
Abstract
Background
The study of molecular mechanisms characterizing disease progression may be relevant to get insights into systemic sclerosis (SSc) pathogenesis and to intercept patients at very early stage. We aimed at investigating the proteomic profile of preclinical systemic sclerosis (PreSSc) via a discovery/validation two-step approach.
Methods
SOMAcan aptamer-based analysis was performed on a serum sample of 13 PreSSc (discovery cohort) according to 2001 LeRoy and Medsger criteria (characterized solely by Raynaud phenomenon plus a positive nailfold capillaroscopy and SSc-specific antibodies without any other sign of definite disease) and 8 healthy controls (HCs) age, gender, and ethnicity matched. Prospective data were available up to 4±0.6 years to determine the progression to definite SSc according to the EULAR/ACR 2013 classification criteria. In proteins with relative fluorescence units (RFU) > |1.5|-fold vs HCs values, univariate analysis was conducted via bootstrap aggregating models to determine the predicting accuracy (progression vs non-progression) of categorized baseline protein values. Gene Ontologies (GO terms) and Reactome terms of significant proteins at the adjusted 0.05 threshold were explored. Significant proteins from the discovery cohort were finally validated via ELISAs in an independent validation cohort of 50 PreSSc with clinical prospective data up to 5 years. Time-to-event analysis for interval-censored data was used to evaluate disease progression.
Results
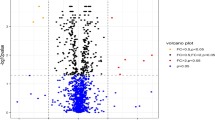
In the discovery cohort, 286 out of 1306 proteins analyzed via SomaScan, were differentially expressed versus HCs. Ten proteins were significantly associated with disease progression; analysis through GO and Reactome showed differentially enriched pathways involving angiogenesis, endothelial cell chemotaxis, and endothelial cell chemotaxis to fibroblast growth factor (FGF). In the validation cohort, endostatin (HR=10.23, CI95=2.2–47.59, p=0.003) was strongly associated with disease progression, as well as bFGF (HR=0.84, CI95=0.709-0.996, p=0.045) and PAF-AHβ (HR=0.372, CI95=0.171–0.809, p=0.013)
Conclusions
A distinct protein profile characterized PreSSc from HCs and proteins associated with hypoxia, vasculopathy, and fibrosis regulation are linked with the progression from preclinical to definite SSc. These proteins, in particular endostatin, can be regarded both as markers of severity and molecules with pathogenetic significance as well as therapeutic targets.
Similar content being viewed by others

Background
Systemic sclerosis (SSc) is a rare disease mainly characterized by vasculopathy, immune-system activation, and fibrosis that can potentially affect any organ. Patients can present different clinical manifestations along with different clinical subsets of disease. The pathogenesis underlying this variety of disease features is complex and still incompletely unrevealed [1]. A limited number of subjects can be intercepted and identified at a preclinical stage of disease presenting solely Raynaud phenomenon (RP), SSc-specific autoantibodies, and/or typical abnormalities at a nailfold video-capillaroscopy (“scleroderma pattern”) without any other clinical fibrotic features [2, 3]. Patients with preclinical SSc (PreSSc) have a risk of disease progression into a definite SSc of about 50% within 5 years of diagnosis [4, 5]. The biological characterization of this group of patients is highly relevant for gaining insight into SSc pathogenesis and mechanisms of disease progression.
Proteins are in closer proximity to pathological processes and therefore are functionally more relevant than information obtained from DNA- or RNA-level studies. Several studies have explored the circulating levels of cytokines, chemokines, and other molecules in SSc subsets. Limited by technologies available, the majority of studies focused only on a small group of proteins chosen a priori, while larger panels of proteins were just analyzed in a few studies [6,7,8]. Cytokines and chemokines related to endothelial dysfunctions, fibrosis, and adhesion molecules clearly emerged as markers of disease severity and stage [9,10,11]. Moreover, the serum proteomic profile of SSc patients is clearly different from that of healthy controls also correlating more closely with molecular dysregulations of affected organs such as skin [6, 8].
In the present project, with a two-step strategy, we aimed at exploring the proteomic profile of a well-characterized group of PreSSc compared with matched healthy controls, using a validated innovative and comprehensive platform based on a library of aptamers. We then performed a validation analysis in a longitudinal cohort of 50 PreSSc through an enzyme-linked immunosorbent assay (ELISA) to confirm the circulating factors that are robustly associated with disease progression.
Methods
Patients and controls
Two different cohorts of patients were examined pursuing a 2-step approach with discovery and validation strategy.
As a first step, 13 PreSSc, defined according to LeRoy and Medsger criteria (Raynaud phenomenon plus a positive nailfold capillaroscopy and SSc-specific auto-antibodies without any other sign of definite disease) [2] with available baseline and subsequent clinical data at approximately 4 years as well as baseline aliquoted serum samples, were included in the discovery cohort. Clinical data, including the occurrence of puffy fingers, sclerodactyly, telangiectasia, lung fibrosis, pulmonary arterial hypertension, scleroderma renal crisis alone or in combination indicative of the progression to definite SSc (thus with a minimum score of 9) according to the EULAR/ACR 2013 classification criteria [12, 13] were retrieved from medical records allowing the evaluation of disease progression as previously described [10]. Patients with a definite SSc but with puffy fingers without skin fibrosis were considered as limited cutaneous SSc (lcSSc). At baseline serum samples were collected and stored at −80 °C; samples from 8 ethnically-, age- and sex-matched healthy controls (HCs) were collected as well. Cases and controls from the discovery cohort were screened via the SomaScan® aptamer analysis to find proteins of interest linked to progression from PreSSc to definite SSc patients.
As a second step, 50 independent PreSSc patients with baseline serum samples and available prospective clinical data at 5 years, were considered as the validation cohort; serum from the validation cohort was aliquoted at a different time from those of the discovery cohort. ELISA (RayBiotech Life, Inc. MyBioSource Cloud-Clone Corp) of serum samples was used to validate the relevant proteins found in the discovery cohort.
The study was performed in accordance with the Declaration of Helsinki and approved by the local ethic committee (approval n. 559_2018) and patients signed informed consent to participate in the study.
Aptamer analysis
Comprehensive targeted proteomics was performed using the SomaScan® assay as described [14] interrogating the levels of 1306 different proteins. All samples were clarified by centrifugation before use and were screened using the SomaScan® aptamer-based screening platform at the Houston Omics Collaborative (https://hoc.bme.uh.edu/). This assay uses aptamer–protein interactions to detect proteins within a sample. In the assay, aptamer-coated streptavidin beads are first added to the sample to allow the aptamers to bind to the proteins. Next, the bound proteins are biotinylated, and the aptamer–protein complexes are cleaved from the streptavidin beads. These aptamer–protein complexes are then conjugated to a second streptavidin bead, and aptamers are separated from the proteins. The aptamers are then collected from the sample and quantitated by hybridization to a DNA microarray. The final output is the relative fluorescence unit (RFU) for each protein; these RFU values were then normalized and statistically analyzed. The limit of detection (LOD) of the aptamer-based scan was determined by spiking proteins into buffer before the assay. The limits of quantitation (LOQ) were established along with the LOD, and the median lower LOQ value is approximately 3-fold higher than the LOD.
Statistical analysis
Predictive accuracy in the discovery cohort
Due to the limited sample size, only proteins whose RFUs were increased or decreased compared to HCs were considered. To this end, a log fold-change (FC) ≥ 0.585 was used as cut-off. Considering 13 cases (Nc = 13) and 8 controls (Nhc = 8), the FC was calculated from individual RFU values.
Bootstrap aggregating (bagging with 100 resamplings) was used to determine the accuracy of categorized baseline protein values in predicting the subsequent status (progression vs non-progression) at the last available observation. In each in-bag sample, the threshold to define the risk of evolution was considered the median value RFU of each protein, whose predictive accuracy was calculated from 2 × 2 tables in the corresponding out-of-bag samples. A 10,000-fold step-down permutation approach (Tmax method) was then used to assess the significance of predictions and to correct for family-wise error rates [15], a nominal 0.05 value was then used. A custom-code written in python by LB on top of the Scikit-learn machine learning libraries [16] was used for the analyses.
Gene ontology (GO) analysis
Enrichment analysis of significant aptamers found in the discovery phase was performed using the ShiniGO web application [17, 18]. To this end, the corresponding genes were used to find significant GOs at the biological process level and to explore Reactome pathways.
Survival analysis in the validation set
To better assess the prognostic implications of individual proteins identified in the discovery set, and to exploit all the available information, prospective data from the validation cohort were used. Time-to-progression was explored using the Cox-regression method for interval-censored data after Box-Cox transformation of data to ensure normality [19]. Significant analytes were categorized after cutpoint estimation on right-censored samples according to the method described by Contal and O’Quigley [20]; the Turnbull method for interval-censored data [21] was used to calculate survival estimates of dicothomized proteins and the corresponding P values were calculated with the generalized logrank test for right-censored failure time data according to Sun [22]. Missing data were first imputed according to Beretta and Santaniello [23] using the rkNN-imputer Scikit-learn library [17] setting the number of neighbors equal to 3. Time-to-event analyses were done using R 4.0.5 [R Core Team, 2021] with the AID [24], icenReg [25], the interval [26], and the survMisc: Miscellaneous Functions for Survival Data [v0.5.5] [27] packages.
Throughout the article, descriptive statistics are presented as mean ± standard deviation except for skewed values that are presented as median and interquartile ranges.
Results
Ten proteins in the discovery cohort associated with fibrogenesis and angiogenesis are associated with progression to definite SSc
The 13 preclinical-SSc subjects included in the discovery cohort aged 53.5 ± 6.3 years, were mostly females (n=10, 76.9%), and all tested positive for antinuclear antibodies (ANA); anticentromere antibodies (ACA) were found in 8 subjects (61.5%), anti-topoisomerase I antibodies (ATA) in 3 (23.1%) and ANA with nucleolar staining in 2 (15.4%). After 4 years, 7 patients (54%) progressed into definite SSc, with lcSSc skin features (6 presenting solely puffy fingers). Non-progressors and progressors were similar regarding overall observation time and baseline characteristics: ACA positivity, 66% vs 57%; forced vital capacity (FVC) % of predicted values, 101.5 ± 12.4 vs 112.7 ± 5.8 diffusing capacity for carbon monoxide (DLco), 95.5 ± 20.3 vs 90.3 ± 11 (Supplemental Table 1).
Two hundred eighty-six proteins out of the 1306 analyzed via the SomaScan® assay were differentially expressed in comparison with 8 matched healthy controls (females, n=7, 87.5%; age 55.8 ± 4.1 years) and were further considered for the analysis. Bagging experiments after resampling with permutation, showed that ten proteins were significantly associated with the development of definite SSc in preclinical samples: NKp30, Endostatin, basic fibroblast growth factor (bFGF), extracellular matrix protein 1 (ECM1), FGF18, phospohexose isomerase (PHI), Fibronectin 1.3 (FN1.3), Ubiquitin +1, platelet-activating factor acetylhydrolase-β subunit (PAF-AHβ), fatty acid binding protein (FABP) (Table 1).
Analysis of GO biological processes showed a few differentially enriched pathways involving angiogenesis, endothelial cell chemotaxis, and endothelial cell chemotaxis to fibroblast growth factor (Fig. 1A). These processes are mostly mediated by binding and activation of FGF receptors (FGFR) as indicated by Reactome enrichment analysis (Fig. 1B).

Enrichment analysis in the discovery set. Enrichment analysis: genes related to the 10 proteins selected in the discovery cohort (see Table 1). A Gene Ontologies (GO) at the biological process level. B Reactome pathways
Validation of proteins with prognostic significance in survival models
The characteristics of the 50 patients included the validation cohort are reported in Table 2.
Twenty subjects (40%) did progress into a definite SSc at the end of the 5-year observation period; the overall estimated 5-year time-to-evolution in the validation cohort is represented in Fig. 2. The prototypical sign of progression was skin involvement, namely puffy fingers in 13 cases (65%) and overt skin fibrosis in 7 with limited cutaneous SSc (lcSSc) (35%); telangiectasia did appear in combination with the above in 6 cases (30%).

Estimated time-to-evolution in the validation cohort. Survival estimates as calculated by the Turnbull’s method, in the validation cohort; T0 = blood draw
Patients with Raynaud’s duration shorter than 10 years (p = 0.0425) at baseline or with reflux disease (p = 0.014) had shorter times to progression while none of the other baseline clinical characteristics was associated with time-to-progression (Supplemental Table 2 and Supplemental Figs. 1 and 2).
ELISA confirmation assay provided technically valuable results for all the 10 analytes selected after step 1, but Ubiquitin +1; hence, step 2 analysis was restricted to 9 molecules. Of these, Endostatin (hazard ratio for transformed data [HR] = 10.23, CI95 = 2.2–47.59, p = 0.003), bFGF (HR = 0.84, CI95 = 0.709–0.996, p = 0.045) and PAF-AHβ (HR = 0.372, CI95 = 0.171–0.809, p = 0.013) were significantly associated with progression after Cox-regression analysis for interval-censored data. Cut-point estimation showed that proteins were found to be associated with a reduced time-to-evolution: endostatin (cut-off ≥ 124 pg/mL, p = 6.65 × 10−4), bFGF (cut-off < 4.9 pg/mL, p = 0.02563), and PAF-AHβ (cut-off < 2.1 ng/mL, p = 0.00145) (Fig. 3). The rough distribution of dicothomized proteins according to the abovementioned thresholds, regardless of the time-to-evolution, in progressors and non-progressors is shown in Fig. 4.

Risk of evolution to definite systemic sclerosis in the validation cohort (replicated proteins). Survival estimates (Turnbull’s method) for dicothomized proteins (high/low serum levels) with prognostic significance in the validation cohort; time, years from blood draw. bFGF, basic fibroblast growth factor; PAF-AHβ, platelet-activating factor acetylhydrolase subunit beta

Distribution of categorized proteins in the validation cohort. Distribution of cases with high or low validated protein levels in preclinical systemic sclerosis patients who did evolve (progressors) or who did not (non-progressors) into definite systemic sclerosis. Dicothomization was performed as described in the main text; for endostatin risk is associated with high serum levels; for the basic fibroblast growth factor (FGF) and platelet-activating factor acetylhydrolase subunit beta (PAFAH1B2) risk is associated with low serum levels; clusterization made on the basis of serum concentrations
Discussion
The study of SSc in its preclinical phase is highly relevant to understand the pathophysiological alterations that sustain the development into a clinically evident fibrotic disease and, consequently, to discover potential avenues of early intervention. Nonetheless, this endeavor has seldom been undertaken and mostly in cross-sectional studies [10, 13, 28,29,30,31,32,33].
The main finding of ours is that 3 proteins with angiogenetic and fibrotic processes regulation significance are differentially expressed in preclinical SSc patients according to the future 5-year progression, and namely endostatin, bFGF, and PAF-AHβ. These findings support the notion that vasculopathy is fundamental in the development of SSc and of its fibrotic manifestations [34] and that microvascular damage is strictly related with the progression of scleroderma [35]. In details, it seems that patients with altered markers of angiogenesis are at risk of progression as compared to subjects with a more indolent circulatory disease, as also testified by the fact that a longer history of RP duration in absence of pivotal signs of definite SSc is also associated with lower rates of progression (Supplemental Fig. 1). Additionally, we showed that the presence of gastro-esophageal reflux was significantly associated with a shorter time of progression, in line with previous findings indicating that in PreSSc patients non-circulatory clinical signs are associated with an increased risk of progression [5, 36].
Among the proteins associated with disease progression, endostatin emerged as the one most strongly related with the passage from preclinical to definite SSc (Fig. 3). Increased serum levels of endostatin have already been described in SSc [37, 38] also correlating with the severity of vascular manifestations [37]. Endostatin is an endogenous inhibitor of proliferation and migration of endothelial cells and angiogenesis [39] that is upregulated in kidney and cardiovascular diseases [40] as well as in patients with peripheral vascular disease [41]. Endostatin is released during ischemia-reperfusion and hypoxia [42,43,44] and hence it may be postulated that in preclinical SSc this molecule is a marker of a more severe form of vasculopathy, as well as an anti-angiogenic factor that promotes disease progression. Endostatin has also anti-fibrogenic functions [45] and its serum increase may also reflect a feedback loop in the attempt to control and reduce upcoming fibrosis and to regulate the collagen turnover.
bFGF is a molecule with pleiotropic effects, mainly promoting angiogenesis, and fibroblast proliferation and that regulates fibrotic processes preventing fibrosis deposition through the inhibition of TGF-β mediated collagen deposition [46]. Its function is upstream of other specialized growth factors, such as vascular endothelial growth factor (VEGF) [47], whose function is tightly regulated by endostatin [48]. bFGF usually increases in response to hypoxic stimuli to promote neoangiogenesis [49] and bFGF levels were found to be mostly undetectably low in patients with SSc [50]. Low bFGF levels in preclinical SSc at risk for progression would mirror a condition of increased vasculopathy and defective response to the hypoxic condition [49, 51, 52]. Conversely, it may be argued that high bFGF levels under hypoxic conditions would act as a protective angiogenetic mechanisms in patients with slow progression rates regulating at the same time collagen deposition.
Shorter times to progression were also observed in patients with low PAF-AHβ serum levels. PAF-AH degrades the platelet-activating factor [53] counteracting its main effect, including leukocyte chemotaxis, adhesion and degranulation, endothelial permeability and dysfunction, vasoconstriction, and the promotion of the release of proinflammatory cytokines, including interleukin (IL)-1 and IL-6 [54]. Notably, PAF-AH may prevent ischemia-reperfusion [55] and is down-regulated in hypoxic rat models [56]. These observations suggest that high levels of PAF-AHβ may be protective against the progression of endothelial dysfunction in scleroderma as well as be a marker of a milder form of vasculopathy that is at lower risk of evolution.
Taken together, our results suggest that soluble factors associated with hypoxia and vasculopathy, are linked with the transition from preclinical to definite SSc and that these may be regarded both as markers of severity and molecules with pathogenetic significance. A quantification of SSc-related vasculopathy, as for instance nailfold video-capillaroscopy (NVC) scores, would have helped to better establish a correlation between endothelial damage, vasculopathy, and circulating markers, yet NVC data were not available in our patients. Nonetheless, NVC scleroderma patterns were found to be predictive of clinical complications of the disease [57, 58] and preclinical patients with severe NVC had shorter times to definite SSc compared with those with less severe patterns [59], indirectly supporting our findings.
The discovery-validation strategy we applied guarantees that our results are reproducible and strongly mitigates the risk of false-positive findings. Nonetheless, we are aware that other potential candidates may have been overlooked because of the selection procedure we used in the discovery phase. Because of the low sample size, we decided to restrict the analysis to a panel of candidates that were differentially expressed as compared to healthy controls. Moreover, statistical results were adjusted for multiple tests and albeit permutations correct type I errors less conservatively than other methods (i.e., Bonferroni or false-discovery rate) [60] the risk of type II errors (e.g. loss of power) is still substantial.
Our cohort was composed uniquely by Caucasian subjects, therefore the generalization of our results to other ethnicities should be assessed by future studies.
Another shortcoming of our study is related to the relatively over-representation of patients with ACAs antibodies and to the long-lasting RP duration. These characteristics clearly underlie the difficulty of intercepting patients with more aggressive disease in favor of subjects that will eventually develop a limited cutaneous form of SSc, as also observed in a multicenter study of very early SSc subjects [5]. This almost unavoidable selection bias clearly warns that caution should be exercised in applying our findings to all PreSSc patients, even if our findings are biologically plausible.
Conclusions
In summary, PreSSc showed a distinct protein profile and proteins that are related to hypoxia, vasculopathy, and collagen turnover, which emerged at characterizing the progression from a preclinical stage of SSc to a definite one. In particular, endostatin was the protein most strongly associated with disease progression, and it is worthwhile to further investigate its mechanistic roles for its possible pathogenetic role in SSc development and its therapeutic potential.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- SSc:
-
Systemic sclerosis
- PreSSc:
-
Preclinical systemic sclerosis
- HCs:
-
Healthy controls
- RFU:
-
Relative fluorescence units
- GO:
-
Gene Ontology
- FGF:
-
Fibroblast growth factor
- ELISA:
-
Enzyme-linked immunosorbent assay
- LOD:
-
Limit of detection
- LOQ:
-
Limits of quantitation
- ANA:
-
Antinuclear antibodies
- ACA:
-
Anticentromere antibodies
- ATA:
-
Anti-topoisomerase I antibodies
- FVC:
-
Forced vital capacity
- DLco:
-
Diffusing capacity for carbon monoxide
- bFGF:
-
Basic fibroblast growth factor
- ECM1:
-
Extracellular matrix protein 1
- PHI:
-
Phospohexose isomerase
- FN1.3:
-
Fibronectin 1.3
- PAF-AHβ:
-
Platelet-activating factor acetylhydrolase-β subunit
- FABP:
-
Fatty acid binding protein
- FGFR:
-
FGF receptors
- VEGF:
-
Vascular endothelial growth factor
- IL:
-
Interleukin
- NVC:
-
Nailfold video-capillaroscopy
References
Varga J, Trojanowska M, Kuwana M. Pathogenesis of systemic sclerosis: recent insights of molecular and cellular mechanisms and therapeutic opportunities. J Scleroderma Relat Disord. 2017;2:137–52 Available from: http://journals.sagepub.com/doi/10.5301/jsrd.5000249.
LeRoy EC, Medsger TA. Criteria for the classification of early systemic sclerosis. J Rheumatol [Internet]. 2001;28:1573–6 Available from: http://www.ncbi.nlm.nih.gov/pubmed/11469464.
van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum [Internet]. 2013;65:2737–47 Available from: https://onlinelibrary.wiley.com/doi/10.1002/art.38098.
Koenig M, Joyal F, Fritzler MJ, Roussin A, Abrahamowicz M, Boire G, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: A twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosi. Arthritis Rheum. 2008;58:3902–12 Available from: https://onlinelibrary.wiley.com/doi/10.1002/art.24038.
Bellando-Randone S, Del Galdo F, Lepri G, Minier T, Huscher D, Furst DE, et al. Progression of patients with Raynaud’s phenomenon to systemic sclerosis: a five-year analysis of the European Scleroderma Trial and Research group multicentre, longitudinal registry study for Very Early Diagnosis of Systemic Sclerosis (VEDOSS). Lancet Rheumatol. 2021;3:e834–43 Available from: https://linkinghub.elsevier.com/retrieve/pii/S2665991321002447.
Bellocchi C, Ying J, Goldmuntz EA, Keyes-Elstein L, Varga J, Hinchcliff ME, et al. Large-Scale Characterization of Systemic Sclerosis Serum Protein Profile: Comparison to Peripheral Blood Cell Transcriptome and Correlations With Skin/Lung Fibrosis. Arthritis Rheumatol (Hoboken, NJ), Available from. 2021;73:660–70 http://www.ncbi.nlm.nih.gov/pubmed/33131208.
Rice LM, Mantero JC, Stifano G, Ziemek J, Simms RW, Gordon J, et al. A Proteome-Derived Longitudinal Pharmacodynamic Biomarker for Diffuse Systemic Sclerosis Skin. J Invest Dermatol. 2017;137:62–70 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27640094.
Farutin V, Kurtagic E, Pradines JR, Capila I, Mayes MD, Wu M, et al. Multiomic study of skin, peripheral blood, and serum: is serum proteome a reflection of disease process at the end-organ level in systemic sclerosis? Arthritis Res Ther. 2021;23:259 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34654463.
Bandinelli F, Del Rosso A, Gabrielli A, Giacomelli R, Bartoli F, Guiducci S, et al. CCL2, CCL3 and CCL5 chemokines in systemic sclerosis: the correlation with SSc clinical features and the effect of prostaglandin E1 treatment. Clin Exp Rheumatol. 30:S44–9 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22691208.
Cossu M, Andracco R, Santaniello A, Marchini M, Severino A, Caronni M, et al. Serum levels of vascular dysfunction markers reflect disease severity and stage in systemic sclerosis patients. Rheumatology (Oxford). 2016;55:1112–6 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26989111.
Riccieri V, Stefanantoni K, Vasile M, Macrì V, Sciarra I, Iannace N, et al. Abnormal plasma levels of different angiogenic molecules are associated with different clinical manifestations in patients with systemic sclerosis. Clin Exp Rheumatol. 29:S46–52 Available from: http://www.ncbi.nlm.nih.gov/pubmed/21586218.
van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65:2737–47 Available from: http://www.ncbi.nlm.nih.gov/pubmed/24122180.
Cossu M, van Bon L, Preti C, Rossato M, Beretta L, Radstake TRDJ. Earliest Phase of Systemic Sclerosis Typified by Increased Levels of Inflammatory Proteins in the Serum. Arthritis Rheumatol (Hoboken, NJ). [Internet]. 2017;69:2359–69 Available from: http://www.ncbi.nlm.nih.gov/pubmed/28859262.
Stanley S, Vanarsa K, Soliman S, Habazi D, Pedroza C, Gidley G, et al. Comprehensive aptamer-based screening identifies a spectrum of urinary biomarkers of lupus nephritis across ethnicities. Nat Commun. 2020;11:2197 Available from: http://www.ncbi.nlm.nih.gov/pubmed/32366845.
Ge Y, Dudoit S, Speed TP. Resampling-based multiple testing for microarray data analysis. Test. 2003;12:1–77 Available from: http://link.springer.com/10.1007/BF02595811.
Pedregosa F, Varoquaux G, Gramfort A, Michel V. Scikit-learn: Machine Learning in Python. J Mach Learn Res. 2011;12:2825–30.
ShiniGO web application [Internet]. [cited 2022 May 19]. Available from: http://bioinformatics.sdstate.edu/go/
Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics [Internet]. 2020;36:2628–9 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31882993.
Cai T, Tian L, Wei LJ. Semiparametric Box: Cox Power Transformation Models for Censored Survival Observations. Biometrika. 2005;92:619–32.
Contal C, O’Quigley J. An application of changepoint methods in studying the effect of age on survival in breast cancer. Comput Stat Data Anal [Internet]. 1999;30:253–70 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0167947398000966.
Turnbull BW. The Empirical Distribution Function with Arbitrarily Grouped, Censored and Truncated Data. J R Stat Soc Ser B (Methodological). 1976;38:290–5.
Sun J. A non-parametric test for interval-censored failure time data with application to AIDS studies. Stat Med [Internet]. 1996;15:1387–95 Available from: http://www.ncbi.nlm.nih.gov/pubmed/8841649.
Beretta L, Santaniello A. Nearest neighbor imputation algorithms: a critical evaluation. BMC Med Inform Decis Mak. 2016;16(Suppl 3):74 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27454392.
Asar Ö, Ilk O, Dag O. Estimating Box-Cox power transformation parameter via goodness-of-fit tests. Commun Stat Simul Comput [Internet]. 2017;46:91–105. https://doi.org/10.1080/03610918.2014.957839.
Anderson-Bergman C. icenReg : Regression Models for Interval Censored Data in R. J Stat Softw [Internet]. 2017:81 Available from: http://www.jstatsoft.org/v81/i12/.
Fay MP, Shaw PA. Exact and Asymptotic Weighted Logrank Tests for Interval Censored Data: The interval R package. J Stat Softw [Internet]. 2010:36 Available from: http://www.ncbi.nlm.nih.gov/pubmed/25285054.
Dardis C. Miscellaneous Functions for Survival Data; 2022.
Cossu M, van Bon L, Nierkens S, Bellocchi C, Santaniello A, Dolstra H, et al. The magnitude of cytokine production by stimulated CD56+ cells is associated with early stages of systemic sclerosis. Clin Immunol. 2016;173:76–80 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27616458.
Brkic Z, van Bon L, Cossu M, van Helden-Meeuwsen CG, Vonk MC, Knaapen H, et al. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann Rheum Dis [Internet]. 2016;75:1567–73 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26371289.
Valentini G, Riccardi A, Vettori S, Irace R, Iudici M, Tolone S, et al. CXCL4 in undifferentiated connective tissue disease at risk for systemic sclerosis (SSc) (previously referred to as very early SSc). Clin Exp Med [Internet]. 2017;17:411–4 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27650429.
Chouri E, Wang M, Hillen MR, Angiolilli C, Silva-Cardoso SC, Wichers CGK, et al. Implication of miR-126 and miR-139-5p in Plasmacytoid Dendritic Cell Dysregulation in Systemic Sclerosis. J Clin Med [Internet]. 2021:10 Available from: http://www.ncbi.nlm.nih.gov/pubmed/33573268.
van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med [Internet]. 2014;370:433–43 Available from: http://www.ncbi.nlm.nih.gov/pubmed/24350901.
Mariotti B, Servaas NH, Rossato M, Tamassia N, Cassatella MA, Cossu M, et al. The Long Non-coding RNA NRIR Drives IFN-Response in Monocytes: Implication for Systemic Sclerosis. Front Immunol [Internet]. 2019;10:100 Available from: http://www.ncbi.nlm.nih.gov/pubmed/30804934.
Asano Y, Sato S. Vasculopathy in scleroderma. Semin Immunopathol [Internet]. 2015;37:489–500 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26152638.
Koenig M, Joyal F, Fritzler MJ, Roussin A, Abrahamowicz M, Boire G, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: a twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosi. Arthritis Rheum [Internet]. 2008;58:3902–12 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19035499.
Valentini G, Cuomo G, Abignano G, Petrillo A, Vettori S, Capasso A, et al. Early systemic sclerosis: assessment of clinical and pre-clinical organ involvement in patients with different disease features. Rheumatology (Oxford) [Internet]. 2011;50:317–23 Available from: http://www.ncbi.nlm.nih.gov/pubmed/20562195.
Hebbar M, Peyrat JP, Hornez L, Hatron PY, Hachulla E, Devulder B. Increased concentrations of the circulating angiogenesis inhibitor endostatin in patients with systemic sclerosis. Arthritis Rheum [Internet]. 2000;43:889–93 Available from: http://www.ncbi.nlm.nih.gov/pubmed/10765935.
Hummers LK, Hall A, Wigley FM, Simons M. Abnormalities in the regulators of angiogenesis in patients with scleroderma. J Rheumatol [Internet]. 2009;36:576–82 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19228661.
O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell [Internet]. 1997;88:277–85 Available from: http://www.ncbi.nlm.nih.gov/pubmed/9008168.
Li M, Popovic Z, Chu C, Krämer BK, Hocher B. Endostatin in Renal and Cardiovascular Diseases. Kidney Dis (Basel, Switzerland). 2021;7:468–81 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34901193.
Golledge J, Clancy P, Hankey GJ, Yeap BB, Norman PE. Serum endostatin concentrations are higher in men with symptoms of intermittent claudication. Dis Markers [Internet]. 2014;2014:298239 Available from: http://www.ncbi.nlm.nih.gov/pubmed/24600079.
Paddenberg R, Faulhammer P, Goldenberg A, Kummer W. Hypoxia-induced increase of endostatin in murine aorta and lung. Histochem Cell Biol [Internet]. 2006;125:497–508 Available from: http://www.ncbi.nlm.nih.gov/pubmed/16465514.
Lauten A, Majos E, Mühlich A, Wahlers T, Weider S, Fischer JH, et al. Ischemia-reperfusion injury activates early extracellular matrix processing and expression of endostatin in the heart with differential effects of temperature. Basic Res Cardiol [Internet]. 2009;104:559–69 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19255800.
Bellini MH, Coutinho EL, Filgueiras TC, Maciel TT, Schor N. Endostatin expression in the murine model of ischaemia/reperfusion-induced acute renal failure. Nephrology (Carlton) [Internet]. 2007;12:459–65 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17803469.
Yamaguchi Y, Takihara T, Chambers RA, Veraldi KL, Larregina AT, Feghali-Bostwick CA. A peptide derived from endostatin ameliorates organ fibrosis. Sci Transl Med [Internet]. 2012;4:136ra71 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22649092.
Ichiki Y, Smith EA, LeRoy EC, Trojanowska M. Basic fibroblast growth factor inhibits basal and transforming growth factor-beta induced collagen alpha 2(I) gene expression in scleroderma and normal fibroblasts. J Rheumatol [Internet]. 1997;24:90–5 Available from: http://www.ncbi.nlm.nih.gov/pubmed/9002017.
Murakami M, Simons M. Fibroblast growth factor regulation of neovascularization. Curr Opin Hematol [Internet]. 2008;15:215–20 Available from: http://www.ncbi.nlm.nih.gov/pubmed/18391788.
Eriksson K, Magnusson P, Dixelius J, Claesson-Welsh L, Cross MJ. Angiostatin and endostatin inhibit endothelial cell migration in response to FGF and VEGF without interfering with specific intracellular signal transduction pathways. FEBS Lett [Internet]. 2003;536:19–24 Available from: http://www.ncbi.nlm.nih.gov/pubmed/12586331.
Kuwabara K, Ogawa S, Matsumoto M, Koga S, Clauss M, Pinsky DJ, et al. Hypoxia-mediated induction of acidic/basic fibroblast growth factor and platelet-derived growth factor in mononuclear phagocytes stimulates growth of hypoxic endothelial cells. Proc Natl Acad Sci U S A [Internet]. 1995;92:4606–10 Available from: http://www.ncbi.nlm.nih.gov/pubmed/7538678.
İlgen U, Yayla ME, Düzgün N. Low serum fibroblast growth factor 2 levels not accompanied by increased serum pentraxin 3 levels in patients with systemic sclerosis. Clin Rheumatol [Internet]. 2017;36:367–72 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27878407.
Lokmic Z, Musyoka J, Hewitson TD, Darby IA. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int Rev Cell Mol Biol [Internet]. 2012;296:139–85 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22559939.
Kuo Y-L, Jou I-M, Jeng S-F, Chu C-H, Huang J-S, Hsu T-I, et al. Hypoxia-induced epithelial-mesenchymal transition and fibrosis for the development of breast capsular contracture. Sci Rep [Internet]. 2019;9:10269 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31311941.
Karasawa K. Clinical aspects of plasma platelet-activating factor-acetylhydrolase. Biochim Biophys Acta [Internet]. 2006;1761:1359–72 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17049457.
Lordan R, Tsoupras A, Zabetakis I, Demopoulos CA. Forty Years Since the Structural Elucidation of Platelet-Activating Factor (PAF): Historical, Current, and Future Research Perspectives. Molecules [Internet]. 2019:24 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31816871.
Morgan EN, Boyle EM, Yun W, Kovacich JC, Canty TG, Chi E, et al. Platelet-activating factor acetylhydrolase prevents myocardial ischemia-reperfusion injury. Circulation [Internet]. 1999;100:II365–8 Available from: http://www.ncbi.nlm.nih.gov/pubmed/10567331.
Maerz S, Liu C-H, Guo W, Zhu Y-Z. Anti-ischaemic effects of bilobalide on neonatal rat cardiomyocytes and the involvement of the platelet-activating factor receptor. Biosci Rep [Internet]. 2011;31:439–47 Available from: http://www.ncbi.nlm.nih.gov/pubmed/21391918.
Sulli A, Paolino S, Pizzorni C, Ferrari G, Pacini G, Pesce G, et al. Progression of nailfold capillaroscopic patterns and correlation with organ involvement in systemic sclerosis: a 12 year study. Rheumatology (Oxford) [Internet]. 2020;59:1051–8 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31750929.
Cutolo M, Pizzorni C, Sulli A, Smith V. Early Diagnostic and Predictive Value of Capillaroscopy in Systemic Sclerosis. Curr Rheumatol Rev [Internet]. 2013;9:249–53 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26932289.
Vigone B, Santaniello A, Marchini M, Montanelli G, Caronni M, Severino A, et al. Role of class II human leucocyte antigens in the progression from early to definite systemic sclerosis. Rheumatology (Oxford) [Internet]. 2015;54:707–11 Available from: http://www.ncbi.nlm.nih.gov/pubmed/25231181.
Camargo A, Azuaje F, Wang H, Zheng H. Permutation - based statistical tests for multiple hypotheses. Source Code Biol Med [Internet]. 2008;3:15 Available from: http://www.ncbi.nlm.nih.gov/pubmed/18939983.
Acknowledgements
We acknowledge GILS (Gruppo Italiano per la Lotta alla Sclerodermia) for all support.
Funding
This work was funded by Gruppo Italiano per la Lotta alla Sclerodermia GILS Bando Giovani Ricercatori (Sep 2017) and the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (R61AR078078, R56AR078211).
This study was partially funded by the Italian Ministry of Health, Ricerca Corrente RC-2021.
Author information
Authors and Affiliations
Contributions
CB, LB, and SA: conceptualization and methodology. CB, LB, SA, ML, MM, CM, and AS: study design, data analysis curation. CB and LB: writing—original draft preparation. CB, LB, SA, AS, ML, MM, and CM: writing—review and editing. LB and SA: supervision. All authors read and approved the final manuscript and account for the accuracy and integrity of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The present study involved human participants and was reviewed and approved by Comitato Etico Milano Area 2. The patients/participants provided their written informed consent to participate in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplemental Table 1.
Clinical characteristics of patients in the discovery (step 1) cohort. Supplemental Table 2. Clinical variables associated with disease progression in the validation cohort. Supplemental Figure 1. Survival estimates in relation to the duration of Raynaund’s phenomenon at baseline in the validation cohort. Supplemental Figure 2. Survival estimates in relation to the presence of reflux disease at baseline in the validation cohort.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bellocchi, C., Assassi, S., Lyons, M. et al. Proteomic aptamer analysis reveals serum markers that characterize preclinical systemic sclerosis (SSc) patients at risk for progression toward definite SSc. Arthritis Res Ther 25, 15 (2023). https://doi.org/10.1186/s13075-023-02989-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-023-02989-w