucleotide level and over ≥ 60% of their length. To identify their potential autonomous partners, all significant hits ranging from 1000 bp to 10-kb bearing some similar sequences with those of eight MITEs were retrieved from viral and their host genomes. These longer elements were characterized using the following four methods: (1) TIRs were predicted using fastPCR [50]; (2) Open reading frame was analyzed using FGENESH [51] or GENSCAN (http://genes.mit.edu/GENSCAN.html); (3) The NCBI Conserved Domain Database (CDD) was employed to identify their putative functional domains; (4) Their nucleotide or protein sequences were used as queries to search against Repbase [22].
Nomenclature
In this study, both several MITEs and their autonomous elements were found in the viral hosts. To avoid confusion, each MITE and the corresponding autonomous element were named X-NA# (where X represents a superfamily, NA represents nonautonomous elements, # is a letter representing the corresponding numbers of transposons) and X-#, respectively (Additional file 1: Table S1). We also noted that both a MITE identified in P. salinus and its autonomous element found in its cellular host were named Submariner_PI and Submariner_Ac1/Submariner_Ac2, respectively [6]. We have now found this MITE and the autonomous element were widespread in viruses and their hosts (Additional file 1: Table S1). For these reasons, we decide to introduce the name Submariner-NA for this MITE and Submariner for its corresponding autonomous element.
Sequence analysis
To identify hATm-like elements in other species, we carried out BLASTp and tBLASTn searches using the predicted hATm transposase sequence from the blood sucking bug R. prolixus as a query against the NCBI Non-redundant protein and Nucleotide collection database, respectively. All significant hits with an identity ≥ 30% and coverage ≥ 60% with an e-value ≤ 5 × 10− 20 were obtained [25]. The detail information is shown in Additional file 12: Table S5 and Additional file 13: Table S6. Copies of all retrieved sequences were extracted with 5000 bp flanking sequences from their host genomes to determine the boundary as well as TSD. Sequence logos were created by WebLogo [52] using 30 nt (15 nt upstream and 15 nt downstream) of their flanking sequences including TSD.
Transposase amino acid sequences from 102 hAT elements have been aligned using T-Coffee [53], with expresso mode (structural alignment). Ambiguous/uninformative alignment positions were, then, removed through Noisy v. 1.5 (Dress et al., 2008), with cutoff value = 0.8 and distance = GTR. A final alignment of 2210 amino acids was used to perform phylogenetic analyses. The best-fit model of evolution was estimated through ProtTest v.3 [54] based on BIC score, and the LG model with gamma distribution (+G) was selected as best substitution model. Maximum Likelihood tree was obtained through RAxML v. 8 [55], using 500 rapid bootstrap replicates for node support.
To determine the relationship of MITEs found in viruses and their hosts, multiple alignments of full-length copies of one MITE from one species were created using MUSCLE [56]. All ambiguous sites were removed, then a neighbor-joining tree was built using MEGA 4 [49] (Pairwise deletion, Maximum composite likelihood model, 1000 bootstrap replicates). Clade credibility values (> 70%) are shown at each node. Fifty full-length copies were randomly selected to include in this analysis for one MITE with more than fifty full-length copies in one species.
To assess misassemble (chimera) regions around MITEs, the PacBio reads that were used to assemble the P. salinus and P. dulcis genomes were mapped on the assembled genomes using BLASR [57]. The coverage was computed as the number of reads for each genomic position and was plotted for 2-kb fragments centered at the position of each MITE site.
Gene sequences of P. dulcis, P. salinus and P. inopinatum as well as a gff file for positions of predicted genes were downloaded from NCBI. Then, pairwise comparison of gene sequences from three pandoraviruses were performed using BLASTp [58], and gene synteny and collinearity were analyzed using MCScanX [59]. MITEs were mapped on the genomes of three pandoraviruses according to each genomic position of copies.
Polymerase chain reaction (PCR) and sequencing
To confirm the presence of MITEs and the potential autonomous element computationally identified in the viral genomes, we designed the PCR primers using the flanking sequences of these MITEs. Primers were listed in Additional file 14: Table S7. For P. inopinatum, genomic DNA was extracted using EZ1 DNA Tissue Kit (QIAGEN) through automated extraction system: EZ1 Advanced XL (QIAGEN). PCR amplification was performed in a 25-μl volume including12.5 μl AmpliTaq Gold 360 Master Mix, 5 μl Pandoravirus genomic DNA, 5.5 μl H2O, 1 μl forward primer and 1 μl reverse primer. PCR runs were carried out with an initial denaturation step for 15 min at 95 °C followed by 39 cycles of denaturation at 95 °C for 30 s, annealing at 58 °C for 30 s, and elongation at 72 °C for 1 min, ending with a 5 min elongation step at 72 °C. Products of PCR were assayed using agarose gel electrophoresis, and were sequenced directly. For GfIV, viral DNA extraction was carried out as described previously [60, 61] and 13 ng of DNA was used for PCR amplification (in 1× PCR buffer) supplemented with 0.33 mM each deoxynucleoside triphosphate (dNTP), 0.25 μM each primer, and 1 unit of Taq DNA polymerase in a 30 μl reaction volume. The amplification was performed for 35 cycles consisting of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min, with a final 5 min extension step at 72 °C. Five microliters per reaction was analyzed on a 1% agarose gel electrophoresis to confirm single amplicon before Sanger sequencing using the same primers used for amplification (Genome Sequencing and Genotyping platform of the CHUL, in Quebec City, Quebec, Canada). For CcBV, PCR reactions were performed on genomic DNA extracted from individuals using GoTaq (Promega, France) in a final volume of 25 μl containing 50 ng of male wasp genomic DNA, 25 U of GoTaq, 3 mM MgCl2 and 20 pmole of each specific primer with the following cycling conditions: 4 min of initial denaturation at 94 °C, followed by 35 cycles of denaturation at 94 °C for 40 s, primer hybridization at 58 °C for 40 s, extension at 72 °C for 60 s, and final extension at 72 °C for 10 min. PCR products were purified using adsorption columns and sequenced by Sanger technology by GATC Biotech (Germany) in both strands using the same forward and reverse primers as for PCR.
Abbreviations
- CDD:
-
Conserved Domain Database
- CMC :
-
Chapaev–Mirage–CACTA
- dNTP:
-
Deoxynucleoside triphosphate
- GfIV:
-
Glypta fumiferanae ichnovirus
- HTs:
-
Horizontal transfers
- MITEs:
-
Miniature inverted-repeat transposable elements
- MSA:
-
Multiple sequence alignment
- NCBI:
-
National Center for Biotechnology Information
- PCR:
-
Polymerase chain reaction
- PDV:
-
Polydnaviridae
- TEs:
-
Transposable elements
- TIRs:
-
Terminal inverted repeats
- TSD:
-
Target site duplication
References
Craig NL, Craigie R, Gellert M, Lambowitz AM. Mobile DNA II. Washington. DC: American Society for Microbiology Press; 2002.
Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–68.
Feschotte C, Zhang X, Wessler SR. Mobile DNA II. Washington. DC: American Society for Microbiology Press; 2002.
Bureau T, Ronald P, Wessler S. A computer-based systematic survey reveals the predominance of small inverted-repeat elements in wild-type rice genes. Proc Natl Acad Sci U S A. 1996;93:8524–9.
Chen J, Hu Q, Zhang Y, Lu C, Kuang H. P-MITE: a database for plant miniature inverted-repeat transposable elements. Nucleic Acids Res. 2014;42:D1176–81.
Sun C, Feschotte C, Wu Z, Mueller RL. DNA transposons have colonized the genome of the giant virus Pandoravirus salinus. BMC Biol. 2015;13:38.
Shen J, Liu J, Xie K, Xing F, Xiong F, Xiao J, Li X, Xiong L. Translational repression by a miniature inverted-repeat transposable element in the 3′ untranslated region. Nat Commun. 2017;3:14651.
Han MJ, Zhou QZ, Zhang HH, Tong X, Lu C, Zhang Z, Dai F. iMITEdb: the genome-wide landscape of miniature inverted-repeat transposable elements in insects. Database (Oxford). 2016.
Abrusan G, Krambeck HJ. Competition may determine the diversity of transposable elements. Theor Popul Biol. 2006;70:364–75.
Roberston HM. Mobile DNA II. Washington. DC: American Society for Microbiology Press; 2002.
Schaack S, Gilbert C, Feschotte C. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol. 2010;25:537–46.
Dupuy C, Periquet G, Serbielle C, Bézier A, Louis F, Drezen JM. Transfer of a chromosomal maverick to endogenous bracovirus in a parasitoid wasp. Genetica. 2011;139:489–96.
Fraser MJ, Smith GE, Summers MD. Acquisition of host cell DNA sequences by Baculoviruses: relationship between host DNA insertions and FP mutants of Autographa californica and galleria mellonella nuclear polyhedrosis viruses. J Virol. 1983;47:287–300.
Drezen JM, Bézier A, Lesobre J, Huguet E, Cattolico L, Periquet G, Dupuy C. The few virus-like genes of Cotesia congregata bracovirus. Arch Insect Biochem Physiol. 2006;61:110–22.
Jehle JA, Nickel A, Vlak JM, Backhaus H. TC14.7: a novel lepidopteran transposon found in Cydia pomonella granulosis virus. Virology. 1995;207:369–79.
Gilbert C, Chateigner A, Ernenwein L, Barbe V, Bézier A, Herniou EA, Cordaux R. Population genomics supports baculoviruses as vectors of horizontal transfer of insect transposons. Nat Commun. 2014;5:3348.
Marquez CP, Pritham EJ. Phantom, a new subclass of Mutator DNA transposons found in insect viruses and widely distributed in animals. Genetics. 2010;185:1507–17.
Piskurek O, Okada N. Poxviruses as possible vectors for horizontal transfer of retroposons from reptiles to mammals. Proc Natl Acad Sci U S A. 2007;104:12046–51.
Thomas J, Schaack S, Pritham EJ. Pervasive horizontal transfer of rolling-circle transposons among animals. Genome Biol Evol. 2010;2:656–64.
Stano M, Beke G, Klucar L. viruSITE-integrated database for viral genomics. Database (Oxford). 2016.
Han Y, Wessler SR. MITE-hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010;38:e199.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7.
Philippe N, Legendre M, Doutre G, Couté Y, Poirot O, Lescot M, Arslan D, Seltzer V, Bertaux L, Bruley C. Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science. 2013;341:281–6.
Arensburger P, Hice RH, Zhou L, Smith RC, Tom AC, Wright JA, Knapp J, O'Brochta DA, Craig NL, Atkinson PW. Phylogenetic and functional characterization of the hAT transposon superfamily. Genetics. 2011;188:45–57.
Rossato DO, Ludwig A, Deprá M, Loreto EL, Ruiz A, Valente VL. BuT2 is a member of the third major group of hAT transposons and is involved in horizontal transfer events in the genus drosophila. Genome Biol Evol. 2014;6:352–65.
Feschotte C, Mouches C. Evidence that a family of miniature inverted-repeat transposable elements (MITEs) from the Arabidopsis thaliana genome has arisen from a pogo-like DNA transposon. Mol Biol Evol. 2000;17:730–7.
Clark KJ, Carlson DF, Leaver MJ, Foster LK, Fahrenkrug SC. Passport, a native Tc1 transposon from flatfish, is functionally active in vertebrate cells. Nucleic Acids Res. 2009;37:1239–47.
Jiang SH, Li GY, Xiong XM. Novel miniature inverted-repeat transposable elements derived from novel CACTA transposons were discovered in the genome of the ant Camponotus floridanus. Genes Genom. 2016;38:1189–99.
Yuan YW, Wessler SR. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc Natl Acad Sci U S A. 2011;108:7884–9.
Djoumad A, Stoltz D, Béliveau C, Boyle B, Kuhn L, Cusson M. Ultrastructural and genomic characterization of a second banchine polydnavirus confirms the existence of shared features within this ichnovirus lineage. J Gen Virol. 2013;94:1888–95.
Bézier A, Annaheim M, Herbinière J, Wetterwald C, Gyapay G, Bernard-Samain S, Wincker P, Roditi I, Heller M, Belghazi M, et al. Polydnaviruses of braconid wasps derive from an ancestral nudivirus. Science. 2009;323:926–30.
Kawakami K. Tol2: A versatile gene transfer vector in vertebrates. Genome Biol. 2007;8(Suppl 1):S7.
Colson P, La Scola B, Levasseur A, Caetano-Anollés G, Raoult D. Mimivirus: leading the way in the discovery of giant viruses of amoebae. Nat Rev Microbiol. 2017;15:243–54.
López-Madrigal S, Latorre A, Porcar M, Moya A, Gil R. Complete genome sequence of “Candidatus Tremblaya princeps” strain PCVAL, an intriguing translational machine below the living-cell status. J Bacteriol. 2011;193:5587–8.
Rao Q, Wang S, Zhu DT, Wang XW, Liu SS. Draft genome sequence of rickettsia sp. strain MEAM1, isolated from the whitefly Bemisia tabaci. J Bacteriol. 2012;194:4741–2.
Filée J, Chandler M. Gene exchange and the origin of giant viruses. Intervirology. 2010;53:354–561.
Filée J, Siguier P, Chandler M. I am what I eat and I eat what I am: acquisition of bacterial genes by giant viruses. Trends Genet. 2007;23:10–5.
M B, Yutin N, Pagnier I, Barrassi L, Fournous G, Espinosa L, Robert C, Azza S, Sun S, Rossmann MG, et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc Natl Acad Sci U S A. 2009;106:21848–53.
Bézier A, Louis F, Jancek S, Periquet G, Thézé J, Gyapay G, Musset K, Lesobre J, Lenoble P, Dupuy C, et al. Functional endogenous viral elements in the genome of the parasitoid wasp Cotesia congregata: insights into the evolutionary dynamics of bracoviruses. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368:20130047.
Espagne E, Dupuy C, Huguet E, Cattolico L, Provost B, Martins N, Poirié M, Periquet G, Drezen JM. Genome sequence of a polydnavirus: insights into symbiotic virus evolution. Science. 2004;306:286–9.
Whitfield JB. Estimating the age of the polydnavirus/braconid wasp symbiosis. Proc Natl Acad Sci U S A. 2002;21:7508–13.
Greub G, Raoult D. Microorganisms resistant to free-living amoebae. Clin Microbiol Rev. 2004;17:413–33.
Moliner C, Fournier PE, Raoult D. Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol Rev. 2010;34:281–94.
Bertelli C, Greub G. Lateral gene exchanges shape the genomes of amoeba resisting microorganisms. Front Cell Infect Microbiol. 2012;2:110.
Dotto BR, Carvalho EL, Silva AF, Duarte Silva LF, Pinto PM, Ortiz MF, Wallau GL. HTT-DB: horizontally transferred transposable elements database. Bioinformatics. 2015;31:2915–7.
Peccoud J, Loiseau V, Cordaux R, Gilbert C. Massive horizontal transfer of transposable elements in insects. Proc Natl Acad Sci U S A. 2017;114:4721–6.
Xia X, Xie Z. DAMBE: software package for data analysis in molecular biology and evolution. J Hered. 2001;92:371–3.
Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15.
Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9.
Kalendar R, Lee D, Schulman AH. FastPCR software for PCR, in silico PCR. and oligonucleotide assembly and analysis Methods Mol Biol. 2014;1116:271–302.
Solovyev V, Kosarev P, Seledsov I, Vorobyev D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006;7(Suppl 1):S10.
Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2014;14(6):1188–90.
Notredame C, Higgins DG, Heringa J. 2000. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–17.
Darriba D, Taboada GL, Doallo R, Posada D. 2011.ProtTest 3: fast selection pf best-fit models of protein evolution. Bioinformatics. 2011;27:1164–5.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracyand high throughput. Nucleic Acids Res. 2004;32:1792–7.
Chaisson M, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): theory and application. BMC Bioinformatics. 2012;13:238.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49.
Krell PJ, Stoltz DB. Virus-like particles in the ovary of an ichneumonid wasp: purification and preliminary characterization. Virology. 1980;101:408–18.
Stoltz DB, Guzo D, Cook D. Studies on polydnavirus transmission. Virology. 1986;155:120–31.
Acknowledgments
We thank Cedric Feschotte and Clement Gilbert for his valuable suggestions to improve our manuscript. We thank Karine Musset for performing amplification and sequencing of bracovirus sequences. We also thank Gaelen R Burke for demonstrating that CMC-NA_1 identified in Microplitis mediator bracovirus is false-positive using PCR amplification and sequencing of PCR products.
Funding
This work was supported by the National Natural Science Foundation of China (31560308 and 31700318 to H.H.Z, 31471197 to Z.Z.), the Hi-Tech Research and Development 863 Program of China (2013AA102507 to F.Y.D.), the Natural Science Foundation of Jiangxi Province (20171BAB204016 to H.H.Z.) and State Key Laboratory of Silkworm Genome Biology (sklsgb161718–8 to H.H.Z).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Author’s contributions
HHZ, MJH, ZZ, XGZ and FYD designed and supervised the study. HHZ, QZZ, PLW, AL, SS, JMD, MC and CS helped with data collection and bioinformatic analyses. AL and CB performed the PCR and sequencing. HHZ, QZZ, PLW, XMX, AL, DR, CA, ML, JMD, MC, SHJ, HOB, TEB, PFC, MJH, ZZ, XGZ and FYD wrote and revised the manuscript. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Characteristics of ten MITEs and its autonomous elements identified in this study. (DOC 144 kb)
Additional file 2:
Table S2. Nucleotide sequences of twelve elements whose TSD could not be determined. (XLSX 11 kb)
Additional file 3:
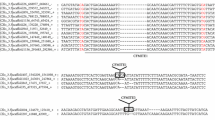
Figure S1. Characteristics of ten MITEs from the viral genomes. A Multiple alignments of full-length copies as well as the flanking sequences of each MITE. TSD is shown in red and the boundary is indicated using black shading. B One empty paralogous site of CMC-NA_1_GfIV. C Secondary structure of ten MITEs. D Length distribution of MITEs with more than 14 copies in one viral genome. E Nucleotide genetic distances between each MITE copy and the corresponding consensus sequences. Only MITEs with reliable consensus sequences in one virus were included in this analysis. Because more than 50% copies of hAT-NA5_PD were shorter than 50% length of the consensus sequence, and it was also excluded in this analysis. Species abbreviations of viruses were listed in Additional file 1: Table S1. (PDF 3703 kb)
Additional file 4:
Figure S2. Gene synteny and collinearity of three Pandoraviruses and distribution of six MITEs on its genomes. A Circle plot showing patterns of synteny and collinearity of three Pandoraviruses. MITEs on the genomes of three Pandoraviruses are shown using different colored lines. B One example of the present of one copy of MITEs in orthologous genomic site of P. salinus and P. dulcis but absent in that of P. inopinatum. C One example of the absence of one copy of MITEs orthologous genomic site of P. inopinatum and P. dulcis but present in that of P. salinus. (PDF 431 kb)
Additional file 5:
Figure S3. Phylogeny of three known families (Buster, Ac and Tip) of the hAT transposons with the hATm family and characteristics of TSD of two members of the hATm family. A Phylogenetic analysis of hAT transposases, performed through Maximum Likelihood method. Numbers at nodes represent bootstrap support after 500 replicates. Four members (hAT5-AC, hAT5_AP, hAT5_APo and hAT5_AQ) of the Ac family were found in this study. The detail information of the hATm transposons is listed in Additional file 1: Table S2. B TSD of hATm-1_CuQ and hATm-1_BeT. The results of sequence logos and empty paralogous site analysis indicated that hATm-1_CuQ and hATm-1_BeT created a 9-bp TSD upon insertion. C Multiple alignments of the 5′ and 3′ terminal sequences of 28 hATm transposons identified in this study. Because hATm transposons are generally flanked by long TIRs (e.g. TIRs of hATm-6_RP are 417 bp), and only 40 bp of both termini of these transposons were shown arrows. (PDF 1651 kb)
Additional file 6:
Table S3. Target site duplications of hATm transposons. (DOC 59 kb)
Additional file 7:
Figure S4. Determination of the boundary and TSD of seven MITEs identified in viral hosts or species related to their hosts. A Multiple alignments of full-length copies as well as the flanking sequences of each MITE. TSD is shown using red color and the boundary is indicated usingi black shading. B Empty paralogous sites of five MITEs identified in viral cellular hosts. (PDF 2219 kb)
Additional file 8:
Table S4. Pairwise comparison of nucleotide sequence identity of seven MITEs involved in HTs between viruses and their cellular hosts or species closely related to their hosts. (DOC 68 kb)
Additional file 9:
Figure S5. Multiple alignments of seven MITEs involved in HTs between viruses and its cellular hosts or species related to their hosts. (PDF 5206 kb)
Additional file 10:
Figure S6. Mapping MITEs on the P. salinus and P. dulcis genomes. The 2 kb fragments coverage centered on each MITE copy including hAT-NA1, hAT-NA2, hAT-NA3, hAT-NA4, hAT-NA5 and Submariner-NA for P. salinus (A) and hAT-NA1, hAT-NA2, hAT-NA3, hAT-NA4, hAT-NA5 for P. dulcis (B) The blue horizontal line corresponds to zero coverage, the red line is the mean coverage and the green line is the median. (PDF 376 kb)
Additional file 11:
Figure S7. Phylogenies of full-length copies of five MITEs involved in HTs between viruses and their hosts. (PDF 482 kb)
Additional file 12:
Table S5. Number of significant hits of hATm-6-RP retrieved using BlastP and TBlastN Tools in NCBI and the percent average identity find with the query. (DOC 44 kb)
Additional file 13:
Table S6. Detail information of significant hits of proteins related to hATm-6-RP retrieved using BlastP and TBlastN Tools in NCBI. (DOC 109 kb)
Additional file 14:
Table S7. The detail information of primers to amplify MITEs and their potential autonomous partner in viruses. (DOC 43 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, HH., Zhou, QZ., Wang, PL. et al. Unexpected invasion of miniature inverted-repeat transposable elements in viral genomes. Mobile DNA 9, 19 (2018). https://doi.org/10.1186/s13100-018-0125-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13100-018-0125-4