
Abstract
Background
Klebsiella pneumoniae (Kp) Gram-negative bacteria cause nosocomial infections and rapidly acquire antimicrobial resistance (AMR), which makes it a global threat to human health. It also has a comparatively rare hypervirulent phenotype that can lead to severe disease in otherwise healthy individuals. Unlike classic Kp, canonical hypervirulent strains usually have limited AMR. However, after initial case reports in 2015, carbapenem-resistant hypervirulent Kp has increased in prevalence, including in China, but there is limited understanding of its burden in other geographical regions.
Methods
Here, we examined the largest collection of publicly available sequenced Kp isolates (n=13,178), containing 1603 different sequence types (e.g. ST11 15.0%, ST258 9.5%), and 2174 (16.5%) hypervirulent strains. We analysed the plasmid replicons and carbapenemase and siderophore encoding genes to understand the movement of hypervirulence and AMR genes located on plasmids, and their convergence in carbapenem-resistant hypervirulent Kp.
Results
We identified and analysed 3034 unique plasmid replicons to inform the epidemiology and transmission dynamics of carbapenem-resistant hypervirulent Kp (n=1028, 7.8%). We found several outbreaks globally, including one involving ST11 strains in China and another of ST231 in Asia centred on India, Thailand, and Pakistan. There was evidence of global flow of Kp, including across multiple continents. In most cases, clusters of Kp isolates are the result of hypervirulence genes entering classic strains, instead of carbapenem resistance genes entering canonical hypervirulent ones.
Conclusions
Our analysis demonstrates the importance of plasmid analysis in the monitoring of carbapenem-resistant and hypervirulent strains of Kp. With the growing adoption of omics-based technologies for clinical and surveillance applications, including in geographical regions with gaps in data and knowledge (e.g. sub-Saharan Africa), the identification of the spread of AMR will inform infection control globally.
Similar content being viewed by others

Background
Klebsiella pneumoniae (Kp) is a Gram-negative pathogen increasingly capable of causing severe organ and life-threatening disease. Kp is classified into two main virulence phenotypes: hypervirulent (hvKp) and classical (non-hvKp). Classical Kp is the most common phenotype and normally a nosocomial infection that occurs in patients with additional co-morbidities [1]. Less common is hvKp, which is characterised by invasive infection within the community setting in healthy individuals and a rapid metastatic spread. Epidemiologically, hvKp is more common in East and Southeast Asia, but it is also an emerging threat in Europe. HvKp associated with carbapenemase-producing clones is particularly concerning [1,2,3,4] as it will make infection control more difficult. It is therefore important to use genomic data and analyses from local, regional, and global studies to monitor its emergence and spread.
The main driver of carbapenem resistance in Kp is the acquisition of genes that encode carbapenemases such as KPC, NDM, some variants of OXA and others [5, 6]. The encoding genes are usually located on small mobile genetic elements such as transposons or insertion sequences. These elements are themselves usually embedded in plasmids, which mediate the transfer of genes between bacterial cells via horizontal gene transfer [7,8,9].
Similarly, hypervirulence is normally associated with aerobactin (iuc) and salmochelin (iro) gene loci carried on plasmids. The iro and iuc loci are frequently accompanied by additional genes (e.g. rmpA, rmpA2, and rmpC) associated with a hypermucoviscous capsule phenotype [10, 11]. K1 and K2 are the dominant capsular genotypes among hvKp [12]. The iuc and iro loci usually reside on large (>200kbp) plasmids, but are occasionally located on chromosomes via integrated chromosomal elements [1].
Since reports of carbapenem-resistant hypervirulent Kp (CRhvKp) in a tertiary hospital in Beijing in 2015 [13], the cases have steadily increased in number and geographic range [14,15,16,17]. The incidence of CRhvKp in China was estimated at ~13% based on the presence of rmpA, rmpA2 or iutA biomarkers [14], but estimates vary regionally within the country. Another large-scale study in China reported that 36% of screened carbapenem-resistant (CRKp) strains carried the hypervirulence-associated siderophore aerobactin locus [17]. Other regional studies focused on Singapore [18], India [19] and the USA [20], while the global landscape has also been investigated [21]. Our work extends the previous reports by exploring in detail the plasmid replicons associated with the CRhvKp genotype, globally. With increased human mobility and volumes of trade and tourism, knowledge of the global genomic diversity of plasmids underlying CRhvKp strains will assist with monitoring the emergence and spread of CRhvKp infections and inform their clinical management.
In this study, we analysed plasmid replicons and carbapenemase and siderophore encoding genes across 12,468 geographically diverse Kp isolates to understand the movement of hypervirulence and antimicrobial resistance genes on plasmids, and their convergence in CRhvKp. Despite a reliance on using all available Kp assemblies instead of a targeted dataset, we found a growing prevalence of CRhvKp assigned to nine clusters, seven of which are statistically robust. These clusters included two previously reported outbreaks identified in China [14, 17, 21, 22]; however, they also include additional outbreaks with one spanning Asia, Africa, and Europe. Our findings demonstrate the utility of large-scale data for understanding the epidemiology of CRhvKp, with the potential to assist to clinical and surveillance investigations.
Methods
All publicly available assemblies labelled as Kp (n=13,178) in the NCBI RefSeq database (as of September 2021) were downloaded [23]. Kleborate software (v2.2.0) was used to confirm species, identify sequence types (ST), find antimicrobial resistance (AMR) and virulence genes, and determine capsular and O-antigen types [21] (Additional file 1: Fig. S1, Additional file 2: Data S1). Isolates were classified with a hypervirulent Kp (hvKp) genotype if they contained either aerobactin (iuc), salmochelin (iro) or both gene loci [10, 11]. Carbapenem resistance can occur without carbapenemase encoding genes, but we conservatively defined a CRKp genotype as isolates that carry a carbapenemase encoding gene. Plasmid replicons were identified using PlasmidFinder software (v2.1.1) [24] with default cut-offs (≥60% coverage and ≥90% identity). The isolate data included complete, contig and scaffold level assemblies.
PlasmidFinder compares assemblies to a database of nucleotide sequences of genes encoding replication control and initiation proteins. The software returns assembly sub-sequences (not necessarily whole contigs) that pass similarity thresholds as either nucleotide sequences or their hashes. The returned sub-sequences were summarised in a binary data matrix (1 = present, 0 absent), where the rows represented individual isolates, and columns (n=3034) were unique replicon nucleotide sequences represented by their hashes. The hashes are more convenient for this task due to their shorter length (32 characters) compared to full nucleotide sequences, the longest of which was 1029nt. This binary matrix was used to calculate the Russell-Rao distance between pairs of isolates as implemented in sklearn software (v0.24.2) [25]. For a pair of samples u and v, the distance is defined as: \(\frac{N-\sum_{i=1}^N\left(1\ if\ u\left[i\right]=v\left[i\right]=1\ else\ 0\right)\ }{N}\), where N is the total number of features. In the context of presence/absence data, the Russell-Rao distance has an advantage over the Jaccard metric, as it takes the total number of features (N) into account. Further, it is an improvement over the Euclidean metric because it returns a distance of one to those samples that have none of the features, instead of zero under the Euclidean metric.
We used the UMAP (v0.5.1) algorithm to project the full 3034-dimensional binary matrix into two-dimensional space [25,26,27]. UMAP aims to project multi-dimensional data into fewer dimensions while preserving some global and local data topology. The approach has similarity to principal components analysis (PCA) and multi-dimensional scaling (MDS or PCoA), where the former uses a covariance matrix and the latter uses a matrix of pairwise distances. Neither PCA nor MDS are intended for binary matrices, though MDS can accommodate ordinal data [28, 29]. UMAP belongs to the family of manifold learning algorithms, which perform well at dimensional reduction of binary data [30,31,32]. We applied HDBSCAN software (v0.81) (min_cluster_size=10 and cluster_selection_epsilon=0.5) to the 2-D projection generated by UMAP to identify clusters of isolates [33]. HDSCAN is a density-based clustering algorithm which does not require all data samples to be part of a cluster, and we have labelled such isolates as “unassigned”. In our context, the algorithm identifies how data from the UMAP 2-D projection is distributed in space, and determines gaps in density between groups of isolates. The minimum cluster size (“min_cluster_size”) parameter determines the density gap. The data points lying in regions of the same density are defined as clusters [33]. This adaptive approach to density, as opposed to a cut-off determined approach of DBSCAN, is the distinguishing feature of HDBSCAN. If, based on cluster density, a sample is too far to be part of the cluster, it becomes an outlier [33].
Unlike PCA and MDS, UMAP is a stochastic algorithm; so we assessed the robustness of clustering based on UMAP projection. For this purpose, we created a Monte Carlo simulation by repeating the UMAP projection and cluster detection 500 times. The clusters identified in each of 500 iterations formed columns of a data matrix, which was itself projected using UMAP with a Hamming distance metric. The clusters in this projection were again determined using HBSCAN (Additional file 1: Fig. S1). These latter clusters aggregate 500 individual runs, and to determine the consistency of aggregated versus each underlying cluster, we performed chi-squared tests, implemented in sklearn software. We labelled the aggregate cluster as robust if less than 1% of chi-squared test p-values were below 10-20 (Additional file 1: Fig. S1).
We also compared replicons found in our dataset to those characterised in 35 historic bacterial isolates [34] (Murray collection) sourced between the years 1920 and 1949 and classified as Kp based on Kleborate software typing.
Results
Isolates and genotypes
While all isolates (n=13,178) were Kp according to metadata, Kleborate screening identified them as Kp (n=11,820; 90%), K. quasipneumoniae (n=604, 5%), K. variicola (n=428; 3%), K. aerogenes (n=299; 2%) and other subspecies of Klebsiella (n=27; 0.2%), i.e. some isolates may have incorrect species data in the NCBI database (Additional file 1: Fig. S1). We did not restrict the analysis to K. pneumoniae sensu stricto, but we removed 710 isolates in which PlasmidFinder did not identify any replicons. These removed isolates covered 331 STs, with ST3910 (n=21; 3%) being most frequent. They also had a high abundance of non Kp sensu stricto (K. quasipneumoniae n=140, 19.7%; K. aerogenes n=115, 16.2%; K. variicola (n=109, 15.4%). Notably, these isolates had limited AMR carriage with only 9 isolates carrying carbapenemase encoding genes, among which blaOXA-48 was the most frequent (n=4). There were also few fluroquinolone gyrA mutations (n=65/710) and aminoglycoside resistance enzymes (n=35/710). Only four isolates carried an aerobactin locus. However, 20 isolates had a truncated iro3 locus and 109 K. aerogenes samples had a chromosomally carried salmochelin locus.
The final isolate dataset (n=12,468) contained 1603 different STs (based on 7 chromosomal loci) with the most frequent being ST11 (15%), ST258 (10%), ST15 (4%), ST512 (4%) ST307 (3%) and ST147 (3%) (Additional file 1: Fig. S1). The number of hypervirulent strains was 1881 (15%). The two canonical hypervirulent strains ST23 and ST86 accounted for 1.8% and 0.7%, respectively. The study had global representation, with isolates from 103 countries, including China (20%), USA (14%), Italy (7%), UK (6%), Thailand (4%), and Germany (4%); however, only 2% of all isolates were from sub-Saharan Africa. While the convenience nature of the sampling may make the dataset unsuitable for the estimation of the prevalence of CRhvKp genotypes, the large sample size presents a likely accurate assessment of regional and temporal trends.
Replicons
An important cornerstone of our analysis is the distinction between the replicon family or name (e.g. IncFII(K), IncFIB(K) and ColRNAI) and the underlying unique nucleotide sequences of the replicons belonging to a specific family. For example, across the 12,468 isolates, there were 3034 unique replicon nucleotide sequences, of which 1096 occurred in more than one isolate. The most frequent replicon nucleotide sequence was the PlasmidFinder reference version of IncFIB(K), which occurred in 3123 (24%) isolates. However, the replicons from an IncFIB(K) family occurred 7052 times with eight distinct sequences occurring in more than 100 Kp isolates (Fig. 1). The family and nucleotide sequence diversity are summarised in Additional file 2: Data S2.

Frequency of the main nucleotide sequences from the IncFIB(K) replicon family. The eight nucleotide sequences are provided in Additional file 2: Data S2
The average number of replicons per isolate was 4.6 (range: 1 to 29). A total of 6783 isolates (55%) with identified replicons had carbapenemase encoding genes, and 1881 (15%) had either iuc or iro loci. A total of 1028 (8%) isolates with identified replicons had both carbapenemase encoding and hypervirulence (CRhvKp) loci (Additional file 1: Fig. S1). Therefore, the numbers of isolates determined to be CRhvKp, CRKp (non- hvKp) and hvKp (non-CRKp) were 1028 (8%), 5755 (46%), and 853 (7%), respectively (Additional file 1: Fig. S1). The majority of the CRhvKp were from China (n=696) representing 27% of isolates from that country with isolates collected in Russia (n=64), Thailand (n=44), Italy (n=39) and India (n=25) being the next most common.
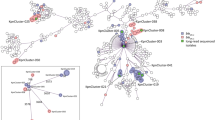
To examine the geographical and temporal trends of carbapenem-resistant hypervirulent genotypes, we assessed the replicon-based clustering of CRKp, hvKP and CRhvKp isolates across 695 plasmid replicon nucleotide sequences that were present in multiple CRKp, hvKp and CRhvKp isolates (Additional file 1: Fig. S1). The UMAP-based analysis of population structure revealed that clustering was not driven solely by geography or ST (Fig. 2). This contrasts with chromosomal-based cluster analysis, which revealed clustering of isolates by ST (data not shown). We applied the HDBSCAN clustering algorithm on the two-dimensional UMAP projection of CRhvKp isolates (n=1028) and identified 9 groups (Clusters A – I; Table 1, Fig. 2), with 79 (8%) isolates not assigned to any group (see Additional file 2: Data S3 for all assignments). Of these 9 clusters, only two (B and E) did not have strong statistical support of their robustness.

Clustering of CRKp, hvKp, and CRhvKp Kp isolates based on replicon sequences coloured by A major sequence types (STs), B country, and C genotype. In D, non-CRhvKp isolates are hidden and only CRhvKp isolates are visible and coloured by plasmid cluster. Axes are dimensionless. Underlying data is presented in Additional file 2: Data S3
Replicon clusters
Cluster A (n=560, Table 1) consisted mostly of isolates from China and accounted for the majority of CRhvKp isolates from that country (n=517/696). This cluster also included isolates with travel links to China [35]. The first isolate from Cluster A was collected in 2012 with the frequency increasing over time (Fig. 3). ST11 was the dominant sequence type in this cluster (n=499/560), which also contained the majority of ST11 CRhvKp isolates (n=499/582). Hypervirulence was driven by iuc1 in nearly all isolates (n=554/560) and 168 isolates additionally had iro1 (77 truncated or incomplete), following the nomenclature established previously [36]. The carbapenemase encoding genes were dominated by blaKPC-2 (n=516/560), with blaOXA-48 the next most common (n=16) (Table 1, Additional file 2: Data S3).

Relative abundance and geographic diversity of carbapenem-resistant hypervirulent Kp (CRhvKp) isolates. Both the total number of all genotyped isolates in each year and percentage that are CRhvKp are presented at the top of the figure
The dominant replicons in Cluster A were ColRNAI (n=943), which occurred multiple times in most isolates, followed by repB (n=555), IncFII (pHN7A8) (n=501), IncHI1B(pNDM-MAR) (n=489) and IncR (n=486) (Additional file 2: Data S2). By examining assembly contigs for any co-presence of carbapenemase, virulence genes and replicons, we were able to link replicon and carbapenemase genes in 74 isolates and replicon and siderophore genes in 341 isolates (Table 1). No carbapenemase or hypervirulence-linked siderophore genes were found on chromosomes. In cases where siderophores were located on the same contig as replicons, the vast majority (n=290/343) had a IncHI1B(pNDM-MAR) replicon (Additional file 2: Data S4). Of these, 125 contigs had both repB and IncHI1B(pNDM-MAR) replicons. In addition, 42 contigs had simultaneously repB replicon and siderophores. The majority (n=52/76) of contigs which carried replicon and carbapenemase genes had an IncFII (pHN7A8) replicon, and 49 of these also had an IncR replicon. Two contigs had both siderophores and carbapenemase genes. The first one had repB, IncFIB (pKPHS1) and IncHI1B(pNDM-MAR) replicons. The second had repB and IncHI1B(pNDM-MAR). Both contigs carried blaKPC-2 and iuc1 loci.
Cluster B (n=83) consisted mainly of isolates from China (n=81/83) and the ST11 sequence type (n=74/83). This cluster did not have strong statistical support for its robustness, and based on visual examination of results (Fig. 2) it is related to, but distinct from, Cluster A isolates. In particular, the isolates in Cluster B lack repB and IncHI1B(pNDM-MAR) replicons, characteristic of Cluster A.
Cluster C (n=82) is geographically diverse, with 32 isolates sourced from Russia, 16 from China, 9 from Egypt, and 8 from Germany. There were 15 STs of which ST147 (n=32; 39%) and ST395 (n=24; 29%) were the most frequent. While ST147 had broad geographic distribution, the majority of ST395 isolates in this cluster were collected in Russia (n=20/24; 83%). The most frequent carbapenemases were blaOXA-48 (n=51) and blaNDM-1 (n=23), again without any strong geographic links. Five isolates had both genes. More broadly, 78 isolates had either a blaOXA or blaNDM gene, with blaKPC-2 (n=2) and blaVIM-1 (n=1) accounting for the rest (Additional file 2: Data S2). The dominant hypervirulence siderophore was iuc1 (n=78/82), while an unassigned salmochelin lineage was present in two isolates. Despite geographic and ST diversity, three replicons accounted for the majority of isolates: Col(pHAD28) (n=91), IncHI1B(pNDM-MAR) (n=80), and IncFIB(pNDM-Mar) (n=79) (Table 1). Out of 14 isolates in which a replicon and iuc1 locus were on the same contig, ten had both IncFIB(pNDM-Mar) and IncHI1B(pNDM-MAR) replicons, and a further two had IncHI1B(pNDM-MAR). Replicons linked to carbapenemase encoding genes (n=16) were diverse, and IncL linked to blaOXA-48 (n=4) was the most frequent association.
Cluster D (n=61) consisted of isolates mainly from South(east) Asia (Thailand, n=27; India, n=17; Pakistan, n=8). The first isolate was collected in Malaysia in 2013, while the most recent six isolates were collected in India in 2019. Nearly all isolates belong to ST231 (n=60/61), which shares only two MLST alleles with canonical hypervirulent ST23. The dominant carbapenemase was blaOXA-232 (n=59/61), with blaOXA-181 and blaOXA-48 in the other two isolates. Unusually, hypervirulence was driven by iuc5 (n=60/61) and accounts for nearly all iuc5 carrying CRhvKp (n=60/75). Salmochelin (iro1) was only present in a single isolate. This cluster had near universal carriage of seven replicons: Col440I, IncFIB(pQil), IncFII(K), IncFIA, IncFII(pAMA1167-NDM-5), ColKP3, and Col(pHAD28) (Table 1). IncFII(pAMA1167-NDM-5) was unique to this cluster and IncFIA only occurred in nine further CRhvKp isolates. We were able to link replicons to a carbapenemase gene in 60 isolates, as well as to a siderophore in 8 isolates. Neither siderophore nor carbapenemase genes occurred on chromosomes. In all cases, blaOXA-232 was linked to the ColKP3 replicon, while iuc5 was co-located on a contig with IncFIA and IncFII(pAMA1167-NDM-5).
Cluster E (n=50) contained isolates mostly from Asia (China, n=16; Singapore, n=14; Thailand, n=13), but unlike Clusters A and B, this one also had five samples from three European countries (Latvia, n=2; Greece, n=2; France, n=1). The first isolates were collected in 2013 in Singapore (n=7) and the most recent was from China in 2020. Sixteen isolates were of the canonical hypervirulent strain ST86 and collected in six countries. This cluster also had ST65 (n=14) and ST23 (n=4) isolates. The former shares only two alleles with ST86. The dominant siderophores in this cluster were iuc1 (n=44) and iro1 (n=40) with 40 isolates carrying both. While blaKPC-2 was the most frequent carbapenemase gene (n=32), some isolates (n=13) carried blaOXA-232 just like the isolates from Cluster B. All these blaOXA-232 carrying isolates were collected as part of the same Thai study in 2016; however, they belonged to 8 different STs [37]. Unlike Clusters A and B, this one had only two near universal replicons: repB (n=50) and IncHI1B(pNDM-MAR) (n=49). We were able to link replicon to carbapenemase genes in 19 isolates and to a siderophore in 32 isolates. Nearly all linked iuc loci were located on a contig with either IncHI1B(pNDM-MAR) (n=4), repB (n=1) or both (n=22) (Additional file 2: Data S4). The blaOXA-232 gene was co-located with a ColKp3 replicon in all blaOXA-232 carrying isolates, like Cluster B. The replicons linked to blaKPC-2 were much more diverse. Out of 6 contigs three had a IncFII(K) replicon, two IncFII(pHN7A8), and one IncX6. None of the examined contigs carried both carbapenemase and hypervirulence genes, nor were any of these genes on chromosomes.
Cluster G consists of 32 isolates collected every year between 2015 and 2020 with the majority isolated in China (n=26) (Table 1). While this cluster includes two ST23 isolates and one ST11, it is dominated by ST15 (n=26/32). The main siderophore locus was iuc1 (n=32/32) with two isolates concurrently carrying iro1. The dominant carbapenemase was blaOXA-232 (n=27/32) with blaNDM-1 (n=3/32) next most common. The most interesting aspect of this cluster was the diversity of frequent replicons: repB (n=31), ColRNAI (n=31), IncHI1B (pNDM-MAR)(n=30), IncFIB (pKPHS1)(n=28), IncFII(K) (n=28), ColKP3(n=28), and Col(pHAD28)(n=28). We linked replicon to carbapenemase in 32 isolates and to a siderophore in 7 isolates. The iuc1 hypervirulent locus was linked to IncHI1B(pNDM-MAR) (n=6) and blaOXA-232 was linked to ColKP3 (n=28) as in cluster D, but that cluster has iuc5 linked to both IncFIA and IncFII(pAMA1167-NDM-5). Part of this cluster has been described previously [22], reinforcing the robustness of our approach.
Salmochelin carrying K. aerogenes
Surprisingly, nearly all K. aerogenes isolates (n=288/299) carried a salmochelin locus, but none had aerobactin. In all 41 complete K. aerogenes assemblies this locus was chromosomal (contig > 4,500,000nt). These salmochelin genes had nucleotide identity between 74% and 86% to Kp sensu stricto salmochelin genes. The nearest amino acid sequences outside K. aerogenes were iroB, iroC, iroB and iroN in Enterobacter oligothropicus with 93%, 90% and 81% identity, respectively. A small portion of K. aerogenes isolates had both salmochelin and carbapenemase genes (n=40), among which carbapenemase genes blaKPC-2 (n=13) and blaOXA-48 (n=6) were the most common. Most of the K. aerogenes without carbapenemase genes (n=231/248) did not have any extended-spectrum beta-lactamase (ESBL) encoding loci, despite most of them (n=171/231) being collected after 2010 — a period in which ESBL encoding genes were common in Kp. PlasmidFinder identified replicons in 174 salmochelin carrying K. aerogenes. Their replicons formed a cluster of samples consisting mainly of USA isolates (n=73), with a few from Germany (n=9), Lebanon (n=6) and other countries. Apart from one small study [38], we believe this is the first major report of the widespread presence of salmochelin in K. aerogenes.
Comparison to replicons of historic isolates
Plasmid sequences of 35 Kp isolates from the historic Murray collection were identified and compared to the 3034 unique replicons in our whole collection (n=12,468). Remarkably, there was a substantial overlap between these unique replicon sequences. For example, the same repB sequences occurred in 1124 of all isolates and in 19 Murray collection isolates. Three further replicon sequences [IncFIB(K), IncFII(pKP91) and IncHI1B(pNDM-MAR)] that occurred once each in the Murray collection occurred in more than 240 general isolates (Additional file 2: Data S5). The co-existence of multiple variants of identical replicons up to 90 years ago requires further investigation into the evolution of plasmid replicon sequences, including their mutation rates and any selective pressure.
IncHI1B(pNDM-MAR) replicon
The Cluster A version of the repB gene differs from PlasmidFinder’s IncHI1B(pNDM-MAR) by 4nt and from the closest sequence in our collection by 3nt (out of 570nt). The latter sequence occurs frequently (n=395) in our whole dataset (Additional file 2: Data S2). More importantly, the first 96nt of this replicon’s sequence (those preceding a repB sequence) are almost unique to Cluster A. These leading 96nt have no similarity using BLAST (word size 7) [39] to any other IncHI1B(pNDM-MAR) variant in our dataset. While this version of replicon does occur outside Cluster A, it is rare among CRhvKp (n=3/1028) and CRKp (n=4/5763) isolates, but more common among hvKp (n=184/988). The Cluster A variant has a strong geographic and ST bias. It is found mostly in China (n=561/676), followed by Russia (n=22/676) and 18 other countries. KL64 (n=343/676) and KL1 (n=190/676) are the dominant serotypes, but these are likely the consequence of ST specificity being common in ST23 and ST11 types. Of all ST23 and ST11 isolates with any IncHI1B(pNDM-MAR) replicon, nearly all carried the Cluster A version of the replicon (ST23 197/216; ST11 461/525). In contrast, all isolates of the canonical hypervirulent ST86 (n=84/84) carried the PlasmidFinder’s reference replicon variant.
The IncHI1B(pNDM-MAR) replicon was nearly monophyletic in Cluster A, with 473 samples with identical replicon sequences and a further 7 replicon sequences among 18 samples. However, across the entire dataset, the IncHI1B(pNDM-MAR) replicon consisted of five main nucleotide sequences with only six mutation differences (Additional file 2: Data S2). Based on the context of Cluster A’s blaKPC-2 and IncHI1B(pNDM-MAR) replicon nucleotide sequence, there is little evidence that this cluster has generated an epidemic in countries outside of China that are well represented in our dataset. However, within China, Cluster A has been identified in at least 13 provinces since its first isolates were identified in year 2013. The monitoring of the core replicon signatures of such clusters can therefore assist with identifying the spread of CRhvKp forms.
Discussion
In this study, we examined plasmid replicons in a large global dataset of 12,468 Kp isolates with the aim of understanding the global distribution of CRhvKp isolates. Our analysis revealed there is structure among the 1028 CRhvKp isolates, with most belonging to multi-country clusters, and at least nine clusters present globally. One such cluster (denoted as Cluster A) involved the spread of ST11 CRhvKp isolates and was detected by a surveillance system in China [17], and appears to have been contained in that country. The data also revealed the potential simultaneous spread of a smaller cluster in China involving ST15 strains with plasmid replicons identical to those found in smaller studies from Hangzou and Shanghai regions of China [22, 40]. Other clusters reveal outbreaks in multiple countries, where the exact dynamics of spread are more difficult to determine due to the limited geographic coverage.
We have focused on the plasmid replicons instead of sequence types (STs), because both hypervirulence-associated and carbapenemase-encoding genes are frequently found on plasmids and transfer horizontally. Hypervirulence-associated genes exist almost exclusively on plasmids; while carbapenemase-encoding genes are generally located on small insertion sequences or transposons embedded in plasmids [7, 15]. While STs are important in the context of outbreaks, we think that plasmid replicons provide greater insights into the spread of hypervirulence and carbapenemases, because plasmids are vectors that transfer AMR and virulence genes between bacterial strains. Due to the relative stability of replicon sequences, they can be used as signatures or barcodes of CRhvKp forms for clinical management, surveillance, and infection control activities.
Recent work on CRhvKp epidemiology using genomic data has focused on single hospitals or geographical regions [15, 41]. However, with increased human mobility and travel worldwide, large-scale analyses of all available sequences can provide insights from a global to local resolution. These activities can assist infection control decision-making through identifying transmission hotspots, blind spots of sampling, and informing resource allocation. Despite the global nature of our analysis, there are some geographical gaps due the convenience nature of the sampling, especially with lower numbers of samples contributed from sub-Saharan Africa. While it is difficult to estimate the prevalence of plasmids and replicons, our analysis reveals the presence of their types and diversity, and determines the spread of clusters. The number of actual circulating clusters is likely to be an underestimate and the dynamics of their spread incomplete. Large-scale routine and timely sequencing globally can provide a more complete picture. This process should involve reviewing and updating the core clusters and their signatures, potentially using our statistical approach, which includes consideration of cluster robustness. For identification of links between AMR or hypervirulence loci that are transferred both horizontally and clonally, our methodology could be complementary to the more general and widespread application of phylogenetic trees, which may not be appropriate for Klebsiella and other pathogens with horizontal gene transfer [42, 43].
Based on our analysis, heavily represented geographies with clusters of CRhvKp isolates (Thailand, Germany, Italy, USA, Russia) have not demonstrated rapid growth of a single cluster within a country. Instead, there were several small geographically diverse clusters, including one (Cluster H) which has a similar number of cases from Russia, Germany, and Egypt, while another (Cluster I) has isolates mainly from Italy, Russia, and China. Travel linked to tourism to an under-represented country is a plausible explanation. Relatedly, isolates collected across three Russian cities were present in six of nine CRhvKp clusters. This pattern may be linked to travel or importation events.
Another interesting finding was the presence of K. aerogenes isolates with salmochelin homologues. This Klebsiella species is not common in a clinical setting and generally has a low level of AMR [38], but it may represent a potential reservoir of hypervirulent bacteria, especially in the USA and Germany where it appears most common. Clinical isolates dominate most collections, but the collection and sequencing of samples from environmental or domestic animal sources may provide insights into AMR transmission. Relatedly, the comparison of the replicons from modern and Murray collection isolates showed perfectly matching replicon sequences in several cases, often from isolates spanning 70 to 90 years. While there may be sample fidelity issues arising from long-term storage, this may also be a result of a low mutation rate in replicon sequences. This possibility in turn raises important questions about diversity of plasmids outside clinical environments. Insights into the diversity of plasmids, as well as the emergence and spread of AMR, will be provided through the future widespread application of cost-effective WGS or alternative amplicon sequencing-based approaches across countries and sampling sources, combined with the development of informatic data platforms and large-scale analysis tools.
Conclusion
Our large-scale analysis of Kp isolates revealed nine global clusters of CRhvKp, including two expanding within China. We found multiple smaller multi-regional clusters that did not have a clear trend in available data. The benefits of the surveillance and monitoring of infections using genomic data have been exemplified by global efforts during the recent COVID-19 pandemic. The generation of sequence data in real time can provide an early warning to infection control decision makers and provide clinical guidance to reduce burden. Relatedly, the presence of multi-country clusters reinforces that the sharing and joint analysis of international data provides important insights into the epidemiology of CRhvKp and other AMR infections, including the identification of countries experiencing outbreaks but potentially without extensive monitoring programmes of their own. The goal would be to identify reservoirs of infections, assist the surveillance activities of infection control agencies, and ultimately reduce the burden of Kp AMR.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI repository. No new isolates were sequenced during this study. Assemblies used are listed in Additional file 2: Data S1. The data was visualised and quantified using a Python Notebook, which is available at https://github.com/AntonS-bio/KpReplicons/ [44].
Abbreviations
- AMR:
-
Antimicrobial resistance
- Kp:
-
Klebsiella pneumoniae
- cKp:
-
Classic K. pneumoniae
- hvKp:
-
Hypervirulent Kp, defined as carrying either salmochelin or aerobactin locus, but not carbapenemase encoding genes
- CRKp:
-
Carbapenemase-resistant Kp, defined as carbapenemase gene carrying Kp, but not salmochelin or aerobactin locus
- CRhvKp:
-
Carbapenemase-resistant hypervirulent Kp
- MLST:
-
Multilocus sequence type
- ST:
-
Sequence type.
References
Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019;32(3):e00001–19.
Zhang Y, Zhao C, Wang Q, Wang X, Chen H, Li H, et al. High prevalence of hypervirulent Klebsiella pneumoniae infection in China: Geographic distribution, clinical characteristics, and antimicrobial resistance. Antimicrob Agents Chemother. 2016;60:6115–20.
Struve C, Roe CC, Stegger M, Stahlhut SG, Hansen DS, Engelthaler DM, et al. Mapping the evolution of hypervirulent Klebsiella pneumoniae. mBio. 2015;6(4):e00630.
European Centre for Disease Prevention and Control. Emergence of hypervirulent Klebsiella pneumoniae ST23 carrying carbapenemase genes in EU/EEA countries. 17 March 2021. ECDC: Stockholm; 2021.
Logan LK, Weinstein RA. The epidemiology of Carbapenem-resistant Enterobacteriaceae: The impact and evolution of a global menace. J Infect Dis. 2017;215(Suppl 1):S28–36.
Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M, et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis. 2013, 13;(9):785–96. https://doi.org/10.1016/S1473-3099(13)70190-7.
Partridge SR, Kwong SM, Firth N, Jensen SO. Mobile genetic elements associated with antimicrobial resistance. Clin Microbiol Rev. 2018;31(4):e00088.
Partridge SR, Iredell JR. Genetic Contexts of blaNDM-1. Antimicrob Agents Chemother. 2012;56(11):6065.
Bush K, Bradford PA. Epidemiology of β-Lactamase-Producing Pathogens. Clin Microbiol Rev. 2020;33(2):e00047.
Russo TA, Olson R, Fang CT, Stoesser N, Miller M, MacDonald U, et al. Identification of Biomarkers for Differentiation of Hypervirulent Klebsiella pneumoniae from Classical K. pneumoniae. J Clin Microbiol. 2018;56(9):e00776.
Catalán-Nájera JC, Garza-Ramos U, Barrios-Camacho H. Hypervirulence and hypermucoviscosity: Two different but complementary Klebsiella spp. phenotypes? Virulence. 2017; 8(7), 1111-1123
Choby JE, Howard-Anderson J, Weiss DS. Hypervirulent Klebsiella pneumoniae – clinical and molecular perspectives. J Intern Med. 2020;287(3):283.
Yao B, Xiao X, Wang F, Zhou L, Zhang X, Zhang J. Clinical and molecular characteristics of multi-clone carbapenem-resistant hypervirulent (hypermucoviscous) Klebsiella pneumoniae isolates in a tertiary hospital in Beijing. China. Int J Infect Dis. 2015;1(37):107–12.
Yang X, Sun Q, Li J, Jiang Y, Li Y, Lin J, et al. Molecular epidemiology of carbapenem-resistant hypervirulent Klebsiella pneumoniae in China. Emerg Microbes Infect. 2022;11(1):841–9.
Wyres KL, Nguyen TNT, Lam MMC, Judd LM, van Vinh CN, Dance DAB, et al. Genomic surveillance for hypervirulence and multi-drug resistance in invasive Klebsiella pneumoniae from South and Southeast Asia. Genome Med. 2020;12(1):11.
Wyres KL, Wick RR, Judd LM, Froumine R, Tokolyi A, Gorrie CL, et al. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019;15(4):e1008114.
Liu C, Dong N, Chan EWC, Chen S, Zhang R. Molecular epidemiology of carbapenem-resistant Klebsiella pneumoniae in China, 2016-20. Lancet Infect Dis. 2022;22(2):167–8.
Yong M, Chen Y, Oo G, Chang KC, Chu WHW, Teo J, et al. Dominant Carbapenemase-Encoding Plasmids in Clinical Enterobacterales Isolates and Hypervirulent Klebsiella pneumoniae. Singapore. Emerg Infect Dis. 2022;28(8):1578–88.
Banerjee T, Wangkheimayum J, Sharma S, Kumar A, Bhattacharjee A. Extensively Drug-Resistant Hypervirulent Klebsiella pneumoniae From a Series of Neonatal Sepsis in a Tertiary Care Hospital, India. Front Med (Lausanne). 2021;8(8):186.
Kochan TJ, Nozick SH, Medernach RL, Cheung BH, Gatesy SWM, Lebrun-Corbin M, et al. Genomic surveillance for multidrug-resistant or hypervirulent Klebsiella pneumoniae among United States bloodstream isolates. BMC Infect Dis. 2022;22:1–21.
Lam MMC, Wick RR, Watts SC, Cerdeira LT, Wyres KL, Holt KE. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat Commun. 2021;12(1):1–16.
Shu L, Dong N, Lu J, Zheng Z, Hu J, Zeng W, et al. Antimicrob Agents Chemother. 2019;63(3):e02246.
Agarwala R, Barrett T, Beck J, Benson DA, Bollin C, Bolton E, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016;44(D1):D7–19.
Carattoli A, Zankari E, Garciá-Fernández A, Larsen MV, Lund O, Villa L, et al. In Silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58(7):3895–903.
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine Learning in Python. J Mac Learn Res. 2011;12(85):2825–30.
McInnes L, Healy J, Melville J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv [Internet]. 2018 Feb 9 [cited 2021 Apr 22]; Available from: http://arxiv.org/abs/1802.03426
van der Maaten L, Hinton G. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–605.
Wickelmaier F. An introduction to MDS. Sound Quality Research Unit, Aalborg University, Denmark. 46(5):1-26.
Lin T, Zha H. Riemannian manifold learning. IEEE Trans Pattern Anal Mach Intell. 2008;30(5):796–809.
Diaz-Papkovich A, Anderson-Trocmé L, Gravel S. A review of UMAP in population genetics. J Hum Genet. 2021;66(1):85–91.
Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2018;37(1):38–47.
Yang Y, Sun H, Zhang Y, Zhang T, Gong J, Wei Y, et al. Dimensionality reduction by UMAP reinforces sample heterogeneity analysis in bulk transcriptomic data. Cell Rep. 2021;36(4):109442.
Campello RJ, Moulavi D, Sander J. Density-based clustering based on hierarchical density estimates. In: Pacific-Asia conference on knowledge discovery and data mining, vol. 14. Berlin, Heidelberg: Springer; 2013. p. 160–72.
Baker KS, Burnett E, McGregor H, Deheer-Graham A, Boinett C, Langridge GC, et al. The Murray collection of pre-antibiotic era Enterobacteriacae: a unique research resource. Genome Med. 2015;7(1):97.
Bonnin RA, Jousset AB, Chiarelli A, Emeraud C, Glaser P, Naas T, et al. Emergence of new non-clonal group 258 high-risk clones among Klebsiella pneumoniae Carbapenemase-Producing K. Pneumoniae Isolates, France. Emerg Infect Dis. 2020;26(6):1212–20.
Lam MMC, Wyres KL, Judd LM, Wick RR, Jenney A, Brisse S, et al. Tracking key virulence loci encoding aerobactin and salmochelin siderophore synthesis in Klebsiella pneumoniae. Genome Med. 2018;10(1):1–5.
Loraine J, Heinz E, de Sousa AJ, Milevskyy O, Voravuthikunchai SP, Srimanote P, et al. Complement Susceptibility in Relation to Genome Sequence of Recent Klebsiella pneumoniae Isolates from Thai Hospitals. MSphere. 2018;3(6):e00537.
Passarelli-Araujo H, Palmeiro JK, Moharana KC, Pedrosa-Silva F, Dalla-Costa LM, Venancio TM. Genomic analysis unveils important aspects of population structure, virulence, and antimicrobial resistance in Klebsiella aerogenes. FEBS J. 2019;286(19):3797–810.
Boratyn GM, Schäffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL. Domain enhanced lookup time accelerated BLAST. Biol Direct. 2012;7(1):1–4.
Zhang Y, Chen C, Wu J, Jin J, Xu T, Zhou Y, et al. Sequence-Based Genomic Analysis Reveals Transmission of Antibiotic Resistance and Virulence among Carbapenemase-Producing Klebsiella pneumoniae Strains. MSphere. 2022. https://doi.org/10.1128/msphere.00143-22.
Hawkey J, Wyres KL, Judd LM, Harshegyi T, Blakeway L, Wick RR, et al. ESBL plasmids in Klebsiella pneumoniae: diversity, transmission and contribution to infection burden in the hospital setting. Genome Med. 2022;14(1):97.
Sakoparnig T, Field C, van Nimwegen E. Whole genome phylogenies reflect the distributions of recombination rates for many bacterial species. Elife. 2021;1(10):1–61.
Stott CM, Bobay LM. Impact of homologous recombination on core genome phylogenies. BMC Genomics. 2020;21(1):1–10.
Spadar A. KpReplicons [Internet]. Github; [cited 2022 Dec 20]. Available from: https://github.com/AntonS-bio/KpReplicons
Acknowledgements
Not applicable
Funding
TGC and SC are funded by the Medical Research Council UK grants (Reference no. MR/M01360X/1, MR/N010469/1, MR/R025576/1, MR/R020973/1, and MR/X005895/1).
Author information
Authors and Affiliations
Contributions
AS and TGC designed the study. AS analysed the data under the supervision of JP, SC and TGC. AS wrote the first draft of the manuscript, with contributions from JP, SC and TGC. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research conformed to the principles of the Helsinki Declaration. No ethics approval or consent to participate was required, as all data used in the study were already publicly available.
Consent for publication
Not required, as all data used in the study were already publicly available.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Analysis work flow, jpg image.
Additional file 2:
Data S1. Samples AMR profile. Data S2. Plasmid sequences and their distribution. Data S3. Assignments of samples to clusters. Data S4. Contig IDs and Replicons. Data S5. Murray Collection Replicons
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Spadar, A., Perdigão, J., Campino, S. et al. Large-scale genomic analysis of global Klebsiella pneumoniae plasmids reveals multiple simultaneous clusters of carbapenem-resistant hypervirulent strains. Genome Med 15, 3 (2023). https://doi.org/10.1186/s13073-023-01153-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-023-01153-y