
Abstract
Background
While many women with rheumatoid arthritis (RA) improve during pregnancy and others worsen, there are no biomarkers to predict this improvement or worsening. In our unique RA pregnancy cohort that includes a pre-pregnancy baseline, we have examined pre-pregnancy gene co-expression networks to identify differences between women with RA who subsequently improve during pregnancy and those who worsen.
Methods
Blood samples were collected before pregnancy (T0) from 19 women with RA and 13 healthy women enrolled in our prospective pregnancy cohort. RA improvement/worsening between T0 and 3rd trimester was assessed by changes in the Clinical Disease Activity Index (CDAI). Pre-pregnancy expression profiles were examined by RNA sequencing and differential gene expression analysis. Weighted gene co-expression network analysis (WGCNA) was used to identify co-expression modules correlated with the improvement/worsening of RA during pregnancy and to assess their functional relevance.
Results
Of the 19 women with RA, 14 improved during pregnancy (RAimproved) while 5 worsened (RAworsened). At the T0 baseline, however, the mean CDAI was similar between the two groups. WGCNA identified one co-expression module related to B cell function that was significantly correlated with the worsening of RA during pregnancy and was significantly enriched in genes differentially expressed between the RAimproved and RAworsened groups. A neutrophil-related expression signature was also identified in the RAimproved group at the T0 baseline.
Conclusion
The pre-pregnancy gene expression signatures identified represent potential biomarkers to predict the subsequent improvement/worsening of RA during pregnancy, which has important implications for the personalized treatment of RA during pregnancy.
Similar content being viewed by others


Background
It is well established that rheumatoid arthritis (RA), though incurable, can improve naturally during pregnancy in a substantial proportion (50–75%) of women and that it may worsen or remain unchanged in others [1, 2]. Unfortunately, thus far, no biomarkers have been identified that can predict, at the pre-pregnancy stage, whether a woman with RA will subsequently improve or worsen during pregnancy. Consequently, women with RA who are considering a pregnancy and do not wish to take medications during their pregnancy, are often concerned about whether their disease will worsen during the pregnancy if they choose to stop their medications.
We had previously reported that, at the pre-pregnancy baseline in our unique prospective pregnancy cohort, women with RA who subsequently improved during pregnancy (RAimproved) and those who worsened (RAworsened) exhibited different RA-associated expression signatures when compared to healthy women [3]. We have now built on those differential gene expression findings in a larger sample of our cohort, using co-expression network analysis [4] as a systems-based approach to dissect the complexity of our pre-pregnancy transcriptome data. Thus, in this hypothesis-generating study, we aimed to build densely connected sub-networks (modules) of genes with highly correlated expression and to determine whether any of these modular repertoires were correlated with our trait of interest, namely the improvement/worsening of RA during pregnancy. We sought to assess whether such a module would be enriched in genes that exhibited significant differential expression between the RAimproved and RAworsened women. Furthermore, since genes co-expressed within a module tend to be functionally related and co-regulated [5], we performed functional analysis of modules to gain insight into the underlying biological differences between the RAimproved and RAworsened women at the pre-pregnancy stage.
Subjects and methods
Study subjects
Healthy women and women with RA of Danish descent who were planning a pregnancy were enrolled in our pregnancy cohort in Denmark and were prospectively followed, as previously described [6, 7]. A set of 19 women with RA and 13 healthy women from this cohort were included in the present study; these included the 9 RA and 5 healthy women from our previous study [3]. The study was approved by the Ethics Committee for Region Hovedstaden (Protocol #: H-2–2009-150) and the Danish Data Protection Agency (Data processing ID: RH-2015–02; record #: i-suite 03601) in Denmark, the Children’s Hospital Oakland Research Institute Institutional Review Board (IRB number: 2009–073), and the Northwestern University IRB (IRB number: STU00217093). All subjects provided written informed consent prior to enrollment.
Assessment of RA disease activity
RA disease activity was assessed using the Clinical Disease Activity Index (CDAI) [8], because it does not include acute phase reactants such as C-reactive protein (CRP) whose levels are known to fluctuate during pregnancy. The change in CDAI (ΔCDAI) from before pregnancy (T0) to the pregnancy time point where improvement/worsening was maximal (second trimester (T2) for 2 women who improved and third trimester (T3) for all others) was used to determine whether disease activity improved or worsened. Patients were categorized as having improved during pregnancy (RAimproved), if their ΔCDAI fit the criteria for a minimum clinically important difference (MCID) based on baseline (T0) disease activity; ΔCDAI values of 12, 6, and 1 were used as a threshold when disease activity at T0 was high, moderate, or low, respectively [9]. Those women with an increase in CDAI from T0 to T3, satisfying the MCID criteria for worsening of disease activity, were included in the “worsened” subset, referred to as RAworsened.
Sample collection, processing, and RNA sequencing
Only pre-pregnancy samples were used in the present study. Blood samples were drawn into PAXgene tubes and frozen. Total RNA was manually extracted using the PAXgene Blood RNA Kit according to the manufacturer’s protocol, and RNA integrity was assayed using a 2100 Bioanalyzer. 250 ng of total RNA were first depleted of ribosomal RNAs and globin mRNAs using the KAPA RiboErase kit (Roche) and KAPA globin depletion hybridizing oligos (Roche), respectively. Barcoded and stranded cDNA libraries prepared using the KAPA RNA HyperPrep kit were pooled and sequenced on an Illumina NovaSeq 6000 instrument, targeting an average of 100 million 150 bp paired-end reads.
Bioinformatics analysis
Rigorous quality control (QC) of the raw data was performed using FASTQC, Picard, and HTSTREAM. Raw FASTQ reads were trimmed using Cutadapt (v2.4) and aligned to the human genome (GRCh38; Ensembl v98) using HISAT2 (v 2.1.0). Multi-mapped reads were filtered using Samtools. Aligned reads were assembled into transcripts and merged using StringTie (v 2.1.1).
Novel lncRNAs in our assembled transcripts were assessed by removing transcripts that (1) overlapped with any known transcript on the same strand (Bedtools v 2.28.0); (2) had open reading frames (ORFs) > 100 amino acids (TransDecoder v 5.5.0); (3) when translated, had similarity to known proteins/protein domains [blastx hits to the RefSeq Protein or Pfam databases] (Blast + v 2.7.1, flags: -strand plus -max_target_seqs 1 -evalue 1E−5); (4) classified as coding by at least one of 3 tools for detecting coding potential [CPAT (v 3.0.2), CPC (v 2.0), and FEELnc (v 0.2)]; and (5) were single-exon. The remaining transcripts were classified as “novel lncRNAs” and appended to the Ensembl v98 gtf file. The resulting annotation file (gtf) containing all known and newly discovered (from our data) transcripts was used as a reference to obtain gene-level counts for all known genes and lncRNAs as well as novel lncRNAs with featureCounts (v 2.0.0, flags: -s 2 -p).
Raw counts were loaded into R, and “rRNA” and “pseudogene” gene types were removed, along with gene types “misc_RNA,” “Mt_tRNA,” “scaRNA,” “snRNA,” “snoRNA,” and “TEC.” Low-expression genes were removed by keeping only genes with CPM > 10/L in 6 or more samples, where L is the minimum library size in millions. Library size was normalized in edgeR (v 3.30.3) with the trimmed mean of M-values (TMM) method using the calcNormFactors function and normalized counts were exported for downstream statistical analyses.
Deconvolution of bulk RNA-seq data
To estimate cell type proportions in each sample, raw reads were aligned to the Ensembl v98 transcriptome using kallisto (v 0.46.1) and aggregated to gene-level data using tximport (v 1.18) in R. Gene-level data for each sample were deconvolved using CIBERSORTx and the accompanying LM22 signature matrix that is based on 22 human immune cell types [10]. We used principal components analysis (PCA) to condense the information about changes in all 22 estimated cell type proportions into principal components (PCs) as proposed by Kong et. al [11] and tested them for association with gene expression.
Differential gene expression analysis
To compare normalized T0 gene-level counts between the groups (RAimproved vs RAworsened; RAimproved or RAworsened vs healthy), differential gene expression analysis was performed using generalized linear model (GLM) likelihood ratio tests and the contrast argument of the glmLRT function in edgeR. Correction for multiple testing was performed using the false discovery rate (FDR) method. An FDR value threshold of 0.05, in combination with a fold change (FC) of at least 1.5, was used to assess significance.
Co-expression network analysis
Co-expression analysis of normalized gene-level counts from the RAimproved, RAworsened, and healthy women was performed in R using the weighted gene co-expression network analysis (WGCNA) package (v1.69) [12]. The following specifications were used: power = 5, networkType = signedHybrid, corType = bicor, maxPOutliers = 0.1, and mergeCutHeight = 0.25. Each module was examined for (i) correlation with the clinical trait of improvement/worsening during pregnancy, (ii) the presence of genes that were differentially expressed (DE) between the RAimproved and RAworsened groups at T0 and enrichment of DE genes, and (iii) hub genes, by selecting the top 10% genes based on network adjacency. Furthermore, since genes co-expressed within a module tend to be co-regulated and functionally related, functional analysis of the modules was performed to gain insight into the potential functions of the genes and lncRNAs being co-expressed within specific modules. Enrichment of genes differentially expressed between the RAimproved and RAworsened groups within co-expression modules was assessed by a hypergeometric test. Protein–protein interaction (PPI) among the hub genes was examined using the Search Tool for the Retrieval of Interacting Genes (STRING) database [13].
Functional enrichment
Enrichment of Gene Ontology (GO) terms and KEGG and Reactome pathways were assessed using WebgestaltR [14]. Interactions between proteins encoded by the significant genes were based on data from the STRING database [15] and visualized in Cytoscape (v3.8.1) [16].
Transcription factor analysis
Enrichment of transcription factor targets was performed using the fora function in the fgsea R package [17]. Transcription factor-target regulons were pulled from the DoRothEA database, using confidence levels A, B, and C [18].
Identification of B cell-related genes
Expression in B cells was based on expression patterns reported in The Human Protein Atlas [19] and the Immunological Genome (ImmGen) Project [20], as well as from a previous report of an 85-gene B cell signature identified from freshly isolated B cells [21].
Results
Study subjects
Of the 19 women with RA, 14 subsequently improved during pregnancy (RAimproved) while 5 worsened (RAworsened), based on MCID thresholds. Nevertheless, at the pre-pregnancy (T0) baseline, the mean CDAI was similar between the two groups [RAimproved (mean ± S.D.): 16.8 ± 11.5; RAworsened: 16.9 ± 7.6, p = 0.9] (Fig. 1). Other patient characteristics at the T0 baseline were also comparable between the two RA groups, including RA duration [RAimproved: 7.6 ± 6.8 years; RAworsened: 8.5 ± 4.4 years, p = 0.4], and age at the T0 visit [RAimproved: 31.0 ± 5.1 years; RAworsened: 33.2 ± 3.3 years, p = 0.4]. Characteristics of the healthy women have been described elsewhere [22]; one woman (of 14) was excluded due to missing pre-pregnancy data. The mean age of the healthy women at the T0 visit was 28.7 ± 3.7 years. The medications that the women with RA were taking at or before the T0 time point are summarized in Table 1.

RA disease activity before and during pregnancy. At the pre-pregnancy (T0) baseline, the mean disease activity scores (CDAI) were similar between the 14 women with RA who subsequently improved during pregnancy (RAimproved) and the 5 women who worsened (RAworsened)
Data QC
After rigorous QC, the gene expression (RNA-seq) data were visualized on a PCA plot (Additional file 1: Fig. S1). There was a small degree of overlap between the RAimproved and RAworsened clusters, and the healthy women cluster was mostly within that area of overlap.
Differences in cell type proportions between the 2 RA groups at T0
Deconvolution of the RNA-seq data using CIBERSORTx produced relative cell type proportions, limited to the 22 immune cell types present in the LM22 reference panel [10]. No significant differences in these cell type proportions were observed between the RAimproved and RAworsened groups at T0 (Additional file 2: Fig. S2).
Differential gene expression
At T0, 448 protein-coding genes and 137 lncRNAs were differentially expressed (FDR < 0.05, fold change [FC] ≥ 1.5) between the RAimproved and RAworsened women (Fig. 2A). The protein-coding genes were enriched among GO terms such as myeloid leukocyte migration (FDR = 0.003), innate immune response (FDR = 0.003), regulation of leukocyte chemotaxis (FDR = 0.01), and B cell proliferation (FDR = 0.01). Furthermore, some neutrophil-related genes (SERPINB10, CAMP, CXCL6, MMP9, PADI4, NECAB2) were over-expressed (FC: 1.9–3.4) among the RAimproved women. Numerous B cell-related genes (such as CD19, CD22, CD40, CD72, CD79A, BLNK, IL7, MS4A1, PAX5, BLK, and several immunoglobulin heavy chain genes) were over-expressed (FC: 1.5–4.1) among the RAworsened women. The differential expression output for all genes analyzed (n = 19,468) is provided in Additional file 3: Table S1.

RA gene expression signatures at the pre-pregnancy baseline. A A total of 448 protein-coding genes and 137 lncRNAs were differentially expressed (FDR < 0.05, fold change [FC] ≥ 1.5) between the RAimproved and RAworsened women at the pre-pregnancy baseline, as shown in the volcano plot. B When T0 expression profiles of each of the RA groups were compared to that of the same 13 healthy women, the RA-associated gene expression signatures observed showed little overlap with each other. Only 42 coding genes, 24 lncRNAs, and 1 miRNA were common to both “RA signatures”
When each RA group was compared to the healthy women [RAimproved (or RAworsened) vs healthy], the genes with RA-associated expression (RAimproved: 545 coding genes, 276 lncRNAs, and 16 miRNAs; RAworsened: 160 coding genes, 73 lncRNAs, and 1 miRNA; FDR < 0.05, FC ≥ 1.5) were mostly specific to each group, with little overlap between them (42 coding genes and 24 lncRNAs, 1 miRNA) (Fig. 2B). As was observed in the results of the RAimproved vs RAworsened analysis, an over-expressed neutrophil signature (FC: 1.5–5.6) was apparent in the RAimproved group (vs healthy), enriched in genes involved in inflammatory response (FDR = 1.6E−11), neutrophil-mediated immunity (FDR = 2.7E−10), and myeloid cell activation involved in immune response (FDR = 2.7E−10), among others. On the other hand, the differentially expressed genes in the RAworsened group included an over-expressed B cell signature (FC: 1.5–2.4), although, overall, the differentially expressed genes were not enriched within any specific GO terms.
Co-expression network analysis
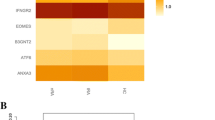
WGCNA identified 27 modules or sub-networks of coding genes and lncRNAs with tightly correlated intra-module expression (Fig. 3). Of these, 3 modules (midnightblue, light yellow, and salmon) were significantly enriched in genes that were differentially expressed between the RAimproved and RAworsened groups, and the eigengene expression patterns within these modules were also significantly correlated with the clinical trait of interest, i.e., subsequent improvement or worsening during pregnancy (midnightblue: r = 0.65; p = 5E−05; light yellow: r = − 0.45, p = 0.01; salmon: r = 0.38, p = 0.03).

Co-expression of protein-coding genes and long non-coding RNAs within functional modules identified by WGCNA at the pre-pregnancy baseline. Using the gene-level counts from the RAimproved, RAworsened and healthy women at the pre-pregnancy baseline, weighted gene co-expression network analysis (WGCNA) identified 27 modules of coding genes and lncRNAs with highly correlated intra-module expression. The different modules (with color labels) are shown on the left. For each module, the five columns to the right indicate, in respective order, (1) the correlation between module eigengene expression and subsequent improvement/worsening during pregnancy (RA group), (2) total number of genes (among all genes analyzed) that clustered within the module, (3) the number of genes (coding/lncRNAs) differentially expressed between the two RA groups that are co-expressed within the module, (4) fold enrichment of these differentially expressed genes within the module, and (5) the FDR value for the enrichment analysis. The midnightblue, light yellow, and salmon modules were significantly enriched in genes differentially expressed between the RAimproved and RAworsened groups, and their eigengene expression patterns were significantly correlated with subsequent improvement or worsening of RA during pregnancy. The grey module represents genes that were not co-expressed and were not assigned to any of the co-expression modules
The midnightblue module
The midnightblue module consisted of 173 members that were tightly co-expressed, 107 (62%) of which (84 protein-coding genes, 21 lncRNAs, and 2 miRNAs) were differentially expressed between the RAimproved and RAworsened groups. Thus, there was a significant enrichment (2^4.4, i.e., 21-fold, FDR = 1.7E−118) of the differentially expressed genes within this module (Fig. 3). The 84 protein-coding genes included numerous B cell-related genes (such as BLK, BLNK, CD19, CD22, CD72, CD79A, CD79B, CD180, CXCR5, FCRLA, FCRL1, FCRL2, LARGE1, MS4A1, PAX5, TNFRSF13B, TNFRSF13C, and some immunoglobulin heavy chain genes), all of which were over-expressed in RAworsened by 1.5- to 2.8-fold; the 21 lncRNAs included FAM30A and COPDA1, both of which have been implicated in B cell regulation [both over-expressed in RAworsened by 1.7- and 3.7-fold, respectively]; the 2 miRNAs, MIR4537 and MIR4539, were 1.6- and 1.7-fold over-expressed in RAworsened group, respectively.
Within a WGCNA module, those genes with the highest degree of network connections—also known as hub genes—tend to have more biological relevance. In the midnightblue module, we identified 18 hub genes (17 protein-coding and 1 lncRNA) (Fig. 4A). Data on protein–protein interactions (PPI) from the STRING database showed that most of the protein-coding hub genes (14 of 18) encoded proteins that interacted with one another functionally (Fig. 4B). Furthermore, because genes clustered within a module are strongly co-expressed, the modules are often related to biological function. Functional enrichment analysis revealed that GO terms related to B cell function were enriched in genes co-expressed in the midnightblue module as well as in hub genes (Fig. 5). Interestingly, a significant correlation was observed between the midnightblue module eigengenes and our estimated proportions of naïve B cells in each of the RAimproved, RAworsened, and healthy women at T0, with expression increasing as naïve B cell proportions increased (r = 0.8, FDR = 3E−07).

Midnightblue module hub genes. A A total of 18 hub genes were identified within the midnightblue module, by selecting the top 10% of genes having the highest degree of intra-modular connectivity. These included 17 protein-coding genes (circles) and 1 lncRNA (diamond). All of the hub genes, except for TSPAN3, were significantly over-expressed (FC: 1.5–2.0) among the RAworsened women, compared to the RAimproved. B Data on protein–protein interactions (PPI) from the STRING database showed that most of the protein-coding hub genes (14 of 18) encoded proteins that interacted with one another functionally

Gene Ontology (GO) terms enriched in genes that were co-expressed within the midnightblue module. Genes that were co-expressed within the midnightblue module were significantly enriched in GO terms related to B cell function
The salmon, light yellow, and blue modules
Of the 221 members of the salmon module, 50 were differentially expressed between the RAimproved and RAworsened groups. These included CD40, IFI35, PARP10/12/14, SP140, STAT2, TAP2, and TLR7, among others. Genes in this module were functionally involved in GO terms such as innate immune response (FDR < 2.2E − 16) and type I interferon signaling pathway (FDR < 2.2E − 16). Of the 125 members of the light yellow module, 25 were differentially expressed between the RAimproved and RAworsened groups. However, the light yellow module overall was not enriched in any specific GO terms. Additionally, while numerous neutrophil-related genes differentially expressed between the RAimproved and RAworsened groups were co-expressed within the blue module, and the blue module eigengenes were under-expressed in the RAworsened women, this module was not significantly associated with improvement or worsening of RA during pregnancy.
Transcription factor analysis
Transcription factor target enrichment analysis revealed that the genes that were co-expressed within the midnight blue module were significantly enriched among the target genes of the transcription factors PAX5, RFX5, GATA3, MEF2B, and RUNX3 (Additional file 4: Table S2). The genes that were co-expressed within the salmon module were significantly enriched among the target genes of the transcription factors STAT2, STAT1, IRF1, IRF2, IRF9, and SPIB.
Discussion
In the present study, we used co-expression network analysis as a novel approach to examine pre-pregnancy transcriptomes from women with RA who subsequently improved or worsened during pregnancy in order to gain insight into the underlying biological differences. We show that, in our cohort of Danish women, a sub-network of highly co-expressed B cell-related genes was significantly correlated with the worsening of RA during pregnancy, while a sub-network of neutrophil-related genes was correlated with the improvement of RA. These findings are novel. There are no previous studies that have examined pre-pregnancy gene expression profiles in relation to subsequent pregnancy-induced improvement or worsening of RA, other than our previous report of differing RA-associated differential expression signatures between the two groups [3].
Although numerous genes were significantly differentially expressed between the RAimproved and RAworsened women at T0, these findings relate to each gene independently of other genes, overlooking the fact that genes interact in complex biological networks. Co-expression network analysis, on the other hand, characterizes the correlation in the expression patterns among genes across samples in a dataset, clustering genes that are highly co-expressed within “modules.” In our data, of the 27 co-expression modules identified by WGCNA, only three (midnightblue, salmon, and light yellow) were both significantly enriched in genes that were differentially expressed between the RAimproved and RAworsened women and were significantly correlated with our trait of interest, i.e., improvement/worsening during pregnancy. Furthermore, co-expressed genes within a module tend to be functionally related. Thus, the midnightblue and salmon modules were associated with GO terms related to B cell function and type I interferon signaling pathway, respectively, while the light yellow module was not functionally related to any specific GO term. In the midnightblue module, the large proportion of B cell-related genes (at least 44) among the differentially expressed protein-coding genes (n = 84) was striking. These B cell signature genes were involved in various B cell functions, such as antigen processing and presentation [HLA-DOA, HLA-DOB] [23], B cell receptor signaling [BANK1 [24], BLK [25], BLNK [26], CD19 [27], CD22 [28], CD72 [29], CD79A, CD79B [30], MS4A1 [31], NIBAN3 (also known as BCNP1) [32]], Fc receptors [FCRLA, FCRL1, FCRL2], transcription factor [E2F5 [33], EBF1 [34], LINC00926 [35], PAX5 [36], POU2AF1 [37], SPIB [38]], and other B cell functions including development, differentiation, migration, and others [COBLL1, CXCR5, FAM30A, TNFRSF13B]. Additionally, there were numerous genes of unknown function [e.g., AFF3, CORO2B, GNG7, OSBPL10, P2RX5, and SYNPO] that are known to be expressed in B cells based on expression data from The Human Protein Atlas [19] or from the validated B cell signature reported by Henning et. al. [21]. Furthermore, all of the midnightblue hub genes, which represent the genes with greater biological relevance within the module, have been involved in B cell function, including lncRNA LINC00926. Comparisons to healthy women revealed that the B cell signature was specific to the RAworsened women. Thus, at the pre-pregnancy stage, the two groups of RA women differed significantly from each other in terms of B cell function.
We do not know at this stage if the observed pre-pregnancy B cell signature was due to the differences in the proportions of specific B cell sub-populations between the RAimproved and RAworsened groups. Naïve B cells, memory B cells, and plasma cells were the only B cell sub-populations for which we could estimate cell proportions, and although naïve and memory B cell proportions were higher among the RAworsened (vs RAimproved) women, these differences were not statistically significant. Nevertheless, we found the estimated proportions of naïve B cells to be correlated with the midnightblue module eigengene expression, which suggests that naïve B cell proportions were contributing to the eigengene expression levels, which in turn were correlated with the worsening of RA during pregnancy. The involvement of naïve B cells in RA is supported by previous observations that activated autoreactive naïve B cells were present in the circulation of RA patients [39] and that naïve B cells were activated prior to an RA flare [40]. Hence, we speculate that the naïve B cells in the RAworsened group may be autoreactive and that their proportion in the circulation may be a contributing factor in the worsening of RA during pregnancy.
Since co-expressed genes within a module tend to be co-regulated, another interesting finding was that 23 differentially expressed lncRNAs were co-expressed with the protein-coding genes within the midnightblue module, one of which was even identified as a hub gene. There is increasing evidence that lncRNAs may function as epigenetic regulators of immune-related gene expression in general and in RA [41, 42]. Co-expression analysis has previously been used to infer the function of some lncRNAs [43, 44], based on the premise that they would be functionally related to the protein-coding genes that they are co-expressed with (guilt-by-association), and whose expression they could potentially be regulating. In fact, some of the lncRNAs co-expressed in the midnightblue module have been previously associated with B cell function, and their increased expression have been associated with pro-inflammatory states. For example, hub gene LINC00926 has been implicated in regulating CD22 expression during B cell differentiation into plasma cells [35]. LINC00926 was co-expressed with TNFRSF13C and CD19 in breast cancer [45], as we observed in our data. In post-traumatic stress disorder (PTSD), increased expression of LINC00926 resulted in increased expression of pro-inflammatory cytokines [46]. Similarly, in a study of blood transcriptomes from vaccine cohorts, increased expression of lncRNA FAM30A in B cells was correlated with the expression of immunoglobulin (Ig) genes within the Ig heavy chain gene cluster on chromosome 14 (14q32.33) where FAM30A maps [47]. This supports our own observations that FAM30A was over-expressed (1.7-fold) in RAworsened women, as were the immunoglobulin genes (1.6–1.9-fold). COPDA1, another lncRNA co-expressed in the midnightblue module, has been shown to upregulate the expression of MS4A1 [48], which encodes the B cell differentiation antigen CD20 [49]. In our data, lncRNA COPDA1 and MS4A1 were 3.7- and 1.8-fold over-expressed, respectively, in RAworsened women. However, while anti-CD20 therapy (such as rituximab) has been used successfully to treat RA [50], it is not clear why CD20 expression was significantly higher in the RAworsened group at the pre-pregnancy stage, when disease activity was similar to that in the RAimproved group. The co-expression of lncRNAs that have already been implicated in the regulation of expression of B cell-related genes, such as LINC00926, FAM30A, and COPDA, within the midnightblue module, and the enrichment of genes with midnightblue module membership among target genes for transcription factors such as PAX5, RFX5, MEF2B, RUNX3, and GATA3 suggest that at least a portion of the differentially expressed B cell-related genes are under transcriptional regulation. There is also a possibility that, for some genes, the differential expression could be due to a combination of differences in cell type proportions and transcriptional regulation.
The neutrophil signature observed among the RAimproved women at the T0 baseline was not as prominent as the B cell signature in the RAworsened group. There was also no specific co-expression module in which the genes from the neutrophil signature were enriched, although some were co-expressed in the blue module. However, the expression patterns of genes in this module were not correlated with improvement or worsening during pregnancy.
The strengths of our study are as follows: Disease activity measures documented before and during pregnancy enabled us to assess clinical improvement or worsening of RA, based on MCID thresholds [9]. To our knowledge, this is the only pregnancy cohort that has clinical data from the same women both at a pre-pregnancy time point and during pregnancy, as well as biological samples available for gene expression assays. Furthermore, the homogeneous Danish background of the women in our cohort is advantageous in terms of minimizing heterogeneity in gene expression due to ethnicity. Using RNA-seq data from total RNA to evaluate gene expression enabled us to examine lncRNAs alongside the protein-coding RNAs. Our study also has limitations. First, sample sizes were small, especially for the RAworsened group. Nonetheless, given the difficulties associated with identifying women with RA at the pre-pregnancy stage and given that there are no other such pre-pregnancy data available from women with RA, these pre-pregnancy gene expression data presented are unique and provide a valuable contribution to the field. Nonetheless, these findings need to be validated in an independent cohort of larger sample size. Second, because total RNA from the whole blood was used, the expression profiles of neutrophils could have dominated the observed expression patterns. Yet, although a neutrophil signature was observed among the RAimproved women, the sensitivity of RNA-seq technology enabled us to detect transcripts that were not neutrophil-specific, including a prominent B cell signature. Third, technical bias and/or batch effects could have been introduced in the data. However, we randomized sample order prior to sample processing, used a block design for sequencing, and at the data processing step, we used sample replicates to ensure that there were no batch effects. Fourth, we did not adjust for dosage and/or specific medications that may have an effect on the immune system due to the lack of variation in medication use within each of the RA groups. Additionally, although there is a possibility that changes in medications may have contributed to improvement or worsening during pregnancy, that is not entirely clear given that two women who discontinued Infliximab and all medication, respectively, improved during pregnancy, while another woman worsened despite maintaining biologic therapy throughout pregnancy.
Conclusions
In our Danish pregnancy cohort, differential gene expression analysis showed little overlap in RA-associated pre-pregnancy (T0) gene expression signatures between the RAimproved and RAworsened women, suggesting that the two groups of women differ significantly in their T0 expression profiles. Using co-expression gene network analysis as a system-based approach to build on the differential expression data, we identified a co-expression module related to B cell function that was correlated with subsequent worsening of RA during pregnancy. This module was also significantly enriched in a set of B cell-related genes that were differentially expressed between the RAimproved and RAworsened groups at T0. Additionally, several neutrophil-related genes were significantly over-expressed among the RAimproved women at the T0 baseline. These pre-pregnancy expression signatures associated with the subsequent improvement or worsening of RA during pregnancy represent potential predictive biomarkers that could have important implications in terms of a personalized approach to the treatment of RA during pregnancy.
Availability of data and materials
The datasets presented in this article are not readily available because the data and materials are protected by the General Data Protection Regulation (GDPR) of the European Union (2016/679) and by the Danish Data Protection Act enacted in May 2018 to supplement the GDPR. However, the authors commit to making the relevant anonymized expression level data available on reasonable request. Such requests should be directed to the corresponding author, Dr. Damini Jawaheer.
Abbreviations
- CDAI:
-
Clinical Disease Activity Index
- CRP:
-
C-reactive protein
- FC:
-
Fold change
- FDR:
-
False discovery rate
- GLM:
-
Generalized linear model
- GO:
-
Gene Ontology
- lncRNAs:
-
Long non-coding RNAs
- MCID:
-
Minimum clinically important difference
- PC:
-
Principal components
- PCA:
-
Principal component analysis
- PPI:
-
Protein-protein interaction
- RNA-seq :
-
RNA sequencing
- RA:
-
Rheumatoid arthritis
- RAimproved :
-
Women with RA who subsequently improved during pregnancy
- RAworsened :
-
Women with RA who subsequently worsened during pregnancy
- STRING:
-
Search Tool for the Retrieval of Interacting Genes
- T0:
-
Pre-pregnancy
- T3:
-
Third trimester
- TF:
-
Transcription factor
- TMM:
-
Trimmed mean of M-values
- WGCNA:
-
Weighted gene co-expression network analysis
References
Ostensen M, Nelson JL. Bits and pieces in a puzzle–rheumatoid arthritis and pregnancy. Br J Rheumatol. 1995;34(1):1–3.
de Man YA, Dolhain RJ, van de Geijn FE, Willemsen SP, Hazes JM. Disease activity of rheumatoid arthritis during pregnancy: results from a nationwide prospective study. Arthritis Rheum. 2008;59(9):1241–8.
Pathi A, Wright M, Smed MK, Nelson JL, Olsen J, Hetland ML, et al. The rheumatoid arthritis gene expression signature among women who improve or worsen during pregnancy: a pilot study. J Rheumatol. 2021;48(7):985–91.
van Dam S, Vosa U, van der Graaf A, Franke L, de Magalhaes JP. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform. 2018;19(4):575–92.
Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P. Coexpression analysis of human genes across many microarray data sets. Genome Res. 2004;14(6):1085–94.
Mittal A, Pachter L, Nelson JL, Kjaergaard H, Smed MK, Gildengorin VL, et al. Pregnancy-induced changes in systemic gene expression among healthy women and women with rheumatoid arthritis. PLoS ONE. 2015;10(12): e0145204.
Goin DE, Smed MK, Pachter L, Purdom E, Nelson JL, Kjaergaard H, et al. Pregnancy-induced gene expression changes in vivo among women with rheumatoid arthritis: a pilot study. Arthritis Res Ther. 2017;19(1):104.
Mierau M, Schoels M, Gonda G, Fuchs J, Aletaha D, Smolen JS. Assessing remission in clinical practice. Rheumatology (Oxford). 2007;46(6):975–9.
Curtis JR, Yang S, Chen L, Pope JE, Keystone EC, Haraoui B, et al. Determining the minimally important difference in the clinical disease activity index for improvement and worsening in early rheumatoid arthritis patients. Arthritis Care Res (Hoboken). 2015;67(10):1345–53.
Steen CB, Liu CL, Alizadeh AA, Newman AM. Profiling cell type abundance and expression in bulk tissues with CIBERSORTx. Methods Mol Biol. 2020;2117:135–57.
Kong Y, Rastogi D, Seoighe C, Greally JM, Suzuki M. Insights from deconvolution of cell subtype proportions enhance the interpretation of functional genomic data. PLoS ONE. 2019;14(4): e0215987.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, et al. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33(Database issue):D433-7.
Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47(W1):W199–205.
Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Database issue):D412-6.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 060012. https://doi.org/10.1101/060012.
Garcia-Alonso L, Holland CH, Ibrahim MM, Turei D, Saez-Rodriguez J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019;29(8):1363–75.
Uhlen M, Karlsson MJ, Zhong W, Tebani A, Pou C, Mikes J, et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science. 2019;366(6472):eaax9198.
Shay T, Kang J. Immunological Genome Project and systems immunology. Trends Immunol. 2013;34(12):602–9.
Henning AN, Green D, Baumann R, Grandinetti P, Highfill SL, Zhou H, et al. Immunomagnetic B cell isolation as a tool to study blood cell subsets and enrich B cell transcripts. BMC Res Notes. 2021;14(1):418.
Wright ML, Goin DE, Smed MK, Jewell NP, Nelson JL, Olsen J, et al. Pregnancy-associated systemic gene expression compared to a pre-pregnancy baseline, among healthy women with term pregnancies. Front Immunol. 2023;14:2023.
Welsh R, Song N, Sadegh-Nasseri S. What to do with HLA-DO/H-2O two decades later? Immunogenetics. 2019;71(3):189–96.
Gomez Hernandez G, Morell M, Alarcon-Riquelme ME. The role of BANK1 in B cell signaling and disease. Cells. 2021;10(5):1184.
Bernal-Quiros M, Wu YY, Alarcon-Riquelme ME, Castillejo-Lopez C. BANK1 and BLK act through phospholipase C gamma 2 in B-cell signaling. PLoS ONE. 2013;8(3): e59842.
Miyazaki A, Yogosawa S, Murakami A, Kitamura D. Identification of CMTM7 as a transmembrane linker of BLNK and the B-cell receptor. PLoS ONE. 2012;7(2): e31829.
Yan J, Wolff MJ, Unternaehrer J, Mellman I, Mamula MJ. Targeting antigen to CD19 on B cells efficiently activates T cells. Int Immunol. 2005;17(7):869–77.
Meyer SJ, Steffensen M, Acs A, Weisenburger T, Wadewitz C, Winkler TH, et al. CD22 controls germinal center B cell receptor signaling, which influences plasma cell and memory B cell output. J Immunol. 2021;207(4):1018–32.
Kumanogoh A, Watanabe C, Lee I, Wang X, Shi W, Araki H, et al. Identification of CD72 as a lymphocyte receptor for the class IV semaphorin CD100: a novel mechanism for regulating B cell signaling. Immunity. 2000;13(5):621–31.
Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193–205.
Pavlasova G, Mraz M. The regulation and function of CD20: an “enigma” of B-cell biology and targeted therapy. Haematologica. 2020;105(6):1494–506.
Patel SJ, Trivedi GL, Darie CC, Clarkson BD. The possible roles of B-cell novel protein-1 (BCNP1) in cellular signalling pathways and in cancer. J Cell Mol Med. 2017;21(3):456–66.
Gaubatz S, Lindeman GJ, Ishida S, Jakoi L, Nevins JR, Livingston DM, et al. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell. 2000;6(3):729–35.
Nechanitzky R, Akbas D, Scherer S, Gyory I, Hoyler T, Ramamoorthy S, et al. Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates in committed cells. Nat Immunol. 2013;14(8):867–75.
Tschumper RC, Hoelzinger DB, Walters DK, Davila JI, Osborne CA, Jelinek DF. Stage-specific non-coding RNA expression patterns during in vitro human B cell differentiation into antibody secreting plasma cells. Noncoding RNA. 2022;8(1):15.
Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. 2007;8(5):463–70.
Montecino-Rodriguez E, Fice M, Casero D, Berent-Maoz B, Barber CL, Dorshkind K. Distinct genetic networks orchestrate the emergence of specific waves of fetal and adult B-1 and B-2 development. Immunity. 2016;45(3):527–39.
Schmidlin H, Diehl SA, Nagasawa M, Scheeren FA, Schotte R, Uittenbogaart CH, et al. Spi-B inhibits human plasma cell differentiation by repressing BLIMP1 and XBP-1 expression. Blood. 2008;112(5):1804–12.
Lu DR, McDavid AN, Kongpachith S, Lingampalli N, Glanville J, Ju CH, et al. T cell-dependent affinity maturation and innate immune pathways differentially drive autoreactive B cell responses in rheumatoid arthritis. Arthritis Rheumatol. 2018;70(11):1732–44.
Orange DE, Yao V, Sawicka K, Fak J, Frank MO, Parveen S, et al. RNA identification of PRIME cells predicting rheumatoid arthritis flares. N Engl J Med. 2020;383(3):218–28.
Elling R, Chan J, Fitzgerald KA. Emerging role of long noncoding RNAs as regulators of innate immune cell development and inflammatory gene expression. Eur J Immunol. 2016;46(3):504–12.
Messemaker TC, Frank-Bertoncelj M, Marques RB, Adriaans A, Bakker AM, Daha N, et al. A novel long non-coding RNA in the rheumatoid arthritis risk locus TRAF1-C5 influences C5 mRNA levels. Genes Immun. 2016;17(2):85–92.
Hao Y, Wu W, Shi F, Dalmolin RJ, Yan M, Tian F, et al. Prediction of long noncoding RNA functions with co-expression network in esophageal squamous cell carcinoma. BMC Cancer. 2015;15:168.
Ren ZH, Shang GP, Wu K, Hu CY, Ji T. WGCNA co-expression network analysis reveals ILF3-AS1 functions as a CeRNA to regulate PTBP1 expression by sponging miR-29a in gastric cancer. Front Genet. 2020;11:39.
Ma W, Zhao F, Yu X, Guan S, Suo H, Tao Z, et al. Immune-related lncRNAs as predictors of survival in breast cancer: a prognostic signature. J Transl Med. 2020;18(1):442.
Bam M, Yang X, Ginsberg JP, Aiello AE, Uddin M, Galea S, et al. Long non-coding RNA LINC00926 regulates WNT10B signaling pathway thereby altering inflammatory gene expression in PTSD. Transl Psychiatry. 2022;12(1):200.
de Lima DS, Cardozo LE, Maracaja-Coutinho V, Suhrbier A, Mane K, Jeffries D, et al. Long noncoding RNAs are involved in multiple immunological pathways in response to vaccination. Proc Natl Acad Sci U S A. 2019;116(34):17121–6.
Zheng M, Hong W, Gao M, Yi E, Zhang J, Hao B, et al. Long noncoding RNA COPDA1 promotes airway smooth muscle cell proliferation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2019;61(5):584–96.
Liang Y, Tedder TF. Identification of a CD20-, FcεRIβ-, and HTm4-related gene family: sixteen new MS4A family members expressed in human and mouse. Genomics. 2001;72(2):119–27.
Soliman MM, Hyrich KL, Lunt M, Watson KD, Symmons DP, Ashcroft DM, et al. Effectiveness of rituximab in patients with rheumatoid arthritis: observational study from the British Society for Rheumatology Biologics Register. J Rheumatol. 2012;39(2):240–6.
Acknowledgements
We are immensely grateful to the study subjects for their participation in the study. We acknowledge our gratitude towards the leadership team at the Juliane Marie Center in Denmark for supporting this project. The rheumatology departments at the following hospitals in Denmark facilitated the collection of data and samples: Rigshospitalet (Glostrup), Odense Universitetshospital, Dansk Gigthospital (Sønderborg), Aarhus University Hospital (Skejby), and Regionshospitalet Viborg. We also acknowledge our appreciation for Dr. Hanne Kjærgaard’s efforts in setting up all of the complex logistics for this study in Denmark; Dr. Kjærgaard passed away in 2013.
We thank all members of our project team for making this work possible: Anne-Grethe Rasmussen, Charlotte Schön Frengler, Dorte Heide, Randi Petersen, Tove Thorup Rasmussen, Lone Thomasen, Britta Hvidberg Nielsen, Irene Tarp Jørgensen, Teresa Rozenfeld, Debbie Nadelmann, Kirsten Junker, Lis Kastberg Schubert, Lis Lund, Jette Barlach, Charlotte Drachmann, Dorte Meinke, Helle Bendtsen, Helle Andersen, Marjo Westerdahl, and Jane Alrø Bøtkjær for their contribution with data and sample collection; Rikke Godtkjær Andersen, Mie Rams Rasmussen, Katrine Elmgaard Jensen, Pia Pedersen, Stine Birkelund, Louise Mielke, Andreas Smed, Sofie Ohlsson Svangren, and Emma Sofie Nielsen for the management of data and samples; and Pia Hynne, Majbritt Norman Nielsen, Mie Rams Rasmussen, Emma Victoria Hatley, Thea Guldmann, Trang Xuan Minh Tran, and Isabel Lorenz Gradert for the administrative assistance. We also greatly appreciate the valuable assistance provided by DANBIO personnel.
Dr. Damini Jawaheer accepts responsibility for the integrity and validity of the data collected and analyzed.
Funding
This work was supported by funds from the National Institutes of Arthritis, Musculoskeletal and Skin Diseases (NIAMS), USA (Grants R21AR057931 and R01AR073111); Gigtforeningen, Denmark (grant R87-A1477-B512); and The Juliane Marie Center, Rigshospitalet (Denmark). These funders did not have any role in conducting this study or in the interpretation and reporting of the results.
Author information
Authors and Affiliations
Contributions
MW analyzed the data, interpreted the results and contributed to writing the original draft of the manuscript. JLN, JO and NJ contributed to the conceptualization and design of the study. MS was responsible for acquisition of the data. MH and VZ contributed to the data acquisition. DJ contributed to the conceptualization and design of overall study and the experiments, to the analysis and interpretation of the data, and to writing the original draft of the manuscript. All authors contributed to critically revising the manuscript for important intellectual content and gave final approval of the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee for Region Hovedstaden (Denmark), the Danish Data Protection Agency, the Children’s Hospital Oakland Research Institute Institutional Review Board (IRB number: 2009–073), and the Northwestern University IRB (IRB number: STU00217093). All subjects provided written informed consent prior to enrollment.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
PCA plot of normalized counts for quality control. Following rigorous quality control of the data, log2-transformed TMM-normalized counts data (CPM) from all genes were plotted as a Principal Components Analysis (PCA) plot for pre-pregnancy (T0) samples from 14 RA women who subsequently improved during pregnancy, 5 who worsened and 13 healthy women.
Additional file 2:
Fig. S2. Relative proportions of different cell populations at the pre-pregnancy baseline among the RAimproved, RAworsened and healthy women. The box plots show how the relative proportions of different cell types estimated using CIBERSORTx compared between the RAimproved, RAworsened and healthy women at the pre-pregnancy (T0) baseline. Only cell types included in the LM22 reference dataset are shown. For some LM22 cell types, proportion estimates were not obtained from CIBERSORTx; those are not shown here (resting dendritic cells, activated mast cells, activated NK cells, gamma delta T cells).
Additional file 3:
Table S1. All protein-coding genes, lncRNAs and miRNAs analyzed for differential expression between the RAimproved and RAworsened groups (n=19,468) are included in this table showing the output from the analysis. logFC refers to the log(fold-change) in the RAworsened compared to the RAimproved group. Information on novel lncRNAs identified in our data (names starting with MSTRG) and included in the analyses are available upon request.
Additional file 4:
Table S2. Transcription factor target genes co-expressed within the midnightblue and salmon modules. Several of the genes that were co-expressed within the midnightblue and salmon modules were identified as target genes for transcription factors. Only transcription factors whose target genes were significantly enriched in co-expressed genes within each module are shown. Target genes in bold represent genes that were also differentially expressed between the RAimproved and RAworsened women.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wright, M., Smed, M.K., Nelson, J.L. et al. Pre-pregnancy gene expression signatures are associated with subsequent improvement/worsening of rheumatoid arthritis during pregnancy. Arthritis Res Ther 25, 191 (2023). https://doi.org/10.1186/s13075-023-03169-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-023-03169-6