
Abstract
Background
Necroptosis is a form of regulated necrosis that is involved in various autoimmune diseases. Mixed lineage kinase domain-like pseudokinase (MLKL) has been identified as a key executor of necroptosis; however, the significance of MLKL in peripheral blood mononuclear cells (PBMCs) of systemic lupus erythematosus (SLE) has not been investigated. In this study, we aimed to determine the mRNA level of MLKL in PBMCs and examine its relationship with clinical features and serological parameters in SLE.
Methods
Real-time transcription-polymerase chain reaction (RT-PCR) analysis was used to determine the expression of MLKL mRNA in PBMCs from 59 patients with SLE, 25 patients with rheumatoid arthritis (RA), and 30 age- and sex-matched healthy controls (HC). Spearman’s correlation test was performed to assess the correlation of MLKL mRNA with clinical variables. The receiver operating characteristic (ROC) curve was created to evaluate the diagnostic value.
Results
Our results showed MLKL mRNA in PBMCs was upregulated in SLE patients compared to that in RA and HC individuals. SLE patients positive for antinuclear antibodies had significantly higher MLKL mRNA than antibody-negative patients. In SLE patients, MLKL mRNA was found to be upregulated in patients with lupus nephritis (LN) as compared with patients without LN, and also higher in active patients than in stable patients. MLKL mRNA level was significantly and positively correlated with c-reaction protein (CRP) (r = 0.3577, p = 0.0237), erythrocyte sedimentation rate (ESR) (r = 0.4091, p = 0.0043), serum immunoglobulin G (IgG) concentration (r = 0.3546, p = 0.0289), and the numbers of positive antinuclear antibodies (ANAs) (r = 0.3945, p = 0.0432). ROC analysis showed that MLKL mRNA in PBMCs had an area under the curve of 0.9277 (95% CI 0.8779–0.9775, p < 0.001) to discriminate SLE from controls.
Conclusions
These results suggest that increased MLKL mRNA level in the PBMCs of SLE patients is correlated with renal involvement and disease activity, identifying a subgroup of patients with SLE or LN who may benefit from early diagnosis and therapies targeting MLKL.
Similar content being viewed by others


Background
SLE is a complex, heterogeneous systemic autoimmune disease that attacks various cells and tissues, resulting in chronic inflammation and persistent tissue damage [1]. A notable characteristic of SLE is the production of pathogenic autoantibodies recognizing nucleic acids or proteins binding to nucleic acids [2]. Dysregulated cell death processes and defective clearance of dying cells have been proposed to contribute to autoantigen generation and induction of autoantibodies, as well as other aberrant immune responses in SLE [3].
Necroptosis is a special form of necrosis that is triggered by multiple pathways [4]. In cells where caspase-8 is inhibited, inflammatory signaling via tumor necrosis factor (TNF) super family receptors, interferons (IFNs), toll-like receptor 3 (TLR3), or TLR4 can lead to the phosphorylation of receptor-interacting serine/threonine-protein kinase 1 (RIPK1), RIPK3, and MLKL [5,6,7,8]. The phosphorylated MLKL inserts itself into the cell membrane, disrupts its integrity, and leads to cell death [9]. Various studies have revealed that necroptosis could be implicated in the pathogenesis of many inflammatory and autoimmune diseases, including SLE [6, 10, 11].
The diversity of the SLE might reflect differences in pathogenesis between different subgroups [12]. Approaches are needed to better understand the pathogenesis and to find new targets for various stages of the disease. Considering the role of necroptosis in the pathogenesis and development of SLE [13,14,15,16], we aimed to analyze MLKL mRNA of PBMCs and figure out whether it could serve as a biomarker for disease diagnosis and monitoring.
Subjects and methods
Study cohorts
We enrolled 59 patients with SLE and 25 patients with RA admitted to the Department of Rheumatology and Immunology of the Third Affiliated Hospital, Southern Medical University, China, from July 2019 to December 2019. Thirty age- and sex-matched HC individuals with no history of SLE or other immune disorders were enrolled at the Health Management Center in the same hospital. All the subjects had no infections. The diagnosis of SLE was according to the 1997 revised American College of Rheumatology (ACR) classification criteria [17]. All participants provided written informed consent for blood draw and MLKL mRNA testing. Serum samples were obtained from all participants during the study.
Analyzing subgroups of SLE is increasingly important to better understand the pathogenesis of disease and provide more tailored medic protocols. Then, we sorted SLE patients into different groups based on serological features, renal involvement, and disease activity. Firstly, SLE patients were divided into two groups: positive ANA group (n = 48) and negative ANA group (n = 11). Another variable was renal involvement, defined as fulfilling the ACR classification criteria for renal manifestation of SLE (≥ 0.5 g of proteinuria per day or 3+ protein on urine dipstick analysis) or having evidence of LN on kidney biopsy. SLE patients were divided into two groups: LN patients (n = 23) and non-LN (n = 36) patients. Lastly, SLE patients were evaluated using the SLE Disease Activity Index (SLEDAI) [18] and divided into 2 groups: stable patients (SLEDAI score < 5, n = 32) and active patients (SLEDAI score ≥ 5, n = 27), according to the physicians’ evaluation.
Isolation of PBMCs and RNA extraction
Considering that autoreactive PBMCs, mainly lymphocytes, may participate in the autoimmune inflammatory process, we chose PBMCs as a source for determining MLKL mRNA level in SLE patients. The venous blood samples (4–5 mL) were collected in an EDTA-K2 tube from all the participants before breakfast, and PBMCs separated within 2 h by Ficoll (TBD Science, Tianjin, China) gradient centrifugation for 30 min at 1700 r/min. PBMCs were then transferred into 1 mL TRIzol Reagent in 1.5 mL centrifuge tubes and stored at − 80 °C until RNA extraction.
Total RNA was extracted from PBMCs by using TRIzol Reagent (Invitrogen, CA, USA) according to the manufacturer’s protocol and quantified with the NanoDrop ND-1000 (Thermo Scientific, USA). Approximately 200-800 ng of RNA was obtainted from 1mL of venous blood samples. Samples were used only if the ratio of the absorbance at 260 nm to that at 280 nm (A 260/A 280) was between 1.8 and 2.1. RNA samples with concentrations > 0.2 μg/μL were used for following reverse transcription reaction.
Real-time polymerase chain reaction validations
According to the manufacturer’s recommendations, 20 μL of final reaction mixture was used containing 10 μL of SYBR Green PCR Master Mix (Takara, Dalian, China), 0.8 μL of sense primer, 0.8 μL of antisense primer, 0.4 μL ROX Reference Dye (50×), 6 μL of sterile deionized water, and 2.0 μL of the synthesized cDNA. Primers were designed by Primer Premier 5.0 and synthesized by Sangon Biotech (Sangon, Shanghai, China). Primers targeting MLKL and human 18S-rRNA were used—MLKL, forward: 5′-GCCACTGGAAAGATCCCGTT-3′, reverse: 5′-CAACAACTCGGGGCAATCCT-3′; human 18S-rRNA, forward: 5′-TGGAAATCCCATCACCATCTTCC-3′, reverse: 5-GGTTCACACCCATGACG-3′. The relative expression level of MLKL was normalized to the internal control 18S-rRNA expression and calculated by the comparative CT (△△CT) method. Amplification was performed in 40 cycles (30 s at 95 °C, 5 s at 95 °C, 34 s at 60 °C) by ABI Step One Plus Real-Time PCR system (Applied Biosystems, CA, USA). A melt curve analysis was used to confirm the specificity of amplification.
Serological assays
The serum total ANA was measured by an indirect immunofluorescence assay (Euroimmun, AG) with a titer of > 1:80 scored as positive. The antibodies to 15 antigens including double-stranded DNA (dsDNA), Smith antigen (Sm), and nucleosome (Nuc), SSA/60, SSA/52, SSB/La, ribonucleoprotein (rRNP), centromereprotein B (CENPB), ribosome P protein (Rib-p), histone (His), proliferating cell nuclear antigen (PCNA), Scl-70, Jo-1, and mitochondria (M2) were detected by chemiluminescent immunoassay (CLIA) (HOB, Suzhou, China). Serum complement 1q (C1q), complement 3 (C3), complement 4 (C4), immunoglobulin G (IgG), immunoglobulin M (IgM), and immunoglobulin A (IgA) were detected by immunoturbidimetric assay (Roche, Shanghai, China), and D-dimer concentration was determined with immunoturbidimetric assay (Sysmex, Japan) according to the manufacturer’s instructions.
Statistical analysis
All data were statistically analyzed using GraphPad Prism 5 (version 5.0) software. Quantitative data were expressed as the mean ± SD. Data with a Gaussian distribution was analyzed using an unpaired t test or one-way analysis of variance (ANOVA), and Spearman’s rank was used to analyze the correlation of the numbers of leukocyte, lymphocyte, and monocyte, with the numbers of positive ANA, CRP, ESR, and D-dimer (D-D) levels. The area under the curve (AUC) was used to assess the specificity and sensitivity of using MLKL mRNA as a novel diagnostic tool for the detection of SLE. p values less than 0.05 were considered statistically significant.
Results
Characteristics and subgroups of SLE patients
Characteristics of the 59 SLE patients, 25 RA patients, and 30 matched HC individuals are shown in Table 1. The median age of SLE patients was 33.68 ± 13.55 years, with 96.6% females (57/59). The majority of SLE patients (48/59, 81.4%) were ANA-positive, less than half of the patients diagnosed with LN (23/59, 40.4%), 32 patients classified as stable patients (low disease activity), and 27 patients as active patients (high disease activity).
MLKL mRNA was upregulated in the PBMCs of SLE patients
We detected MLKL mRNA levels in the PBMCs of SLE patients, RA patients, and HC individuals. The levels of MLKL mRNA were significantly higher in SLE patients than in RA patients and HC individuals (p < 0.0001, respectively, Fig. 1).

The increased levels of MLKL mRNA in the PBMCs of SLE patients. RT-PCR was used to determine the relative expression level of MLKL mRNA in the PBMCs of SLE (n = 59), RA (n = 25), and HC individuals (n = 30). ***p < 0.0001
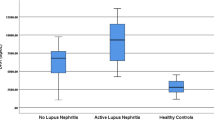
We further analyzed the expression of MLKL mRNA in the subgroups of SLE patients and found that SLE patients positive for ANAs exhibited significantly higher levels of MLKL mRNA than those with negative ANAs (p < 0.05, Fig. 2a). MLKL mRNA was also found to be obviously upregulated in LN patients when compared with patients without LN (p < 0.005, Fig. 2b), and higher in active patients than in stable patients (p < 0.05, Fig. 2c).

The differential expression of MLKL mRNAs in the three subgroups of SLE. a SLE patients with positive ANA (ANA+, n = 48) vs negative ANA (ANA−, n = 11). b LN patients (n = 23) vs non-LN patients (n = 36). c Active patients (SLEDAI score ≥ 5, n = 27) vs stable patients (SLEDAI score < 5, n = 32). *p < 0.05; **p < 0.005
MLKL mRNA was positively correlated with clinical and pathological parameters of SLE
The relationships between the levels of MLKL mRNA in PBMCs and the clinical or pathological characteristics of SLE are assessed and detailed in Table 2. Interestingly, MLKL mRNA level was significantly and positively correlated with ESR (r = 0.4091, p = 0.0043), CRP (r = 0.3571, p = 0.0237), serum IgG concentration (r = 0.3546, p = 0.0289), and the numbers of positive ANAs (r = 0.3597, p = 0.0432) (Fig. 3), but not associated with C3, C4, and other serologic indicators. Taken together, we found that increased MLKL mRNA was correlated with the activity in SLE patients.

Positive correlations of MLKL mRNA levels with clinical parameters of SLE patients
MLKL mRNA in the PBMCs was sensitive for the diagnosis of SLE
ROC analysis was employed to analyze the diagnostic efficiency of the MLKL mRNA for SLE patients. The diagnostic ability of MLKL mRNA achieved very high diagnostic accuracy 0.9277 (95% CI 0.878–0.978) with high sensitivity (81.36%) and specificity (93.3%), implying that MLKL mRNA of PBMCs may be a potential diagnosis biomarker for SLE (Fig. 4).

ROC analysis of MLKL mRNA for SLE patients
Discussion
In this study, we showed that MLKL mRNA level in the PBMCs of SLE patients was significantly upregulated, especially in patients with positive serum ANAs. MLKL mRNA level in the PBMCs was also significantly and positively correlated with ESR (r = 0.4091, p = 0.0043), CRP (r = 0.3571, p = 0.0237), serum IgG concentration (r = 0.3546, p = 0.0289), and the numbers of positive ANAs (r = 0.3597, p = 0.0432). So far as we know, this is the first report that MLKL mRNA level in the PBMCs is increased in SLE patients.
Once phosphorylated, MLKL translocates from the cytosol to the plasma membrane to execute necroptosis. Defective clearance of necroptotic cells has been proposed to initiate inflammatory responses by the release of danger-associated molecular patterns (DAMPs). DNA acts as a major DAMP and is sensed in endolysosomes by toll-like receptor 9 (TLR9) and in the cytoplasm by cyclic GMP–AMP (cGAMP) synthase (cGAS), inducing the production of type I and type III IFNs and eliciting strong inflammatory responses [19, 20]. Several studies have demonstrated that patients with SLE have elevated circulating IFNs [21,22,23], whose signaling contributes to the steady-state expression of MLKL and the initiation of necroptosis, which not only causes tissue damage [6], but may also form a dynamic feedback loop in SLE pathogenesis.
Although SLE is a chronic inflammatory disease that can affect many organs, the kidneys are the mostly attacked [24]. LN is one of the most frequent and serious complications in SLE, and a real challenge for SLE treatment [25]. We surprisingly found that MLKL mRNA was obviously upregulated in the PBMCs of LN patients when compared with patients without LN (p < 0.005). To date, only one paper reported the correlation of necroptosis with LN, showing that PIPK3 and MLKL were activated in podocytes in renal biopsies from patients with LN [11]. Whether there is a crosstalk between the renal parenchymal cells and peripheral blood cells in necroptosis process still needs to be analyzed.
The current understanding of SLE implies autoimmunity to nuclear and cytoplasmic antigens, leading to generation large amount of ANAs [26]. Our findings are that SLE patients positive for ANAs exhibited higher MLKL mRNA levels than serum negative patients and that there are very significantly positive correlations between MLKL mRNA in the PBMCs and the numbers of positive ANAs or serum IgG concentrations, suggesting that necroptosis may play a potential role in the production of ANAs.
Conventional serologic ANAs are of limited sensitivity and/or specificity for diagnosis and monitoring in SLE [27]. Here, we reported that MLKL mRNA in the PBMCs could differentiate between SLE patients and HC individuals, and the AUC was as high as 0.9277 (95% CI 0.878–0.978) with high sensitivity (81.36%) and specificity (93.3%). As PBMCs are easy to obtain, this suggests that MLKL mRNA of PBMCs may be a novel biomarker for the diagnosis and monitoring of disease activity of SLE.
There were also several limitations. Firstly, as the patients in this study are from one hospital, whether there is a difference between patients from different areas is not known. Therefore, a multi-center cohort might be necessary for future implementation of techniques. Secondly, the molecular mechanism that how MLKL is involved in the progression of SLE remains unclear. Lastly, which specific cell of PBMCs expressed high MLKL mRNA level needs to be explored in the future.
Conclusions
This is the first study to point out the upregulation of the MLKL mRNA in the PBMCs of SLE patients. The data presented here may provide certain evidence for the role of necroptosis in the pathogenesis and development of SLE, and also suggest new therapies by blocking signaling of necroptosis pathway in human SLE, especially in LN patients. Importantly, the MLKL mRNA expression levels in PBMCs may be useful in identifying those subgroups of SLE patients that may benefit from necroptotic blocking therapies. Finally, we believe that these findings could be of relevance for understanding the pathogenesis and diversity of SLE.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- MLKL:
-
Mixed lineage kinase domain-like pseudokinase
- PBMCs:
-
Peripheral blood mononuclear cells
- SLE:
-
Systemic lupus erythematosus
- RT-PCR:
-
Real-time transcription-polymerase chain reaction
- RA:
-
Rheumatoid arthritis
- HC:
-
Healthy controls
- ROC:
-
Receiver operating characteristic
- LN:
-
Lupus nephritis
- CRP:
-
C-reaction protein
- ESR:
-
Erythrocyte sedimentation rate
- ANAs:
-
Antinuclear antibodies
- IgG:
-
Immunoglobulin G
- IgM:
-
Immunoglobulin M
- IgA:
-
Immunoglobulin A
- CLIA:
-
Chemiluminescent immunoassay
- TNF:
-
Tumor necrosis factor
- TLR:
-
Toll-like receptor
- IFNs:
-
Interferons
- RIPK:
-
Receptor-interacting serine/threonine-protein kinase
- ACR:
-
American College of Rheumatology
- SLEDAI:
-
SLE Disease Activity Index
- DAMP:
-
Damage-associated molecular pattern
- cGAS:
-
Cytoplasm by cyclic GMP–AMP (cGAMP) synthase
References
Yeoh S, Dias SS, Isenberg DA. Advances in systemic lupus erythematosus. Medicine. 2018;46(2):84–92.
Olsen NJ, Karp DR. Autoantibodies and SLE—the threshold for disease. Nat Rev Rheumatol. 2014;10(3):181–6.
Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716–30.
Visan I. Necroptosis regulator. Nat Immunol. 2015;16(5):447.
He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108(50):20054–9.
Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, Ilyukha V, Sorokin M, Buzdin A, Fitzgerald KA, et al. Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ. 2019;26(2):332–47.
Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014;5(7):e1312.
Someda M, Kuroki S, Miyachi H, Tachibana M, Yonehara S. Caspase-8, receptor-interacting protein kinase 1 (RIPK1), and RIPK3 regulate retinoic acid-induced cell differentiation and necroptosis. Cell Death Differ. 2020;27(5):1539-53.
Weber K, Roelandt R, Bruggeman I, Estornes Y, Vandenabeele P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun Biol. 2018;1(1):6.
Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–20.
Guo C, Fu R, Zhou M, Wang S, Huang Y, Hu H, Zhao J, Gaskin F, Yang N, Fu SM. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J Autoimmun. 2019;103:102286.
Franklyn K, Hoi A, Nikpour M, Morand EF. The need to define treatment goals for systemic lupus erythematosus. Nat Rev Rheumatol. 2014;10(9):567–71.
Salem D, Subang R, Kuwana M, Levine JS, Rauch J. T cells from induced and spontaneous models of SLE recognize a common T cell epitope on β2-glycoprotein I. Cell Mol Immunol. 2019;16(8):685–93.
Morel L. Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol. 2017;13(5):280–90.
Catalina MD, Bachali P, Geraci NS, Grammer AC, Lipsky PE. Gene expression analysis delineates the potential roles of multiple interferons in systemic lupus erythematosus. Commun Biol. 2019;2(1):140.
Fan H, Liu F, Dong G, Ren D, Xu Y, Dou J, Wang T, Sun L, Hou Y. Activation-induced necroptosis contributes to B-cell lymphopenia in active systemic lupus erythematosus. Cell Death Dis. 2014;5(9):e1416.
Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725.
Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35(6):630–40.
Paludan SR, Reinert LS, Hornung V. DNA-stimulated cell death: implications for host defence, inflammatory diseases and cancer. Nat Rev Immunol. 2019;19(3):141–53.
Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38(5):870–80.
Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, Ueda M, Nishide M, Koyama S, Hayama Y, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. 2018;77(10):1507–15.
Liu M, Guo Q, Wu C, Sterlin D, Goswami S, Zhang Y, Li T, Bao C, Shen N, Fu Q, et al. Type I interferons promote the survival and proinflammatory properties of transitional B cells in systemic lupus erythematosus patients. Cell Mol Immunol. 2019;16(4):367–79.
Niewold TB, Hua J, Lehman TJA, Harley JB, Crow MK. High serum IFN-α activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8(6):492–502.
Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J Am Soc Nephrol. 2017;12(5):825–35.
Nnodum B, Dudley L. Class IV lupus nephritis in the setting of serologically quiescent disease and normal urine sediment in a patient with late-onset systemic lupus erythematosus. Case Rep Rheumatol. 2019;2019:1219529.
Scharer CD, Blalock EL, Mi T, Barwick BG, Jenks SA, Deguchi T, Cashman KS, Neary BE, Patterson DG, Hicks SL, et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat Immunol. 2019;20(8):1071–82.
Vasquez-Canizares N, Wahezi D, Putterman C. Diagnostic and prognostic tests in systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2017;31(3):351–63.
Acknowledgements
The authors wish to express gratitude to their colleagues in the research group of Dr. E.W. Sun.
Funding
This work was supported by grants from the National Natural Science Foundation of China (grant nos. 31600732, 81671623, 81873880) and sponsored by Tianhe District of Guangzhou City Science And Technology Special Project (grant no. 201704KW032).
Author information
Authors and Affiliations
Contributions
Mingjiao Zhang and Hongyu Jie carried out most of the experiments, participated in the analysis of data, and drafted the manuscript. Yong Wu participated in the design of the study, data analysis, and interpretation and drafted the manuscript. Xinai Han performed the statistical analysis. Xing Li et al. participated in the RT-PCR assays and revising the manuscript. E.W. Sun participated in the design and coordination of the study and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study has been approved by the Ethics Committee of the Third Affiliated Hospital, Southern Medical University, and all participants signed informed consent forms.
Consent for publication
All authors consented to the publication of this manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, M., Jie, H., Wu, Y. et al. Increased MLKL mRNA level in the PBMCs is correlated with autoantibody production, renal involvement, and SLE disease activity. Arthritis Res Ther 22, 239 (2020). https://doi.org/10.1186/s13075-020-02332-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-020-02332-7