
Abstract
Objectives
Histone deacetylase (HDAC) 6 promotes inflammation. We investigated the anti-arthritic effects of CKD-506, a novel HDAC6 inhibitor, in vitro and in a murine model of arthritis as a novel treatment option for rheumatoid arthritis (RA).
Methods
HDAC6 was overexpressed in mouse peritoneal macrophages and RAW 264.7 cells, and the effects of a HDAC6 inhibitor CKD-506 on cytokine production and activity of NF-κB and AP-1 signaling were examined. Peripheral blood mononuclear cells (PBMCs) from RA patients and fibroblast-like synoviocytes (FLS) were activated in the presence of CKD-506. Next, regulatory T cells (Tregs) were induced from RA patients and co-cultured with healthy effector T cells (Teffs) and cell proliferation was analyzed by flow cytometry. Finally, the effects of the inhibitor on the severity of arthritis were assessed in a murine model of adjuvant-induced arthritis (AIA).
Results
Overexpression of HDAC6 induced macrophages to produce TNF-α and IL-6. The inhibitory effect of CKD-506 was mediated via blockade of NF-κB and AP-1 activation. HDAC6 inhibition reduced TNF-α and IL-6 production by activated RA PBMCs. CKD-506 inhibited production of MMP-1, MMP-3, IL-6, and IL-8 by activated FLS. In addition, CKD-506 inhibited proliferation of Teffs directly and indirectly by improving iTreg function. In AIA rats, oral CKD-506 improved clinical arthritis in a dose-dependent manner. A combination of sub-therapeutic CKD-506 and methotrexate exerted a synergistic effect.
Conclusion
The novel HDAC6 inhibitor CKD-506 suppresses inflammatory responses by monocytes/macrophages, improves Treg function, and ameliorates arthritis severity in a murine model of RA. Thus, CKD-506 might be a novel and effective treatment option for RA.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation and destruction of joints [1]. Activated immune cells produce inflammatory cytokines, including tumor necrosis factor (TNF-α), interleukin (IL)-1β, and IL-6, which drive inflammatory responses and induce fibroblast-like synoviocytes (FLS) to produce tissue-destructive matrix metalloproteinases (MMPs) and chemokines. These inflammatory mediators, in turn, recruit immune cells and potentiate joint destruction [2, 3]. Regulatory T (Treg) cells, which counteract “overshooting” of the immune response, are compromised in RA patients [4]. Therefore, a treatment that targets multiple key steps in RA pathogenesis might be more effective than those that target a single pathway. A potential treatment is inhibition of histone deacetylase (HDAC) [5, 6].
While nuclear HDAC is critical for epigenetic regulation, which ultimately determines cell differentiation and function, cytosolic HDACs are involved in post-translational modification of non-histone proteins in the cytosol, which are essential for cellular functions such as intra-cellular transport and cell migration among others [7]. HDAC activity in RA patients is higher than that in healthy controls, and HDAC inhibitors are effective in murine models of RA [8,9,10,11,12]. Non-specific pan-HDAC inhibitors, which suppress multiple isoforms of HDAC with pleiotropic effects, have anti-inflammatory properties; however, they are also associated with side effects such as fatigue, diarrhea, nausea, and neutropenia [13, 14]. Therefore, selective inhibition of a specific HDAC isoform might offer substantial advantages with a better safety margin. HDAC6 is different from other HDAC isotypes in that it is present almost exclusively in the cytosol and, therefore, is not involved in epigenetic regulation [15, 16]. Overexpression of HDAC6 is associated with increased inflammatory responses [17], and inhibiting it reduces disease activity in murine model of RA and systemic lupus erythematosus [18, 19]. CKD-506 is a potent and selective HDAC6 inhibitor (> 100-fold selectivity for HDAC6 over other HDAC isotypes). It induced tubulin acetylation in a dose-dependent manner without affecting histone H4 acetylation in both human and murine cells [20]. CKD-506 ameliorated acute and chronic murine colitis and lupus nephritis in animal models [20, 21].
The aim of this study was to investigate the therapeutic effects of CKD-506, a novel HDAC6 inhibitor, as a potential drug candidate for the treatment of RA. We show that inhibiting HDAC6 with CKD-506 improves Treg function, suppresses inflammatory responses by macrophages and FLS, and attenuates arthritis in a murine model of adjuvant-induced arthritis (AIA).
Materials and methods
Cell preparation
Resident peritoneal macrophages were isolated from 6-week-old male ICR mice (Experimental Animal Center, Hallym University) as described previously [17]. Peripheral blood mononuclear cells (PBMCs) were isolated from the blood of RA patients by Ficoll-Hypaque density centrifugation and resuspended in RPMI-1640 containing 1% fetal bovine serum and 1% penicillin/streptomycin. FLS, isolated from synovial tissue taken from RA patients, were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin. For all experiments, cells were used between passages 3 and 7. The viability of PBMCs and FLS was evaluated after incubation for 2 h with CCK-8 (DOJINDO, Kumamoto, Japan). Optical density was read at 450 nm.
Transient transfection and luciferase assay
RAW 264.7 cells were transfected with a pcDNA3.1 control vector or a HDAC6 expression vector (pcDNA-HDAC6-FLAG) using Lipofectamine 3000 reagent (Thermo Fisher Scientific). For the reporter assays, cells were co-transfected with a pNF-κB-luc or pAP-1-luc plasmid (Stratagene, La Jolla, CA, USA) and a control (pCMV-β-galactosidase) plasmid using Lipofectamine 3000 reagent as previously described [17]. Cell lysates were prepared, and luciferase and β-galactosidase activity analyzed. The luciferase activity of each sample was normalized to that of β-galactosidase, and the results were expressed as a fold change in transactivation.
Measurement of cytokines in cell culture supernatants
RA PBMCs were treated with HDAC6 inhibitors and then stimulated with LPS. RA FLS were treated with HDAC6 inhibitors and then stimulated with IL-1β. After 24 h, cell culture supernatants were collected and the amounts of TNF-α, IL-1β, IL-6, and IL-10 secreted by PBMCs and the amounts of MMP-1, MMP-3, IL-6, and IL-8 secreted by FLS were measured in enzyme-linked immunosorbent assays (ELISAs).
Cell proliferation assay
CD4+ CD25− T cells were purified from healthy PBMCs by negative selection using a CD4+ T Cell Biotin-Antibody Cocktail (Miltenyi Biotec). Induced Treg (iTreg) cells were generated from CD4+ CD25− T cells of RA patients in the presence of an anti-CD3 antibody (eBioscience, San Diego, CA, USA), an anti-CD28 antibody (BD Pharmingen, San Diego, CA, USA), IL-2 (PEPROTECH), TGF-β (PEPROTECH), and vitamin D3 (SIGMA) [15]. CD4+ CD25− T cells from healthy controls were labeled for 10 min with 5 mM carboxyfluorescein diacetate succinimidyl ester (CFSE) (Life Technologies, Eugene, OR, USA). RA iTreg cells and CFSE-labeled healthy effector T cells were co-cultured for 72 h at a ratio of 0:1, 0.3:1, and 1:1 in the presence of Dynabeads Human T-Activator CD3/CD28 (Invitrogen Dynal AS, Life Technologies, Oslo, Norway). T cell proliferation was measured by flow cytometry.
Induction of experimental arthritis in rats
All animal experiments were approved by the Animal Care and Use Committee. Lewis rats (female, 5 weeks old) were purchased from Central Lab Animal, Inc. (Seoul, Korea). Complete Freund’s adjuvant (CFA) (Chondrex, Seattle, WA, USA) was resuspended vigorously and 100 μl injected subcutaneously into the tail base. Animals were randomized into six groups (0.5% methylcellulose as vehicle, n = 7; CKD-506, 3 mg/kg, n = 8; CKD-506, 10 mg/kg, n = 8; CKD-506, 30 mg/kg, n = 8; CKD-506, 50 mg/kg, n = 8; and CKD-506, 100 mg/kg, n = 8). Each group received vehicle or oral CKD-506 once a day from day − 1 to day 16 relative to the injection. The severity of arthritis was assessed on days 9, 13, and 16 after injection of complete Freund’s adjuvant. Thereafter, rats were sacrificed.
Arthritis assessment
The severity of arthritis was evaluated by scoring each joint (digits, metatarsal bones, and tarsal bones) as follows: 0, no swelling or erythema; 1, slight swelling and/or erythema; 2, low to moderate edema; 3, pronounced edema with limited joint usage; and 4, excess edema with joint rigidity. The clinical scores of four joints were summed to generate a total score for each animal.
Statistical analysis
Data are presented as the mean ± SEM. Group in vitro experiments were compared using t tests. Clinical scores of treatment groups over time were compared using repeated measure analysis of variance (RM-ANOVA). All statistical analyses were performed in Prism software (GraphPad, La Jolla, CA, USA). p values < 0.05 were considered statistically significant.
Results
HDAC6 mediates pro-inflammatory responses via NF-κB and AP-1 signaling
First, we examined the effects of CKD-506 on production of pro-inflammatory cytokines and related signaling pathways in peritoneal macrophages transfected with an HDAC6 expression vector. Overexpression of HDAC6 increased spontaneous production of TNF-α and IL-6. By contrast, CKD-506 decreased production of TNF-α and IL-6 in a dose-dependent manner (Fig. 1a, b). Next, we analyzed the effect of CKD-506 on NF-κB and AP-1 promoter activity in HDAC6-transfected RAW 264.7 cells. Pretreatment of HDAC6-transfected cells with CKD-506 reduced NF-κB and AP-1 promoter activity in a dose-dependent manner (Fig. 1c, d). CKD-506 suppressed production of TNF-α, but not that of IL-6, by PBMCs from RA patients in response to LPS stimulation (Fig. 1e, f). These results suggest that CKD-506 inhibits HDCA6-mediated production of pro-inflammatory cytokines by regulating NF-κB and AP-1 signaling cascades. Of note, tubastatin A, another HDAC6 inhibitor, showed a similar effect.

HDAC6 inhibitors suppress HDAC6-induced inflammatory responses. a, b Primary peritoneal macrophages (n = 3) were pretreated for 1 h with the indicated concentrations with CKD-506 or tubastatin (TBA) and then transfected with a control (pcDNA3.1 [pc]) or HDAC6 expression vector (1 μg/ml). At 48 h post-transfection, the levels of TNF-α (a) and IL-6 (b) in the culture medium were measured by ELISA. c, d RAW 264.7 cells (n = 3) were pretreated for 1 h with CKD-506 and then transiently co-transfected with an NF-κB (c) or AP-1 (d) promoter-luciferase expression vector, a β-galactosidase plasmid (pCMV-lacZ), and a control or HDAC6 expression vector. After 48 h, the luciferase activity in transfected cells was determined. Luciferase activity was normalized to that of β-galactosidase and expressed as -fold change over the control level. Data are expressed as the mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001, compared with HDAC6-transfected cells. e, f PBMCs from RA patients (n = 5) were pretreated for 1 h with increasing concentrations of CKD-506 and then stimulated with LPS (100 ng/ml). Production TNF-α and IL-6 in the supernatant was measured using an ELISA. All data represent the mean value ± SEM. *p < 0.05, **p < 0.01 compared with no CKD-506 treatment. pc, plasmid control; TNF, tumor necrosis factor; IL, interleukin; TBA, tubastatin
CKD-506 suppresses metalloproteinase and cytokine/chemokine production by FLS
RA FLS were pretreated with increasing concentrations of CKD-506 and then stimulated with IL-1β. In response to IL-1β, FLS produced a large amount of metalloproteinases (MMP-1 and MMP-6), IL-6, and IL-8, along with chemokines CXCL10 and CCL2. Pretreatment with CKD-506 significantly reduced production of all of the above (Fig. 2).

CKD-506 suppresses production of tissue-degradative enzymes, inflammatory cytokines, and chemokines by fibroblast-like synoviocytes (FLS). RA FLS (n = 3) were pretreated for 1 h with CKD-506 and then activated for 24 h with IL-1β (10 ng/ml). Then, the amounts of MMP-1 (a), MMP-3 (b), IL-6 (c), IL-8 (d), CXCL10 (e), and CCL2 (f) in the supernatant were measured in an ELISA. Data represent the mean value ± SEM. *p < 0.05
CKD-506 improves Treg function
We investigated whether CKD-506 improves impaired Treg function in RA patients. First, iTregs from RA patients were co-cultured with CFSE-labeled T cells from healthy donors in the presence of increasing concentrations of CKD-506. In the absence of CKD-506, T cell proliferation decreased as the ratio of iTreg to T cells increased. CKD-506 potentiated inhibition of T cell proliferation. Interestingly, CKD-506 inhibited T cell proliferation even in the absence of iTregs. CKD-506 increased expression of CTLA4 by Foxp3+ iTregs and Foxp3− T cells (Fig. 3).

CKD-506 augments Treg-mediated suppression of T cell proliferation. Induced Treg cells (iTreg) from RA patients (n = 3) were co-cultured for 72 h at different ratios with CFSE-labeled T cells from healthy controls in the presence of an HDAC6 inhibitor and CD3/28 Dynabeads, and the proliferation of T cells was examined by FACS. a Representative FACS data from three independent experiments are shown. b T cell proliferation under different ratio of iTreg to T cells. c Changes in expression of CTLA4 by iTreg and T cells after treatment with CKD-506. *p < 0.05, **p < 0.01, ***p < 0.001 vs. no treatment
CKD-506 prevents experimental arthritis in a murine model
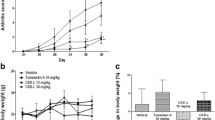
The efficacy of CKD-506 on inflammatory arthritis was evaluated in a murine arthritis model. Rats were treated with daily oral CKD-506 at 3, 10, 30, 50, and 100 mg/kg, or with tofacitinib at 5 mg/kg, from 1 day before to 16 days after CFA injection. The clinical arthritis scores started to rise on day 9 and continued to rise until day 16 (Fig. 4a). CKD-506 reduced the arthritis score on days 13 and 16 in a dose-dependent manner (Fig. 4b). CDK-506 at 100 mg/kg/day, and tofacitinib at 5.0 mg kg/day, had comparable effects with respect to inhibiting synovial inflammation and bone destruction (Fig. 4c, d). Of note, serum levels of anti-CCP antibody on day 16 tended to be lower in rats treated with CKD-506 (Supplementary figure S1).

CKD-506 prevents experimental arthritis in an AIA rat model. a Experimental design. Rats with experimentally induced arthritis were treated with daily oral CKD-506. Severity of arthritis was assessed on days 9, 13, and 16 after CFA injection. Rats were sacrificed thereafter. b Clinical scores over the treatment period are shown according to CKD-506 treatment dose. Groups were compared using repeated measure ANOVA, followed by post hoc analysis with Bonferroni corrections. c Representative 3D reconstructed image of the hind foot. White arrows indicate bone erosions. d The efficacy of CKD-506 at 100 mg/kg QD vs. tofacitinib 5 mg/kg (with respect to the effects on synovial inflammation and thickness) was compared. Data represent the mean value ± SEM. *p < 0.05, **p < 0.01 vs. CKD-506 at 0 mg/kg. CFA, complete Freund’s adjuvant; Tofa, tofacitinib
CKD-506 ameliorates experimental arthritis and has synergistic effect of CDK-506 with methotrexate in a murine model
To test the therapeutic effect of CKD-506, rats were treated with daily CKD-506 at 0, 30, and 100 mg/kg/day from 9 to 16 days after CFA injection (Fig. 5a). CKD-506 ameliorated the arthritis score in a dose-dependent manner (Fig. 5b). AIA rats were treated with a sub-therapeutic dose of CKD-506 (1 mg/kg/day) or methotrexate (MTX; 1 m/kg/week), or both. CKD-506 or MTX alone did not improve the clinical score compared with vehicle treatment. However, the combination of CKD-506 and MTX reduced the arthritis score significantly on days 13 (5.8 + 3.1 vs. 0.3 + 0.2 p < 0.001) and 16 (8.2 + 3.7 vs. 2.9 + 2.3, p = 0.003), respectively (Fig. 5c).

CKD-506 ameliorates experimental arthritis and shows a synergistic effect with MTX in an AIA rat model. a After induction of arthritis, rats were treated with CKD-506 with or without MTX starting 9 days after CFA injection. b Anti-arthritic effects by CKD-506 at 0 (vehicle treatment), 30, and 100 mg/kg/day were depicted. c Anti-arthritic effects of low dose MTX 1 mg/kg/week, low dose CKD 1 mg/kg/day, and their combination treatment were shown. Data represent the mean value ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. vehicle treatment. Groups were compared using repeated measure ANOVA, followed by post hoc analysis with Bonferroni corrections
Discussion
A hallmark of RA is chronic inflammation and destruction of joints. Dysfunction at various steps of the complex network comprising humoral and cellular immune responses results in perpetuation of inflammatory activation during development of RA, in which pro-inflammatory autoimmune responses are not adequately regulated by anti-inflammatory mechanisms [3]. Here, we show that CKD-506, a novel specific HDAC6 inhibitor, suppressed inflammatory responses by macrophages, improved Treg function, and inhibited inflammatory responses and the joint-destructive properties of FLS in vitro. In vivo, CKD-506 attenuated arthritis in a murine RA model with efficacy comparable with that of tofacitinib. In addition, CKD-506 had a synergistic effect when combined with methotrexate (both drugs were used at a dose that was ineffective when each was used alone).
Pan-HDAC inhibitors exhibit strong anti-inflammatory and anti-proliferative properties in several immune-mediated and hematologic malignancies; however, they have a narrow therapeutic margin due to side effects such as memory loss, cytopenia, and diarrhea [7, 14, 22,23,24]. In addition, epigenetic changes might have an unpredictable, long-lasting impact on the immune and non-immune cells even after HDAC inhibition is stopped. HDAC6 is localized predominantly to the cytoplasm where it deacetylates cytoplasmic non-histone proteins such as HSP90 and α-tubulin [15, 25, 26]. Hyperacetylation of α-tubulin has anti-inflammatory effects [23, 24]. CKD-506 is a highly specific inhibitor of HDAC6; as such, it has pleiotropic effects on both immune cells and FLS, which constitute the main cellular infiltrates in inflamed RA joints [20]. Here, we found that CKD-506 decreased secretion of pro-inflammatory cytokines TNF-α and IL-6 by macrophages in part via suppression of the NF-κB and AP-1 pathways (Fig. 1). Overexpression of HDAC6 during an inflammatory response upregulated NADPH oxidase with increased generation of cytosolic reactive oxygen species (ROS). The generated ROS promotes activation of the NF-κB and AP-1 signaling pathways with subsequent transcription and production of inflammatory cytokines [17]. Intriguingly, CKD-506 did not significantly suppress IL-6 production by the LPS-stimulated RA PBMCs (Fig. 1f). This is likely due to the relatively low IL-6 production by PBMCs which results in “less suppressive effect” of CKD-506.
CKD-506 suppressed immune responses directly by suppressing proliferation of activated T cells and indirectly by augmenting the function of regulatory T cells (Fig. 3). Restoration of defective Treg function is crucial for maintenance of self-tolerance and for preventing aggressive autoimmune cells from destroying tissues in RA joints [27]. Transcription factor Forkhead box P3 (Foxp3) is crucial for the development and function of Tregs. In Treg cells, acetylation of Foxp3 is an important post-translational mechanism to regulate its concentration since acetylated Foxp3 is protected from proteosomal degradation. HDAC6 inhibitor therefore increases Foxp3 half-life and so enhances suppressive function of Treg cells [28, 29]. It is possible that T cells become more sensitive to the Treg-mediated inhibition by CKD-506. Therefore, CKD-506 suppresses T cell proliferation by multiple mechanisms.
The inflammatory environment of RA transforms normal resident synoviocytes into tissue-destructive FLS, which produce large amounts of IL-6, IL-8, and extracellular matrix proteinases MMP-1 and MMP-3 [30, 31]. CKD-506 markedly inhibits joint-destructive FLS [18, 29]. The multicellular effects of HDAC6 inhibition by CKD-506 might contribute to the anti-arthritic effects observed in the murine RA model; these effects were comparable with those of tofacitinib, a JAK/STAT signaling inhibitor with proven clinical efficacy (Fig. 4). Of note, CKD-506 exhibited both prophylactic and therapeutic effects; CKD-506 treatment before and after induction of arthritis showed anti-arthritic effects although the prophylactic treatment ameliorated the arthritis score better than therapeutic treatment. Strikingly, the combination of low (sub-therapeutic) dose CKD-506 and low (sub-therapeutic) dose methotrexate had a synergistic therapeutic effect on the arthritis severity in the murine model (Fig. 5). A previous report shows that methotrexate restores Treg function by demethylating the upstream enhancer of FoxP3, which might also potentiate restoration of impaired Treg cell function by CKD-506 [32]. While the mechanism underlying synergism needs further investigation, combination treatment might enable reduction of the dose of CKD-506 and reduce the risk of side effects. Long-term drug safety is of particular interest since patients with RA require life-long treatment [33]. Taken together, inhibiting multiple cell types with the novel HDAC6 inhibitor CKD-506 is promising, and the results are consistent with the observed anti-arthritic effects of other HDAC6 inhibitors [34, 35]. This supports also the anti-inflammatory effect of CKD-506 as a novel therapeutic agent to treat diverse autoimmune diseases such as SLE and inflammatory bowel disease [20, 21].
A main limitation of the study is that we did not investigate the exact mechanism by which the HDAC6 inhibitor affects multiple steps in the inflammatory response; indeed, many cytosolic and non-cytosolic proteins are substrates for HDAC6, so examining the mechanism is important.
Conclusions
In conclusion, CKD-506, a novel HDAC6 inhibitor, regulates innate and adaptive immune responses and ameliorates experimental arthritis. CKD-506 has potential as a novel treatment option for treatment of RA.
Availability of data and materials
Not applicable.
Abbreviations
- AIA:
-
Adjuvant-induced arthritis
- CFSE:
-
Carboxyfluorescein diacetate succinimidyl ester
- ELISA:
-
Enzyme-linked immunosorbent assay
- FLS:
-
Fibroblast-like synoviocytes
- HDAC:
-
Histone deacetylase
- IL:
-
Interleukin
- MMP:
-
Matrix metalloproteinase
- MTX:
-
Methotrexate
- PBMC:
-
Peripheral blood mononuclear cell
- iTreg:
-
Induced regulatory T cell
- RA:
-
Rheumatoid arthritis
- TNF:
-
Tumor necrosis factor
References
Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–61.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19.
McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. 2017;389(10086):2328–37.
Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2008;105(49):19396–401.
Meda F, Folci M, Baccarelli A, Selmi C. The epigenetics of autoimmunity. Cell Mol Immunol. 2011;8(3):226–36.
Choo QY, Ho PC, Lin HS. Histone deacetylase inhibitors: new hope for rheumatoid arthritis? Curr Pharm Des. 2008;14(8):803–20.
Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–8.
Horiuchi M, Morinobu A, Chin T, Sakai Y, Kurosaka M, Kumagai S. Expression and function of histone deacetylases in rheumatoid arthritis synovial fibroblasts. J Rheumatol. 2009;36(8):1580–9.
Ren J, Liao X, Vieson MD, Chen M, Scott R, Kazmierczak J, Luo XM, Reilly CM. Selective HDAC6 inhibition decreases early stage of lupus nephritis by down-regulating both innate and adaptive immune responses. Clin Exp Immunol. 2018;191(1):19–31.
Nishida K, Komiyama T, Miyazawa S, Shen ZN, Furumatsu T, Doi H, Yoshida A, Yamana J, Yamamura M, Ninomiya Y, et al. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheum. 2004;50(10):3365–76.
Nasu Y, Nishida K, Miyazawa S, Komiyama T, Kadota Y, Abe N, Yoshida A, Hirohata S, Ohtsuka A, Ozaki T. Trichostatin A, a histone deacetylase inhibitor, suppresses synovial inflammation and subsequent cartilage destruction in a collagen antibody-induced arthritis mouse model. Osteoarthr Cartil. 2008;16(6):723–32.
Joosten LA, Leoni F, Meghji S, Mascagni P. Inhibition of HDAC activity by ITF2357 ameliorates joint inflammation and prevents cartilage and bone destruction in experimental arthritis. Mol Med. 2011;17(5–6):391–6.
Hancock WW, Akimova T, Beier UH, Liu Y, Wang L. HDAC inhibitor therapy in autoimmunity and transplantation. Ann Rheum Dis. 2012;71(Suppl 2):i46–54.
Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals (Basel). 2010;3(9):2751–67.
Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18(6):291–7.
Spiegel S, Milstien S, Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene. 2012;31(5):537–51.
Youn GS, Lee KW, Choi SY, Park J. Overexpression of HDAC6 induces pro-inflammatory responses by regulating ROS-MAPK-NF-kappaB/AP-1 signaling pathways in macrophages. Free Radic Biol Med. 2016;97:14–23.
Oh BR, Suh DH, Bae D, Ha N, Choi YI, Yoo HJ, Park JK, Lee EY, Lee EB, Song YW. Therapeutic effect of a novel histone deacetylase 6 inhibitor, CKD-L, on collagen-induced arthritis in vivo and regulatory T cells in rheumatoid arthritis in vitro. Arthritis Res Ther. 2017;19(1):154.
Regna NL, Vieson MD, Gojmerac AM, Luo XM, Caudell DL, Reilly CM. HDAC expression and activity is upregulated in diseased lupus-prone mice. Int Immunopharmacol. 2015;29(2):494–503.
Choi EW, Song JW, Ha N, Choi YI, Kim S. CKD-506, a novel HDAC6-selective inhibitor, improves renal outcomes and survival in a mouse model of systemic lupus erythematosus. Sci Rep. 2018;8(1):17297.
Lee JW, Lee SM, Chun J, Im JP, Seo SK, Ha N, Il Choi Y, Kim JS. Novel histone deacetylase 6 inhibitor CKD-506 inhibits NF-kappaB signaling in intestinal epithelial cells and macrophages and ameliorates acute and chronic murine colitis. Inflamm Bowel Dis. 2020;26(6):852–62.
Aldana-Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer. J Biomed Biotechnol. 2011;2011:875824.
Kim DH, Kim M, Kwon HJ. Histone deacetylase in carcinogenesis and its inhibitors as anti-cancer agents. J Biochem Mol Biol. 2003;36(1):110–9.
Sweet MJ, Shakespear MR, Kamal NA, Fairlie DP. HDAC inhibitors: modulating leukocyte differentiation, survival, proliferation and inflammation. Immunol Cell Biol. 2012;90(1):14–22.
Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, Ordentlich P, Wang XF, Counter CM, Yao TP. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008;68(18):7561–9.
Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27(2):197–213.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64.
Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal. 2012;5(229):ra45.
Xiao Y, Li B, Zhou Z, Hancock WW, Zhang H, Greene MI. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr Opin Immunol. 2010;22(5):583–91.
Tolboom TC, Pieterman E, van der Laan WH, Toes RE, Huidekoper AL, Nelissen RG, Breedveld FC, Huizinga TW. Invasive properties of fibroblast-like synoviocytes: correlation with growth characteristics and expression of MMP-1, MMP-3, and MMP-10. Ann Rheum Dis. 2002;61(11):975–80.
Firestein GS. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum. 1996;39(11):1781–90.
Cribbs AP, Kennedy A, Penn H, Amjadi P, Green P, Read JE, Brennan F, Gregory B, Williams RO. Methotrexate restores regulatory T cell function through demethylation of the FoxP3 upstream enhancer in patients with rheumatoid arthritis. Arthritis Rheumatol. 2015;67(5):1182–92.
Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358(9285):903–11.
Lee J, Hong EC, Jeong H, Hwang JW, Kim H, Bae EK, Ahn JK, Choi YL, Han J, Cha HS, et al. A novel histone deacetylase 6-selective inhibitor suppresses synovial inflammation and joint destruction in a collagen antibody-induced arthritis mouse model. Int J Rheum Dis. 2015;18(5):514–23.
Vishwakarma S, Iyer LR, Muley M, Singh PK, Shastry A, Saxena A, Kulathingal J, Vijaykanth G, Raghul J, Rajesh N, et al. Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. Int Immunopharmacol. 2013;16(1):72–8.
Acknowledgements
Not applicable.
Funding
This study was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C1277); the Ministry of Science, ICT and Future Planning (NRF-2015M3A9B6052011, 2019M3A9A8065574); and the Chong Kun Dang Pharmaceutical Corp.
Author information
Authors and Affiliations
Contributions
JJP and YJJ performed all operations and data analysis and prepared the manuscript. YSW conceived the study design, data interpretation, and direction of discussion. BRO, JH, DB, NH, YIC, GSY, JP, EYL, and EBL participated in the design of the study and discussion. YWS conceived of the study design and decided the direction of discussion. All authors read and approved final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained from all participants in accordance with the Declaration of Helsinki. The study was approved by the Institutional Review Board at Seoul National University Hospital (IRB No: H-1507-108-690).
Consent for publication
Not applicable.
Competing interests
Authors declare no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 : Supplementary figure S1.
Serum anti-CCP antibody titers were decreased by CKD-506. Serum anti-CCP levels were measured on Day 16 after CFA injection. Data represent the mean value ± SEM. *p < 0.05, **p < 0.01, ***p < 0.011 vs. CKD-506 0 mg/kg.)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Park, J.K., Jang, Y.J., Oh, B.R. et al. Therapeutic potential of CKD-506, a novel selective histone deacetylase 6 inhibitor, in a murine model of rheumatoid arthritis. Arthritis Res Ther 22, 176 (2020). https://doi.org/10.1186/s13075-020-02258-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-020-02258-0