
Abstract
Background
Low-frequency variants play an important role in breast cancer (BC) susceptibility. Gene-based methods can increase power by combining multiple variants in the same gene and help identify target genes.
Methods
We evaluated the potential of gene-based aggregation in the Breast Cancer Association Consortium cohorts including 83,471 cases and 59,199 controls. Low-frequency variants were aggregated for individual genes’ coding and regulatory regions. Association results in European ancestry samples were compared to single-marker association results in the same cohort. Gene-based associations were also combined in meta-analysis across individuals with European, Asian, African, and Latin American and Hispanic ancestry.
Results
In European ancestry samples, 14 genes were significantly associated (q < 0.05) with BC. Of those, two genes, FMNL3 (P = 6.11 × 10−6) and AC058822.1 (P = 1.47 × 10−4), represent new associations. High FMNL3 expression has previously been linked to poor prognosis in several other cancers. Meta-analysis of samples with diverse ancestry discovered further associations including established candidate genes ESR1 and CBLB. Furthermore, literature review and database query found further support for a biologically plausible link with cancer for genes CBLB, FMNL3, FGFR2, LSP1, MAP3K1, and SRGAP2C.
Conclusions
Using extended gene-based aggregation tests including coding and regulatory variation, we report identification of plausible target genes for previously identified single-marker associations with BC as well as the discovery of novel genes implicated in BC development. Including multi ancestral cohorts in this study enabled the identification of otherwise missed disease associations as ESR1 (P = 1.31 × 10−5), demonstrating the importance of diversifying study cohorts.
Similar content being viewed by others


Background
Breast cancer is the most commonly diagnosed cancer in women worldwide, making up 11.7% of new cancer diagnoses in 2020 [1]. Heritability estimates for breast cancer range from 13% [2] to 30% [3]. Breast cancer follows a predominantly complex genetic architecture, which in large parts remains unsolved to this day [4]. Identifying disease predisposing genes in breast cancer can help understand pathological pathways and discover new clinical biomarkers or drug targets. However, linking single-marker associations identified in genome-wide association studies (GWAS) to target genes is still ongoing [5], precluding better mechanistic disease understanding.
The analysis of data from diverse ancestral groups can uncover new insights about genetic risk factors due to ancestral differences in variant frequency and linkage disequilibrium patterns, especially in the context of low-frequency variants, as well as variation in environmental factors [6,7,8]. Thus, extending genetic studies to diverse populations and groups is a necessary advance to gain a comprehensive understanding of genetic architectures of complex diseases.
In this study, we extend the recently published gene aggregation method combining coding and regulatory variants [9] to large-scale whole-genome genotyping cohorts to uncover novel genes implicated in breast cancer development. We used data from the Breast Cancer Association Consortium (BCAC) which has been studied previously including GWAS [10, 11], candidate gene analysis [12], and polygenic risk score analysis [13, 14].
We employ the following strategies to empower the discovery of novel gene-disease associations using data from BCAC: (1) aggregation of all coding and regulatory variants linked to a single gene, (2) effective utilization of low-frequency variants, (3) exploiting genetic diversity between different ancestral groups, and (4) restricting multiple testing burden to one statistical test per gene (~ 18,500).
Methods
Samples and genotype data
We used data on 142,670 individuals from BCAC. Detailed description of recruitment criteria, sample demographics, genotyping quality control, and imputation of additional markers have been reported previously [10, 15, 16]. In short, 83,471 breast cancer cases and 59,199 controls of diverse ancestry were recruited in 80 studies (see Fig. 1A, Additional file 1: Table S1). For each study, country of origin, and case and control numbers can be found in Additional file 1: Table S2. Samples were genotyped using the OncoArray (Illumina) [17], a custom SNP array enriched for cancer-associated genetic regions.

Study design. A Breast cancer patients and control individuals included in this study originate from 33 different study center countries, and comprise samples of African, Asian, European, or Latin American and Hispanic ancestry. B The mummy implemented extended SKAT-O analysis includes variants located in coding regions with an extended window and variants located in linked regulatory regions. Regulatory regions were identified based on overlap with genetic range of coding features or based on presence of gene-specific eQTLs in GTEx data in those regulatory regions
Quality control of genotype data
Sample quality control based on genotype and imputation quality has been performed previously [10]. In short, samples were genotyped on the custom OncoArray. Genotyped markers failing any of the following quality criteria were excluded: (i) call rate above 98% in all consortia, (ii) MAF < 1%, (iii) no significant deviation from Hardy–Weinberg Equilibrium (controls: P < 10 − 7, cases: P < 10 − 12). Markers were imputed in a two-stage approach using shapeit2 and impute2 (V2) and the October 2014 (version 3) release of the 1000 Genomes dataset as reference panel [10]. The imputation was carried out for 5-Mb segments of the genome and for groups of 10,000 samples to reduce the computation burden. We included only low-frequency variants (minor allele frequency MAF < 0.05). Variants with imputation accuracy scores (generated with IMPUTE version 2) below 0.7 were excluded from analysis.
Selection of genetic elements
Our previously developed analysis pipeline “mummy” [9] was used to identify coding and regulatory regions for individual genes and to prepare input data for robust rare variant SNPset association testing software MONSTER [18]. Aggregation tests were performed for genes defined in GENCODE v25 and with at least three but not more than 5000 low-frequency variants.
For each of these genes, we identified genetic elements that are likely to contain relevant functional or expression variation using the mummy wrapper. These include the exomes and untranslated regions (UTR) of the gene. We selected additional regulatory elements that have been shown to be enriched for complex trait associations [19, 20]: promoter, enhancer, and transcription-factor-binding units if they could be linked to the gene. These elements were identified from the Ensembl build 84 resource. The link of regulatory genetic elements to genes was either based on physical overlap with the coding region, e.g., when an element was located within an intron of the gene, or physical overlap with significantly associated eQTLs for the specific gene (see Fig. 1B). Thus, we included the three types of regulatory elements if there was evidence that they affect expression levels of the gene. This was based on eQTL data for all available cell types from GTEx version 6.
For each gene, all the low-frequency variants in these selected genetic elements were extracted and formatted to the MONSTER required input and weighted using Phred-scaled EigenPC pathogenicity scores [21]. EigenPC scores have been previously shown to offer the best balance between coding and noncoding variants for application in aggregation testing [9].
The original implementation of “mummy” was adapted to allow for the input of genotype data based on DNA microarrays instead of sequencing data in VCF format. The adapted “mummy” code is accessible on github here: https://github.com/stef-mueller/mummy_for_genotypes.
Gene-based aggregation test
MONSTER (Minimum P‐value Optimized Nuisance parameter Score Test Extended to Relatives) was used to perform SNPset variant aggregation tests for the variants selected for each gene [18]. MONSTER generalizes the SKAT-O algorithm to allow for testing of related samples and sample cohorts with underlying population structure using a mixed effects model. SKAT-O is a unified test that combines a variance component and a burden test. The original MONSTER code was adapted to allow for the inclusion of larger sample numbers. The adapted MONSTER code is available on github here: https://github.com/stef-mueller/MONSTER.
Samples were processed in 15 study groupings due to the computational demand. Groups were formed based on study origin and genetic ancestry of samples while ensuring balanced case and control numbers. Additional file 1: Table S3 lists the number of analyzed genes for each cohort. Sample numbers per cohort can be found in Additional file 1: Table S4.
The mixed effects models testing for gene associations included relatedness in the form of a kinship matrix as a random effect. The kinship matrix was derived by, first, creating an LD pruned marker set using plink2 [22] (window size: 50 kb, step size: 5, r2 threshold: 0.5, minor allele frequency threshold: > 0.2), second, calculating a relationship matrix using gemma [23], third, calculating individuals’ inbreeding coefficients using plink2 –ibc command, and fourth, combining relationship matrix and inbreeding coefficients to the MONSTER required input format. Additionally, age and for some cohorts the recruitment study or study country were included in the model as fixed effects (Additional file 1: Table S5).
As is common for SNPset aggregation tests, MONSTER reports as output P-values but not effect sizes or effect directions for linear mixed model aggregation tests. To check for unaccounted population stratification effects, raw aggregation test results per cohort were plotted against the theoretical distribution of P-values using quantile–quantile (QQ) plots (see Additional file 1: Figure S1), and genetic inflation factors lambda and lambda1000 were calculated (see Additional file 1: Table S4). Lambda is dependent on sample size and will be increased for large samples. Lambda1000 has been established to be comparable across studies. It corrects for sample size.
Two of the 15 cohorts, one of European ancestry and the Latin American and hispanic group, were found to have increased genetic inflation factors with lambda1000 metrics of 1.32 and 1.14, respectively. Thus, raw aggregation test P-values for these two cohorts were corrected using the genomic control method.
Meta-analysis of aggregation tests
Two meta-analyses were performed to combine raw aggregation association results from individual cohorts. First, to allow for comparison with the published GWAS [10] results based on the same sample set, all cohorts including samples of predominantly European ancestries (twelve cohorts, all named “eur*”) were combined in an all-European meta-analysis. Next, a second meta-analysis was performed including all cohorts.
The Stouffer [24, 25] method was used to perform the meta-analysis. It combines the z-statistic derived from P-values of the aggregate test for each cohort after weighting with the square root of the respective sample size. For cohorts with increased genetic inflation factor lambda1000, genetic control corrected P-values, rather than the raw P-values, were included in the meta-analysis. The R package metaP (version 1.3) was used to perform Stouffer meta-analysis. No evidence for increased inflation was observed for the meta-analysis results based on QQ plots and inflation estimates (Additional file 1: Figure S2).
Benjamini–Hochberg false discovery rate (FDR) method was used to correct the meta-analysis results for multiple testing. To ensure robust association signals, genes with missing results for the majority of cohorts were excluded from further analysis. Significant hits were defined as those with FDR-corrected P-values < 0.05.
Follow-up on significantly associated genes
We evaluated whether any of the significant gene-based associations with breast cancer overlapped with significant single-marker associations arising from the European ancestry GWAS. The genome-wide association analysis for single markers in the European ancestry samples has been previously described [10]. The comparison was based on coding and regulatory regions of the gene-based hits with a flanking region of 100 kb. The flanking region of 100 kb was chosen to ensure inclusion of the majority of cis-eQTL elements which, based on GTEx data of 44 tissues, have a median distance of 28.9 or 50.1 kb from the transcription start site (TSS) of genes for primary and secondary cis-eQTLs, respectively [26]. Loci that included SNPs with P-values below 5 × 10−8 from the single-marker association analysis in the examined regions were classified as previously identified breast cancer association hits.
We carried out bioinformatic annotations for each significantly associated gene. Four open-source databases were queried for prior evidence of a causal role of the genes in breast cancer pathology specifically as well as any cancer pathology. First, the ClinVar database was used to identify any putative pathogenic, single-gene variants reported previously in the context of the phenotypes of interest. The ClinVar database was queried on the 1st of March 2021. Pathogenic, single-gene ClinVar variant entries with at least one star review status were classified as supportive evidence.
Second, the aggregated gene-disease database MalaCards [27] was used to identify any significant correlation of genes and phenotypes of interest based on 68 different data sources and utilizing NLP (Natural Language Processing) algorithms to include evidence from non-structured data sources like research publications. Supportive evidence of causal role of genes was defined as a MalaCards search relevance score over 1. The MalaCard database was queried on the 1st of March 2021.
Third, the expert-curated Genetics Home Reference data was queried for all genes of interest and examined for evidence of causal role in breast cancer or any cancer. The queried data version was published on the 28th of July 2020.
And fourth, investigating possible roles as driver genes in breast cancer and cancer pathogenicity, we queried the COSMIC Cancer Gene Census data (version 92) which classifies genes as either (1) TIER1: genes with strong evidence of causal role promoting cancer such as documented relevance in cancer and oncogenic mutations, (2) TIER2: genes with substantial indications to play a role in cancer etiology, and (3) untiered genes: genes with no substantial evidence of a causal role.
Results
Gene-wise aggregation analysis was performed in 83,471 breast cancer patients and 59,199 matched controls. Of those 142,670 samples, 83.4% (n = 119,014) were of European ancestry, with 10.7% (n = 15,321) of samples being of Asian, 4.1% (n = 5784) of African, or 1.8% (n = 2551) Latin American and Hispanic ancestry, respectively. Samples were recruited to studies in 33 countries (see Fig. 1A).
All-European meta-analysis finds 14 associated breast cancer genes
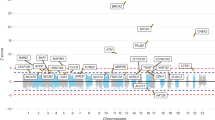
First, we combined gene-wise association results for European cohorts in an all-European meta-analysis. After multiple testing correction, we found 14 genes located in nine different regions to be significantly associated with breast cancer risk (Table 1). Overlap in coding and regulatory regions of genes can cause non-unique mapping of variants to multiple genes for the association aggregation test performed in MONSTER. Thus, four loci were identified containing more than one associated gene. Regional plots for all 14 genes can be found in Additional file 1: Figure S3.
For twelve of the 14 associated genes, the region (gene plus a 100-kB flanking region) contained markers that were individually associated with breast cancer at genome-wide significance (P-value < 5 × 10−8).
Two novel associations
The gene-wise aggregation of low-frequency variants based on coding and regulatory features was able to extend findings of a standard GWAS analysis. The analysis identified two novel gene associations that do not overlap previously reported single-marker-based loci (Fig. 2). The FMNL3 (Formin-Like 3) gene at 12q13.12 was associated with breast cancer risk with a q-value of 0.013. It encodes the Formin-like protein 3, a cytoskeletal regulator, whose overexpression is associated with cancer cell migration, invasion, metastasis, and poor prognosis in multiple cancer types, such as colorectal carcinoma [28], nasopharyngeal carcinoma [29], and tongue squamous cell carcinoma [30].

Regional Plot the FMNL3 Gene on Chromosome 12. Regional plots for the breast cancer association of FMNL3 at 12q13.12. A Depiction of coding regions of all coding genes (data retrieved from Ensembl biomart hg38) within the chromosomal region with FMNL3 highlighted in blue. B Variants included in the aggregation test, plotted according to their chromosomal position and analysis weight. Highlighted in blue are variants exclusively present in the analysis of samples of diverse ancestry. C Single-marker association results based on the same samples [10], with blue solid line denoting P-value for meta-analysis of all cohorts for gene of interest (P = 1.24 × 10−5) in this study and blue dashed line denoting unadjusted P-value for all-European meta-analysis (P = 6.11 × 10−6)
The second novel association was found at 4q12 for AC058822.1 (q-value = 0.020), also named RP11-231C18.3. This lncRNA gene is a scarcely characterized genetic element spanning almost 1 MB.
Gene-based aggregation can help identify the causal genes
To assess whether the gene-based approach can help highlight biologically plausible gene candidates, we assessed whether other evidence, such as genetic epidemiological studies or cell models, supports a role for the significantly associated genes in cancer. We queried different public databases for links to breast cancer and other cancer types for the 14 genes found to be associated with breast cancer in the all-European meta-analysis.
Two genes, MAP3K1 and FGFR2, in addition to being previously identified in breast cancer-associated genetic region in GWAS (see Table 2), are both classified as TIER1 cancer-driving genes in COSMIC Cancer Gene Census. Thus, there is strong evidence that somatic mutations in both genes have a functional involvement in cancer etiology.
To search for previous causal evidence of germline mutations in associated genes, we queried ClinVar, Genetic Home Reference, and MalaCards databases—the last two being an expert-curated gene-disease database and an aggregation database of 68 data sources, respectively. Five genes were implicated in the development of other cancer types: SRGAP2C, MAP3K1, FGFR2, LSP1, and FMNL3.
In addition, the gene ABRAXAS1 codes for a subunit of the BRCA1-A complex [31]. This protein complex plays an important role in DNA damage repair and mutations in the BRCA1 gene predispose to increased risks of cancer [32].
In summary, we found support for aggregated gene associations coinciding with prior causal evidence in breast cancer for two of the nine associated genes and in any cancer for five of them. Among the four associated genes without or very limited prior evidence in cancer pathophysiology is the single-gene locus spanning gene ABRAXAS1—a promising candidate gene for further follow-up owing to its close interactions with protein BRCA1 and its role in DNA damage repair [33].
Including ancestrally diverse samples finds additional gene associations
We furthermore tested gene-based associations in the African (n = 5784), Asian (n = 15,321), and Latin American and Hispanic (n = 2551) ancestry cohorts. There were no significant associations after FDR multiple testing correction. We considered suggestive associations with unadjusted, or in case of the Latin American and Hispanic cohort genetic control corrected, P-values below 1 × 10−4. While no suggestive associations were found in the Latin American and Hispanic cohort, four and five gene associations could be identified in the African and Asian cohort, respectively (Additional file 1: Table S7 and Table S8). This included a suggestive association of gene CBLB (unadjusted P-value: 2.11 × 10−5, Additional file 1: Figure S5) in the African cohort. The E3 Ubiquitin Ligase Cbl-b, coded by oncogene CBLB, has been reported to affect cancer development and progression [34] and has been proposed as a clinical biomarker in breast cancer [35]. No variants located in the coding region of CBLB (plus 100 kb flanking region) were found to be associated in the 2017 large-scale GWAS [10]. None of the variants at this locus have been previously linked to any breast cancer phenotype based on the GWAS Catalog. Thus, the inclusion of diverse ancestry samples shows promise for the identification of new suggestive associations for a plausible candidate gene.
In a second meta-analysis, all 15 sample cohorts, including European ancestry cohorts and cohorts of Asian, African, or Latin American and Hispanic ancestry, were combined (Additional file 1: Table S6). This analysis identified an additional association of gene ESR1 (FDR adjusted P-value in all cohort meta-analysis: 0.0269; Additional file 1: figure S4). The gene ESR1 codes for the estrogen receptor alpha protein and genetic variations in this gene have been reported to be associated with breast cancer [10, 36] and are well described in breast cancer etiology [37] impacting cancer progression [38], treatment success [39], and long term disease outcomes [40].
Discussion
We report the results of a gene-based association analysis in the BCAC resource. Adopting a recently proposed aggregation method that combines variants in coding and regulatory regions, we were able to replicate and extend previously reported findings. This aggregation method helps identify target genes of previously reported single-marker associations and uncovers additional associations that were missed by other methods.
We found 14 genes located in nine loci to be significantly associated with breast cancer risk in samples of European ancestry. Variants near seven of these loci have previously been implicated in breast cancer development based on the 2017 GWAS by Michailidou et al. [10] and we were able to link those single-marker associations to putative target genes. We found independent evidence for a role in breast cancer development for five of the genes. Two of them, MAP3K1 and FGFR2, are long-established risk genes for breast cancer mediated by both germline and somatic mutations [41, 42]. MAP kinase MEKK1, coded by MAP3K1, has been reported to promote cancer cell migration by contributing to an accommodating breast tumor microenvironment [43, 44], while FGFR2 has been identified as a viable drug target in breast cancer [45]. Additionally, the genes SRGAP2C, LSP1, and FMNL3 have been implicated in the etiology of other types of cancer. Although there is currently no functional evidence to substantiate the role of these three genes in breast cancer, sharing of genetic risk factors between different cancers is prevalent [46]. Jiang et al. report a genetic correlation of 0.24, 0.18, and 0.15 for breast cancer with ovarian, lung, and colorectal cancer, respectively [2].
As a further plausible target gene, we have identified ABRAXAS1, which codes for a subunit of the BRCA1 DNA repair protein complex. Differential allelic expression in the genomic region 4q21, in which gene ABRAXAS1 is located, has been previously reported to be associated with breast cancer susceptibility [47]. Interestingly, a recent study using burden testing for rare, protein-truncating or pathogenic variants in ABRAXAS1 based on sequencing data from 60,000 patients and 53,000 controls from the BCAC cohort did not find a significant disease association, with the odds ratio reported as 0.98 (0.50–1.94) [12]. In contrast, our approach focusing on low-frequency coding and regulatory variants identified a significant association of this gene with breast cancer risk. This suggests that our method enables gene discoveries that are missed by other approaches because the local genetic architecture of genes affecting breast cancer susceptibility varies between ancestry groups.
Beyond the identification of putative target genes in loci that have been previously found to harbor disease-associated variants, we report here two new disease associations for genes FMNL3 and AC058822.1. FMNL3 is a member of the diaphanous-related formin family, which represents a family of highly conserved cytoskeletal regulatory proteins [48]. FMNL3 expression is reported to promote migration and invasion of cancer cells and predicts clinical outcome in different solid cancers such as colorectal carcinoma [28, 49], squamous cell carcinoma of the tongue [30], and melanoma [50]. No markers in the proximity of this gene were found to be associated with breast cancer in the 2017 GWAS in the same dataset.
Features of the method that may facilitate discoveries beyond those identified by other approaches include (i) a reduction of multiple testing burden, (ii) boosting signals by aggregating over all genetic regions affecting individual genes expression and function, (iii) inclusion of low-frequency variants often underpowered in other studies, and (iv) ability to synthesize evidence for genetic risk factors in different ancestries regardless of differences in non-disease-associated variational background.
The inclusion of samples of non-European ancestry in genetic studies can advance our understanding of genetic disease landscapes [8]. However, differences between populations in terms of allele frequencies and linkage disequilibrium can lead to heterogeneity and false positive associations in single-marker association analyses. Additionally, different causal variants may be present in different ancestral groups [51] which can be driven by ancestry differences in allele frequencies. Aggregation methods offer a solution because they can accommodate multiple causal variants at a locus. A meta-analysis including all cohorts in this study was able to identify an additional association for ESR1, which was not detected in a European ancestry only analysis. Ancestry-related differences in disease-associated variants and minor allele frequencies in the ESR1 locus (6q25 region) have been previously reported [52, 53]. This ESR1 gene is coding for the estrogen receptor alpha monomer, an established risk factor and promising clinical biomarker in breast cancer pathophysiology [37, 54, 55].
The comparably small sample size of cohorts of non-European ancestry is a limitation of our study. Although no gene reached FDR-corrected significance in these analyses, nine genes were associated at suggestive thresholds, including biologically plausible candidate gene CBLB. This gene codes for the E3 Ubiquitin Ligase Cbl-b, which is a confirmed protagonist in cancer development and progression [56, 57]. There is recently mounting evidence that CBLB expression may be useful as a prognostic factor in breast cancer [35, 58, 59].
We note the following limitations for the adopted method in this study. First, no effect sizes or effect directions are derived. Second, it is not clear how statistical power for identification of associations is affected by gene length, mutational constrictions, number of transcripts, and amount of prior evidence for regulatory elements. Future analyses could deliver insights in this regard. Third, we were not always able to narrow down associations to a single target gene in loci due to overlapping genetic features. This limitation is affected by the LD structure in a specific region and the amount of prior information available in form of eQTL data and regions of overlapping transcripts. Fourth, although we are able to find plausible target genes applying this method to samples of diverse ancestry, there is potential for further optimisation. Regulatory features for genes have been identified using GTEx data, which predominantly is derived from European ancestry samples. Additionally, variants are weighted using Phred-scaled EigenPC pathogenicity scores [21]. These scores are derived using unsupervised learning on a labeled training dataset predominantly based on samples of European descent. Fifth, the current implementation of the method is computationally demanding but nonetheless able to analyze large sample sets (here over 140,000 samples). Sixth, our analysis did not consider different transcripts of genes so our findings are limited to the assigned major transcript. And lastly, the optimal aggregate methods depend on the genetic architecture at a given locus. We used SKAT-O a unified test to capture a range of different architectures. However, the choice of method may impact on the results.
Conclusions
Our findings show that usage of extended gene aggregation methods covering coding and regulatory regions in addition to standard single-marker tests (i.e., GWAS) have the potential to discover novel associations in available datasets. This study helps uncover the role of low-frequency genetic variation in breast cancer susceptibility and empowers gene discovery in ancestrally diverse cohorts.
Availability of data and materials
Gene aggregation results for all genes and all analyses, as well as code used in the analysis for this manuscript, are made available in the following github repository: https://github.com/stef-mueller/BCAC_genotype_aggregation_analysis [60].
Code for running mummy on genotypes available in public github repository here: https://github.com/stef-mueller/mummy_for_genotypes.
An implementation of MONSTER, adapted for analyzing large-scale genotype data, is accessible on github: https://github.com/stef-mueller/MONSTER.
Annotation sources used in this project are (1) ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/; (2) MalaCards, https://www.malacards.org/; (3) Genetics Home Reference, https://medlineplus.gov/genetics/; (4) COSMIC Cancer Gene Census data, https://cancer.sanger.ac.uk/census.
Summary statistics of GWAS data for breast cancer are available through the BCAC website: http://bcac.ccge.medschl.cam.ac.uk. The individual level datasets analyzed during the current study are not publicly available due to protection of participant privacy and confidentiality, and ownership of the contributing institutions, but may be made available in an anonymized form via the corresponding author on reasonable request and after approval of the involved institutions. To receive access to the data, a concept form must be submitted, which will then be reviewed by the BCAC Data Access Coordination Committee (DACC); see http://bcac.ccge.medschl.cam.ac.uk/bcacdata/. This work was carried out under the approved BCAC concept form #595.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. Available from: https://doi.org/10.3322/caac.21660.
Jiang X, Finucane HK, Schumacher FR, Schmit SL, Tyrer JP, Han Y, et al. Shared heritability and functional enrichment across six solid cancers. Nat Commun. 2019;10(1):431. Available from: https://doi.org/10.1038/s41467-018-08054-4.
Möller S, Mucci LA, Harris JR, Scheike T, Holst K, Halekoh U, et al. The heritability of breast cancer among women in the nordic twin study of cancer. Cancer Epidemiol Biomarkers Prev. 2016;25(1):145–50. Available from: https://doi.org/10.1158/1055-9965.EPI-15-0913.
Skol AD, Sasaki MM, Onel K. The genetics of breast cancer risk in the post-genome era: thoughts on study design to move past BRCA and towards clinical relevance. Breast Cancer Res. 2016;18(1):99. Available from: https://doi.org/10.1186/s13058-016-0759-4.
Fachal L, Aschard H, Beesley J, Barnes DR, Allen J, Kar S, et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat Genet. 2020;52(1):56–73. Available from: https://doi.org/10.1038/s41588-019-0537-1.
Kuchenbaecker K, Telkar N, Reiker T, Walters RG, Lin K, Eriksson A, et al. The transferability of lipid loci across African, Asian and European cohorts. Nat Commun. 2019;10(1):4330. Available from: https://doi.org/10.1038/s41467-019-12026-7.
Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570(7762):514–8. Available from: https://doi.org/10.1038/s41586-019-1310-4.
Peterson RE, Kuchenbaecker K, Walters RK, Chen CY, Popejoy AB, Periyasamy S, et al. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell. 2019;179(3):589–603. Available from: https://doi.org/10.1016/j.cell.2019.08.051.
Gilly A, Suveges D, Kuchenbaecker K, Pollard M, Southam L, Hatzikotoulas K, et al. Cohort-wide deep whole genome sequencing and the allelic architecture of complex traits. Nature Commun. 2018;9(1):4674. Available from: https://doi.org/10.1038/s41467-018-07070-8.
Michailidou K, Lindström S, Dennis J, Beesley J, Hui S, Kar S, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551(7678):92–4. Available from: https://doi.org/10.1038/nature24284.
Zhang H, Ahearn TU, Lecarpentier J, Barnes D, Beesley J, Qi G, et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat Genet. 2020;52(6):572–81. Available from: https://doi.org/10.1038/s41588-020-0609-2.
Breast Cancer Association Consortium, Dorling L, Carvalho S, Allen J, González-Neira A, Luccarini C, et al. Breast cancer risk genes - association analysis in more than 113,000 women. N Engl J Med. 2021;384(5):428–39. Available from: https://doi.org/10.1056/NEJMoa1913948.
Kramer I, Hooning MJ, Mavaddat N, Hauptmann M, Keeman R, Steyerberg EW, et al. Breast Cancer Polygenic Risk Score and Contralateral Breast Cancer Risk. Am J Hum Genet. 2020;107(5):837–48. Available from: https://doi.org/10.1016/j.ajhg.2020.09.001.
Mavaddat N, Michailidou K, Dennis J, Lush M, Fachal L, Lee A, et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am J Hum Genet. 2019;104(1):21–34. Available from: https://doi.org/10.1016/j.ajhg.2018.11.002.
Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45(4):353–61, 361e1-2. Available from: https://doi.org/10.1038/ng2563.
Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet. 2015;47(4):373–80. Available from: https://doi.org/10.1038/ng.3242.
Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, et al. The OncoArray Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomarkers Prev. 2017;26(1):126–35. Available from: https://doi.org/10.1158/1055-9965.EPI-16-0106.
Jiang D, McPeek MS. Robust rare variant association testing for quantitative traits in samples with related individuals. Genet Epidemiol. 2014;38(1):10–20. Available from: https://doi.org/10.1002/gepi.21775.
Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47(11):1228–35. Available from: https://doi.org/10.1038/ng.3404.
Nasser J, Bergman DT, Fulco CP, Guckelberger P, Doughty BR, Patwardhan TA, et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593(7858):238–43. Available from: https://doi.org/10.1038/s41586-021-03446-x.
Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48(2):214–20. Available from: https://doi.org/10.1038/ng.3477.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. Available from: https://doi.org/10.1186/s13742-015-0047-8.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4. Available from: https://doi.org/10.1038/ng.2310.
Stouffer SA, Suchman EA, Devinney LC, Star SA, Williams RM Jr. The American soldier: Adjustment during army life. (Studies in social psychology in World War II). Princeton Univ. Press; 1949.
Zaykin DV. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J Evol Biol. 2011;24(8):1836 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3135688/. Cited 30 Mar 2021.
GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–13. Available from: https://www.nature.com/articles/nature24277. Cited 27 Apr 2021.
Rappaport N, Twik M, Plaschkes I, Nudel R, Stein TI, Levitt J, et al. MalaCards: an amalgamated human disease compendium with diverse clinical and genetic annotation and structured search, Nucleic Acids Research. 2017;45(D1):D877–D887. https://doi.org/10.1093/nar/gkw1012.
Zeng YF, Xiao YS, Lu MZ, Luo XJ, Hu GZ, Deng KY, et al. Increased expression of formin-like 3 contributes to metastasis and poor prognosis in colorectal carcinoma. Exp Mol Pathol. 2015;98(2):260–7. Available from: https://doi.org/10.1016/j.yexmp.2015.03.008.
Wu Y, Shen Z, Wang K, Ha Y, Lei H, Jia Y, et al. High FMNL3 expression promotes nasopharyngeal carcinoma cell metastasis: role in TGF-β1-induced epithelia-to-mesenchymal transition. Sci Rep. 2017;7:42507. Available from: https://doi.org/10.1038/srep.
Liu J, Chen S, Chen Y, Geng N, Feng C. High expression of FMNL3 associates with cancer cell migration, invasion, and unfavorable prognosis in tongue squamous cell carcinoma. J Oral Pathol Med. 2019;48(6):459–67. Available from: https://doi.org/10.1111/jop.12857.
Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316(5828):1194–8. https://doi.org/10.1126/science.1139476.
Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317(23):2402–16. Available from: https://doi.org/10.1001/jama.2017.7112.
Solyom S, Aressy B, Pylkäs K, Patterson-Fortin J, Hartikainen JM, Kallioniemi A, et al. Breast cancer-associated Abraxas mutation disrupts nuclear localization and DNA damage response functions. Sci Transl Med. 2012;4(122):122ra23. Available from: https://doi.org/10.1126/scitranslmed.3003223.
Liyasova MS, Ma K, Lipkowitz S. Molecular pathways: Cbl proteins in tumorigenesis and antitumor immunity-opportunities for cancer treatment. Clin Cancer Res. 2015;21:1789–94. Available from: https://doi.org/10.1158/1078-0432.ccr-13-2490.
Liu X, Teng Y, Wu X, Li Z, Bao B, Liu Y, et al. The E3 ubiquitin ligase Cbl-b predicts favorable prognosis in breast cancer. Front Oncol. 2020;10:695. Available from: https://doi.org/10.3389/fonc.2020.00695.
Milne RL, Kuchenbaecker KB, Michailidou K, Beesley J, Kar S, Lindström S, et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat Genet. 2017;49(12):1767–78. Available from: https://doi.org/10.1038/ng.3785.
Dustin D, Gu G, Fuqua SAW. ESR1 mutations in breast cancer. Cancer. 2019;125:3714–28. Available from: https://doi.org/10.1002/cncr.32345.
Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, Cao J, et al. Functional annotation of ESR1 gene fusions in estrogen receptor-positive breast cancer. Cell Rep. 2018;24(6):1434-44.e7. Available from: https://doi.org/10.1016/j.celrep.2018.07.009.
Santo ID, De Santo I, McCartney A, Migliaccio I, Di Leo A, Malorni L. The emerging role of ESR1 mutations in luminal breast cancer as a prognostic and predictive biomarker of response to endocrine therapy. Cancers. 2019;11:1894. Available from: https://doi.org/10.3390/cancers11121894.
Zundelevich A, Dadiani M, Kahana-Edwin S, Itay A, Sella T, Gadot M, et al. ESR1 mutations are frequent in newly diagnosed metastatic and loco-regional recurrence of endocrine-treated breast cancer and carry worse prognosis. Breast Cancer Res. 2020;22(1):16. Available from: https://doi.org/10.1186/s13058-020-1246-5.
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–4. Available from: https://doi.org/10.1038/nature11017.
Easton DF, Pooley KA, Dunning AM, Pharoah PDP, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447(7148):1087–93. Available from: https://doi.org/10.1038/nature05887.
Gentile S, Eskandari N, Rieger MA, Cuevas BD. MEKK1 regulates chemokine expression in mammary fibroblasts: implications for the breast tumor microenvironment. Front Oncol. 2021;11:609918. Available from: https://doi.org/10.3389/fonc.2021.609918.
Cuevas BD, Winter-Vann AM, Johnson NL, Johnson GL. MEKK1 controls matrix degradation and tumor cell dissemination during metastasis of polyoma middle-T driven mammary cancer. Oncogene. 2006;25(36):4998–5010. Available from: https://doi.org/10.1038/sj.onc.1209507.
Chae YK, Hong F, Vaklavas C, Cheng HH, Hammerman P, Mitchell EP, et al. Phase II study of AZD4547 in patients with tumors harboring aberrations in the fgfr pathway: results from the NCI-MATCH Trial (EAY131) Subprotocol W. J Clin Oncol. 2020;38(21):2407–17. Available from: https://doi.org/10.1200/JCO.19.02630.
Rashkin SR, Graff RE, Kachuri L, Thai KK, Alexeeff SE, Blatchins MA, et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat Commun. 2020;11(1):4423. Available from: https://doi.org/10.1038/s41467-020-18246-6.
Hamdi Y, Soucy P, Adoue V, Michailidou K, Canisius S, Lemaçon A, et al. Association of breast cancer risk with genetic variants showing differential allelic expression: Identification of a novel breast cancer susceptibility locus at 4q21. Oncotarget. 2016;7(49):80140–6. Available from: https://doi.org/10.18632/oncotarget.12818.
Katoh M, Katoh M. Identification and characterization of human FMNL1, FMNL2 and FMNL3 genes in silico. Int J Oncol. 2003;22(5):1161–8 Available from: https://www.ncbi.nlm.nih.gov/pubmed/12684686.
Zeng YF, Xiao YS, Liu Y, Luo XJ, Wen LD, Liu Q, et al. Formin-like 3 regulates RhoC/FAK pathway and actin assembly to promote cell invasion in colorectal carcinoma. World J Gastroenterol. 2018;24(34):3884–97. Available from: https://doi.org/10.3748/wjg.v24.i34.3884.
Gardberg M, Heuser VD, Koskivuo I, Koivisto M, Carpén O. FMNL2/FMNL3 formins are linked with oncogenic pathways and predict melanoma outcome. Hip Int. 2016;2(1):41–52. Available from: https://doi.org/10.1002/cjp2.34.
Gelernter J, Sun N, Polimanti R, Pietrzak RH, Levey DF, Lu Q, et al. Genome-wide association study of maximum habitual alcohol intake in >140,000 U.S. European and African American Veterans Yields Novel Risk Loci. Biol Psychiatry. 2019;86(5):365–76. Available from: https://doi.org/10.1016/j.biopsych.2019.03.984.
Fejerman L, Ahmadiyeh N, Hu D, Huntsman S, Beckman KB, Caswell JL, et al. Genome-wide association study of breast cancer in Latinas identifies novel protective variants on 6q25. Nat Commun. 2014;5:5260. Available from: https://doi.org/10.1038/ncomms6260.
Hoffman J, Fejerman L, Hu D, Huntsman S, Li M, John EM, et al. Identification of novel common breast cancer risk variants at the 6q25 locus among Latinas. Breast Cancer Res. 2019;21(1):3. Available from: https://doi.org/10.1186/s13058-018-1085-9.
Dunning AM, Michailidou K, Kuchenbaecker KB, Thompson D, French JD, Beesley J, et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat Genet. 2016;48(4):374–86. Available from: https://doi.org/10.1038/ng.3521.
Carausu M, Bidard FC, Callens C, Melaabi S, Jeannot E, Pierga JY, et al. ESR1 mutations: a new biomarker in breast cancer. Expert Rev Mol Diagn. 2019;19(7):599–611. Available from: https://doi.org/10.1080/14737159.2019.1631799.
Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507(7493):508–12. Available from: https://doi.org/10.1038/nature12998.
Liyasova MS, Ma K, Lipkowitz S. Molecular pathways: cbl proteins in tumorigenesis and antitumor immunity-opportunities for cancer treatment. Clin Cancer Res. 2015;21(8):1789–94. Available from: https://doi.org/10.1158/1078-0432.CCR-13-2490.
Xu L, Zhang Y, Qu X, Che X, Guo T, Cai Y, et al. E3 ubiquitin ligase Cbl-b prevents tumor metastasis by maintaining the epithelial phenotype in multiple drug-resistant gastric and breast cancer cells. Neoplasia. 2017;19(4):374–82. Available from: https://doi.org/10.1016/j.neo.2017.01.011.
CHe X, Zhang Y, Qu X, Guo T, Ma Y, Li C, et al. The E3 ubiquitin ligase Cbl-b inhibits tumor growth in multidrug-resistant gastric and breast cancer cells. Neoplasma. 2017;64(6):887–92. Available from: https://doi.org/10.4149/neo2017610.
Mueller HS, et al. Gene-aggregation results for all genes and all analyses generated in context of this project, github. 2022. Available from: https://github.com/stef-mueller/BCAC_genotype_aggregation_analysis.
Acknowledgements
We would like to thank Dr. Mario Parreno Centeno for insightful comments regarding the COSMIC database and Dr. Arthur Gilly for sharing Phred-scaled EIGEN PC scores.
We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians, and administrative staff who have enabled this work to be carried out. ABCS thanks the Blood bank Sanquin, The Netherlands. ABCTB Investigators: Christine Clarke, Deborah Marsh, Rodney Scott, Robert Baxter, Desmond Yip, Jane Carpenter, Alison Davis, Nirmala Pathmanathan, Peter Simpson, J. Dinny Graham, Mythily Sachchithananthan. Samples are made available to researchers on a non-exclusive basis. The ACP study wishes to thank the participants in the Thai Breast Cancer study. Special thanks also go to the Thai Ministry of Public Health (MOPH), doctors and nurses who helped with the data collection process. Finally, the study would like to thank Dr Prat Boonyawongviroj, the former Permanent Secretary of MOPH and Dr Pornthep Siriwanarungsan, the former Department Director-General of Disease Control who have supported the study throughout. BBCS thanks Eileen Williams, Elaine Ryder-Mills, and Kara Sargus. The BCINIS study would not have been possible without the contributions of Dr. K. Landsman, Dr. N. Gronich, Dr. A. Flugelman, Dr. W. Saliba, Dr. F. Lejbkowicz, Dr. E. Liani, Dr. I. Cohen, Dr. S. Kalet, Dr. V. Friedman, Dr. O. Barnet of the NICCC in Haifa, and all the contributing family medicine, surgery, pathology, and oncology teams in all medical institutes in Northern Israel. The BREOGAN study would not have been possible without the contributions of the following: Manuela Gago-Dominguez, Jose Esteban Castelao, Angel Carracedo, Victor Muñoz Garzón, Alejandro Novo Domínguez, Maria Elena Martinez, Sara Miranda Ponte, Carmen Redondo Marey, Maite Peña Fernández, Manuel Enguix Castelo, Maria Torres, Manuel Calaza (BREOGAN), José Antúnez, Máximo Fraga and the staff of the Department of Pathology and Biobank of the University Hospital Complex of Santiago-CHUS, Instituto de Investigación Sanitaria de Santiago, IDIS, Xerencia de Xestion Integrada de Santiago-SERGAS; Joaquín González-Carreró and the staff of the Department of Pathology and Biobank of University Hospital Complex of Vigo, Instituto de Investigacion Biomedica Galicia Sur, SERGAS, Vigo, Spain. The BSUCH study acknowledges the Principal Investigator, Barbara Burwinkel, and thanks Peter Bugert, Medical Faculty Mannheim. The CAMA study would like to recognize CONACyT for the financial support provided for this work and all physicians responsible for the project in the different participating hospitals: Dr. Germán Castelazo (IMSS, Ciudad de México, DF), Dr. Sinhué Barroso Bravo (IMSS, Ciudad de México, DF), Dr. Fernando Mainero Ratchelous (IMSS, Ciudad de México, DF), Dr. Joaquín Zarco Méndez (ISSSTE, Ciudad de México, DF), Dr. Edelmiro Pérez Rodríguez (Hospital Universitario, Monterrey, Nuevo León), Dr. Jesús Pablo Esparza Cano (IMSS, Monterrey, Nuevo León), Dr. Heriberto Fabela (IMSS, Monterrey, Nuevo León), Dr. Fausto Hernández Morales (ISSSTE, Veracruz, Veracruz), Dr. Pedro Coronel Brizio (CECAN SS, Xalapa, Veracruz), and Dr. Vicente A. Saldaña Quiroz (IMSS, Veracruz, Veracruz). CBCS thanks study participants, co-investigators, collaborators, and staff of the Canadian Breast Cancer Study, and project coordinators Agnes Lai and Celine Morissette. CCGP thanks Styliani Apostolaki, Anna Margiolaki, Georgios Nintos, Maria Perraki, Georgia Saloustrou, Georgia Sevastaki, and Konstantinos Pompodakis. CGPS thanks staff and participants of the Copenhagen General Population Study. For the excellent technical assistance: Dorthe Uldall Andersen, Maria Birna Arnadottir, Anne Bank, Dorthe Kjeldgård Hansen. The Danish Cancer Biobank is acknowledged for providing infrastructure for the collection of blood samples for the cases. COLBCCC thanks all patients, the physicians Justo G. Olaya, Mauricio Tawil, Lilian Torregrosa, Elias Quintero, Sebastian Quintero, Claudia Ramírez, José J. Caicedo, and Jose F. Robledo, and the technician Michael Gilbert for their contributions and commitment to this study. Investigators from the CPS-II cohort thank the participants and Study Management Group for their invaluable contributions to this research. They also acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, as well as cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program. The authors would like to thank the California Teachers Study Steering Committee that is responsible for the formation and maintenance of the Study within which this research was conducted. A full list of California Teachers Study (CTS) team members is available at https://www.calteachersstudy.org/team. DIETCOMPLYF thanks the patients, nurses, and clinical staff involved in the study. The DietCompLyf study was funded by the charity Against Breast Cancer (Registered Charity Number 1121258) and the NCRN. We thank the participants and the investigators of EPIC (European Prospective Investigation into Cancer and Nutrition). ESTHER thanks Hartwig Ziegler, Sonja Wolf, Volker Hermann, Christa Stegmaier, and Katja Butterbach. FHRISK and PROCAS thank NIHR for funding. GC-HBOC thanks Stefanie Engert, Heide Hellebrand, Sandra Kröber and LIFE—Leipzig Research Centre for Civilization Diseases (Markus Loeffler, Joachim Thiery, Matthias Nüchter, Ronny Baber). The GENICA Network: Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, and University of Tübingen, Germany [Hiltrud Brauch, Wing-Yee Lo], German Cancer Consortium (DKTK) and German Cancer Research Center (DKFZ), Partner Site Tübingen, 72074 Tübingen, Germany [Hiltrud Brauch], gefördert durch die Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Exzellenzstrategie des Bundes und der Länder—EXC 2180—390900677 [Hiltrud Brauch], Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany [YDK, Christian Baisch], Institute of Pathology, University of Bonn, Germany [Hans-Peter Fischer], Molecular Genetics of Breast Cancer, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Germany [Ute Hamann], Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, Germany [Thomas Brüning, Beate Pesch, Sylvia Rabstein, Anne Lotz]; and Institute of Occupational Medicine and Maritime Medicine, University Medical Center Hamburg-Eppendorf, Germany [Volker Harth]. HEBCS thanks Johanna Kiiski, Taru A. Muranen, Kristiina Aittomäki, Kirsimari Aaltonen, Karl von Smitten, Irja Erkkilä. HKBCS thanks Hong Kong Sanatorium and Hospital, Dr Ellen Li Charitable Foundation, The Kerry Group Kuok Foundation, National Institute of Health 1R03CA130065 and the North California Cancer Center for support. HMBCS thanks Peter Hillemanns, Hans Christiansen, and Johann H. Karstens. HUBCS thanks Darya Prokofyeva and Shamil Gantsev. We thank all investigators of the KOHBRA (Korean Hereditary Breast Cancer) Study. LMBC thanks Gilian Peuteman, Thomas Van Brussel, EvyVanderheyden, and Kathleen Corthouts. MABCS thanks Milena Jakimovska (RCGEB “Georgi D. Efremov”), Snezhana Smichkoska, Emilija Lazarova, Marina Iljoska (University Clinic of Radiotherapy and Oncology), Dzengis Jasar, Mitko Karadjozov (Adzibadem-Sistina Hospital), Andrej Arsovski, and Liljana Stojanovska (Re-Medika Hospital) for their contributions and commitment to this study. MARIE thanks Petra Seibold, Nadia Obi, Sabine Behrens, Ursula Eilber, and Muhabbet Celik. MBCSG (Milan Breast Cancer Study Group): Paolo Peterlongo, Siranoush Manoukian, Bernard Peissel, Jacopo Azzollini, Erica Rosina, Daniela Zaffaroni, Irene Feroce, Mariarosaria Calvello, Aliana Guerrieri Gonzaga, Monica Marabelli, Davide Bondavalli, and the personnel of the Cogentech Cancer Genetic Test Laboratory. We thank the coordinators, the research staff, and especially the MMHS participants for their continued collaboration on research studies in breast cancer. MSKCC thanks Marina Corines, Lauren Jacobs. MYBRCA thanks study participants and research staff (particularly Patsy Ng, Nurhidayu Hassan, Yoon Sook-Yee, Daphne Lee, Lee Sheau Yee, Phuah Sze Yee, and Norhashimah Hassan) for their contributions and commitment to this study. NBHS and SBCGS thank study participants and research staff for their contributions and commitment to the studies. For NHS and NHS2, the study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. We would like to thank the participants and staff of the NHS and NHS2 for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data. ORIGO thanks E. Krol-Warmerdam, and J. Blom for patient accrual, administering questionnaires, and managing clinical information. The LUMC survival data were retrieved from the Leiden hospital-based cancer registry system (ONCDOC) with the help of Dr. J. Molenaar. PBCS thanks Louise Brinton, Mark Sherman, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao, and Michael Stagner. The ethical approval for the POSH study is MREC /00/6/69, UKCRN ID: 1137. We thank staff in the Experimental Cancer Medicine Centre (ECMC) supported Faculty of Medicine Tissue Bank and the Faculty of Medicine DNA Banking resource. The authors wish to acknowledge the roles of the Breast Cancer Now Tissue Bank in collecting and making available the samples and/or data, and the patients who have generously donated their tissues and shared their data to be used in the generation of this publication. PREFACE thanks Sonja Oeser and Silke Landrith. We thank the SEARCH and EPIC teams. SGBCC thanks the participants and all research coordinators for their excellent help with recruitment, data and sample collection. SKKDKFZS thanks all study participants, clinicians, family doctors, researchers, and technicians for their contributions and commitment to this study. We thank the SUCCESS Study teams in Munich, Düsseldorf, Erlangen and Ulm. UCIBCS thanks Irene Masunaka. UKBGS thanks Breast Cancer Now and the Institute of Cancer Research for support and funding of the Generations Study, and the study participants, study staff, and the doctors, nurses, and other health care providers and health information sources who have contributed to the study. We acknowledge NHS funding to the Royal Marsden/ICR NIHR Biomedical Research Centre.
The following are NBCS Collaborators: Kristine K. Sahlberg (PhD), Anne-Lise Børresen-Dale (Prof. Em.), Lars Ottestad (MD), Rolf Kåresen (Prof. Em.) Dr. Ellen Schlichting (MD), Marit Muri Holmen (MD), Toril Sauer (MD), Vilde Haakensen (MD), Olav Engebråten (MD), Bjørn Naume (MD), Alexander Fosså (MD), Cecile E. Kiserud (MD), Kristin V. Reinertsen (MD), Åslaug Helland (MD), Margit Riis (MD), Jürgen Geisler (MD), OSBREAC, and Grethe I. Grenaker Alnæs (MSc).
The following are ABCTB Investigators: Prof Christine Clarke, Centre for Cancer Research, The Westmead Institute for Medical Research, The University of Sydney, Sydney, NSW, Australia; Prof Deborah Marsh, University of Technology Sydney, Translational Oncology Group, School of Life Sciences, Faculty of Science, Ultimo, NSW, Australia; Prof Rodney Scott, School of Biomedical Sciences, University of Newcastle, Newcastle; Hunter Medical Research Institute and NSW Health Pathology North, Newcastle, Australia; Prof Robert Baxter, Kolling Institute of Medical Research, University of Sydney, St Leonards, NSW, Australia; A/Prof Desmond Yip, Epigenetics & Transcription Laboratory Melanie Swan Memorial Translational Centre, Sci-Tech, University of Canberra, Canberra, ACT; Department of Medical Oncology, The Canberra Hospital, Garran, ACT, Australia; Ms Jane Carpenter, Scientific Platforms, The Westmead Institute for Medical Research, The University of Sydney, Sydney, NSW, Australia; Dr Alison Davis, The Canberra Hospital, Garran, ACT; The Australian National University, ACT, Australia; A/Prof Nirmala Pathmanathan, Westmead Breast Cancer Institute, Western Sydney Local Health District, Westmead, New South Wales, Australia; University of Sydney, Western Clinical School, Westmead, New South Wales, Australia; Dr Peter Simpson, UQ Centre for Clinical Research, Faculty of Medicine, The University of Queensland, Herston, QLD, Australia; Dr Dinny Graham, Centre for Cancer Research, The Westmead Institute for Medical Research, The University of Sydney, Sydney, NSW, Australia; Dr Mythily Sachchithananthan, Centre for Cancer Research, The Westmead Institute for Medical Research, The University of Sydney, Sydney, NSW, Australia.
Funding
This result is part of a project that has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant agreement No. 948561).
BCAC is funded by the European Union’s Horizon 2020 Research and Innovation Programme (grant numbers 634935 and 633,784 for BRIDGES and B-CAST respectively), and the PERSPECTIVE I&I project, funded by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the Ministère de l’Économie et de l'Innovation du Québec through Genome Québec, the Quebec Breast Cancer Foundation. The EU Horizon 2020 Research and Innovation Programme funding source had no role in study design, data collection, data analysis, data interpretation, or writing of the report. Additional funding for BCAC is provided via the Confluence project which is funded with intramural funds from the National Cancer Institute Intramural Research Program, National Institutes of Health.
Genotyping of the OncoArray was funded by the NIH Grant U19 CA148065, and Cancer UK Grant C1287/A16563 and the PERSPECTIVE project supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research (grant GPH-129344), and the Ministère de l’Économie, Science et Innovation du Québec through Genome Québec and the PSRSIIRI-701 grant, and the Quebec Breast Cancer Foundation. Funding for iCOGS came from: the European Community’s Seventh Framework Programme under grant agreement n° 223,175 (HEALTH-F2-2009–223,175) (COGS), Cancer Research UK (C1287/A10118, C1287/A10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112—the GAME-ON initiative), the Department of Defence (W81XWH-10–1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, and Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. The BRIDGES panel sequencing was supported by the European Union Horizon 2020 research and innovation program BRIDGES (grant number, 634,935) and the Wellcome Trust (v203477/Z/16/Z). The ABCS study was supported by the Dutch Cancer Society [grants NKI 2007–3839; 2009 4363]. The Australian Breast Cancer Tissue Bank (ABCTB) was supported by the National Health and Medical Research Council of Australia, The Cancer Institute NSW and the National Breast Cancer Foundation. The ACP study is funded by the Breast Cancer Research Trust, UK. KM and AL are supported by the NIHR Manchester Biomedical Research Centre, the Allan Turing Institute under the EPSRC grant EP/N510129/1. The AHS study is supported by the intramural research program of the National Institutes of Health, the National Cancer Institute (grant number Z01-CP010119), and the National Institute of Environmental Health Sciences (grant number Z01-ES049030). The work of the BBCC was partly funded by ELAN-Fond of the University Hospital of Erlangen. The BBCS is funded by Cancer Research UK and Breast Cancer Now and acknowledges NHS funding to the NIHR Biomedical Research Centre, and the National Cancer Research Network (NCRN). For the BCFR-NY, BCFR-ON, BCFR-PA, BCFR-UT this work was supported by grant UM1 CA164920 from the National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the BCFR. The BCINIS study is supported in part by the Breast Cancer Research Foundation (BCRF). The BREast Oncology GAlician Network (BREOGAN) is funded by Acción Estratégica de Salud del Instituto de Salud Carlos III FIS PI12/02125/Cofinanciado and FEDER PI17/00918/Cofinanciado FEDER; Acción Estratégica de Salud del Instituto de Salud Carlos III FIS Intrasalud (PI13/01136); Programa Grupos Emergentes, Cancer Genetics Unit, Instituto de Investigacion Biomedica Galicia Sur. Xerencia de Xestion Integrada de Vigo-SERGAS, Instituto de Salud Carlos III, Spain; Grant 10CSA012E, Consellería de Industria Programa Sectorial de Investigación Aplicada, PEME I + D e I + D Suma del Plan Gallego de Investigación, Desarrollo e Innovación Tecnológica de la Consellería de Industria de la Xunta de Galicia, Spain; Grant EC11-192. Fomento de la Investigación Clínica Independiente, Ministerio de Sanidad, Servicios Sociales e Igualdad, Spain; and Grant FEDER-Innterconecta. Ministerio de Economia y Competitividad, Xunta de Galicia, Spain. The BSUCH study was supported by the Dietmar-Hopp Foundation, the Helmholtz Society and the German Cancer Research Center (DKFZ). The CAMA study was funded by Consejo Nacional de Ciencia y Tecnología (CONACyT) (SALUD-2002-C01-7462). Sample collection and processing was funded in part by grants from the National Cancer Institute (NCI R01CA120120 and K24CA169004). CBCS is funded by the Canadian Cancer Society (grant # 313,404) and the Canadian Institutes of Health Research. CCGP is supported by funding from the University of Crete. The CECILE study was supported by Fondation de France, Institut National du Cancer (INCa), Ligue Nationale contre le Cancer, Agence Nationale de Sécurité Sanitaire, de l'Alimentation, de l'Environnement et du Travail (ANSES), Agence Nationale de la Recherche (ANR). The CGPS was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council, and Herlev and Gentofte Hospital. COLBCCC is supported by the German Cancer Research Center (DKFZ), Heidelberg, Germany. Diana Torres was in part supported by a postdoctoral fellowship from the Alexander von Humboldt Foundation. The American Cancer Society funds the creation, maintenance, and updating of the CPS-II cohort. The California Teachers Study (CTS) and the research reported in this publication were supported by the National Cancer Institute of the National Institutes of Health under award number U01-CA199277; P30-CA033572; P30-CA023100; UM1-CA164917; and R01-CA077398. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The collection of cancer incidence data used in the California Teachers Study was supported by the California Department of Public Health pursuant to California Health and Safety Code Section 103,885; Centers for Disease Control and Prevention’s National Program of Cancer Registries, under cooperative agreement 5NU58DP006344; the National Cancer Institute’s Surveillance, Epidemiology and End Results Program under contract HHSN261201800032I awarded to the University of California, San Francisco, contract HHSN261201800015I awarded to the University of Southern California, and contract HHSN261201800009I awarded to the Public Health Institute. The opinions, findings, and conclusions expressed herein are those of the author(s) and do not necessarily reflect the official views of the State of California, Department of Public Health, the National Cancer Institute, the National Institutes of Health, the Centers for Disease Control and Prevention or their Contractors and Subcontractors, or the Regents of the University of California, or any of its programs. The University of Westminster curates the DietCompLyf database funded by Against Breast Cancer Registered Charity No. 1121258 and the NCRN. The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by: Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF) (Germany); the Hellenic Health Foundation, the Stavros Niarchos Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Cancer Research UK (14,136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1,000,143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom). Where authors are identified as personnel of the International Agency for Research on Cancer / World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer / World Health Organization. The ESTHER study was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe). FHRISK and PROCAS are funded from NIHR grant PGfAR 0707–10,031. DGE, AH and WGN are supported by the NIHR Manchester Biomedical Research Centre (IS-BRC-1215–20,007). The GC-HBOC (German Consortium of Hereditary Breast and Ovarian Cancer) is supported by the German Cancer Aid (grant no 110837, coordinator: Rita K. Schmutzler, Cologne). This work was also funded by the European Regional Development Fund and Free State of Saxony, Germany (LIFE—Leipzig Research Centre for Civilization Diseases, project numbers 713–241,202, 713–241,202, 14,505/2470, 14,575/2470). The GENICA was funded by the Federal Ministry of Education and Research (BMBF) Germany grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114, the Robert Bosch Foundation, Stuttgart, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, the Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, as well as the Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany. The GEPARSIXTO study was conducted by the German Breast Group GmbH. The GESBC was supported by the Deutsche Krebshilfe e. V. [70492] and the German Cancer Research Center (DKFZ). The HABCS study was supported by the Claudia von Schilling Foundation for Breast Cancer Research, by the Lower Saxonian Cancer Society, and by the Rudolf Bartling Foundation. The HEBCS was financially supported by the Helsinki University Hospital Research Fund, the Sigrid Juselius Foundation and the Cancer Foundation Finland. The HERPACC was supported by MEXT Kakenhi (No. 170150181 and 26,253,041) from the Ministry of Education, Science, Sports, Culture and Technology of Japan, by a Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cancer Control from Ministry Health, Labour and Welfare of Japan, by Health and Labour Sciences Research Grants for Research on Applying Health Technology from Ministry Health, Labour and Welfare of Japan, by National Cancer Center Research and Development Fund, and "Practical Research for Innovative Cancer Control (15ck0106177h0001 and 20ck0106553)" from Japan Agency for Medical Research and development, AMED, and Cancer Bio Bank Aichi. The HMBCS was supported by a grant from the Friends of Hannover Medical School and by the Rudolf Bartling Foundation. The HUBCS was supported by a grant from the German Federal Ministry of Research and Education (RUS08/017), B.M. was supported by grant 17–44-020,498, 17–29-06,014 of the Russian Foundation for Basic Research, D.P. was supported by grant 18–29-09,129 of the Russian Foundation for Basic Research, E.K was supported by the program for support the bioresource collections №007–030,164/2 and by the megagrant from the Government of Russian Federation (2020–220-08–2197), and study was performed as part of the assignment of the Ministry of Science and Higher Education of Russian Federation (№AAAA-A16-116,020,350,032–1). Financial support for KARBAC was provided through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet, the Swedish Cancer Society, The Gustav V Jubilee foundation and Bert von Kantzows foundation. The KARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer. The KBCP was financially supported by the special Government Funding (VTR) of Kuopio University Hospital grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, and by the strategic funding of the University of Eastern Finland. The KOHBRA study was partially supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), and the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea (HI16C1127; 1,020,350; 1,420,190). LMBC is supported by the “Stichting tegen Kanker”. DL is supported by the FWO. The MABCS study is funded by the Research Centre for Genetic Engineering and Biotechnology "Georgi D. Efremov", MASA. The MARIE study was supported by the Deutsche Krebshilfe e.V. [70–2892-BR I, 106,332, 108,253, 108,419, 110,826, 110828], the Hamburg Cancer Society, the German Cancer Research Center (DKFZ) and the Federal Ministry of Education and Research (BMBF) Germany [01KH0402]. MBCSG is supported by grants from the Italian Association for Cancer Research (AIRC). The MCBCS was supported by the NIH grants R35CA253187, R01CA192393, R01CA116167, R01CA176785 a NIH Specialized Program of Research Excellence (SPORE) in Breast Cancer [P50CA116201], and the Breast Cancer Research FoundationThe MEC was supported by NIH grants CA63464, CA54281, CA098758, CA132839 and CA164973. The MISS study is supported by funding from ERC-2011–294,576 Advanced grant, Swedish Cancer Society, Swedish Research Council, Local hospital funds, Berta Kamprad Foundation, Gunnar Nilsson. The MMHS study was supported by NIH grants CA97396, CA128931, CA116201, CA140286 and CA177150. MSKCC is supported by grants from the Breast Cancer Research Foundation and Robert and Kate Niehaus Clinical Cancer Genetics Initiative. MYBRCA is funded by research grants from the Wellcome Trust (v203477/Z/16/Z), the Malaysian Ministry of Higher Education (UM.C/HlR/MOHE/06) and Cancer Research Malaysia. MYMAMMO is supported by research grants from Yayasan Sime Darby LPGA Tournament and Malaysian Ministry of Higher Education (RP046B-15HTM). The NBCS has received funding from the K.G. Jebsen Centre for Breast Cancer Research; the Research Council of Norway grant 193,387/V50 (to A-L Børresen-Dale and V.N. Kristensen) and grant 193,387/H10 (to A-L Børresen-Dale and V.N. Kristensen), South Eastern Norway Health Authority (grant 39,346 to A-L Børresen-Dale) and the Norwegian Cancer Society (to A-L Børresen-Dale and V.N. Kristensen). The NBHS was supported by NIH grant R01CA100374. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The Northern California Breast Cancer Family Registry (NC-BCFR) was supported by grant U01CA164920 from the USA National Cancer Institute of the National Institutes of Health. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The Carolina Breast Cancer Study (NCBCS) was funded by Komen Foundation, the National Cancer Institute (P50 CA058223, U54 CA156733, U01 CA179715), and the North Carolina University Cancer Research Fund. The NGOBCS was supported by the National Cancer Center Research and Development Fund (Japan). The NHS was supported by NIH grants P01 CA87969, UM1 CA186107, and U19 CA148065. The NHS2 was supported by NIH grants UM1 CA176726 and U19 CA148065. The ORIGO study was supported by the Dutch Cancer Society (RUL 1997–1505) and the Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL CP16). The PBCS was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. Genotyping for PLCO was supported by the Intramural Research Program of the National Institutes of Health, NCI, Division of Cancer Epidemiology and Genetics. The PLCO is supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, National Institutes of Health. The POSH study is funded by Cancer Research UK (grants C1275/A11699, C1275/C22524, C1275/A19187, C1275/A15956 and Breast Cancer Campaign 2010PR62, 2013PR044. The SBCGS was supported primarily by NIH grants R01CA64277, R01CA148667, UMCA182910, and R37CA70867. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The scientific development and funding of this project were, in part, supported by the Genetic Associations and Mechanisms in Oncology (GAME-ON) Network U19 CA148065. The SBCS was supported by Sheffield Experimental Cancer Medicine Centre and Breast Cancer Now Tissue Bank. SEARCH is funded by Cancer Research UK [C490/A10124, C490/A16561] and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. The University of Cambridge has received salary support for PDPP from the NHS in the East of England through the Clinical Academic Reserve. SEBCS was supported by the BRL (Basic Research Laboratory) program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2012–0,000,347). SGBCC is funded by the National Research Foundation Singapore, NUS start-up Grant, National University Cancer Institute Singapore (NCIS) Centre Grant, Breast Cancer Prevention Programme, Asian Breast Cancer Research Fund and the NMRC Clinician Scientist Award (SI Category). Population-based controls were from the Multi-Ethnic Cohort (MEC) funded by grants from the Ministry of Health, Singapore, National University of Singapore and National University Health System, Singapore. The Sister Study (SISTER) is supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01-ES044005 and Z01-ES049033). The Two Sister Study (2SISTER) was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01-ES044005 and Z01-ES102245), and, also by a grant from Susan G. Komen for the Cure, grant FAS0703856. SKKDKFZS is supported by the DKFZ. The SMC is funded by the Swedish Cancer Foundation and the Swedish Research Council (VR 2017–00,644) grant for the Swedish Infrastructure for Medical Population-based Life-course Environmental Research (SIMPLER). The TNBCC was supported by: a Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), a grant from the Breast Cancer Research Foundation, a generous gift from the David F. and Margaret T. Grohne Family Foundation. The TWBCS is supported by the Taiwan Biobank project of the Institute of Biomedical Sciences, Academia Sinica, Taiwan. The UCIBCS component of this research was supported by the NIH [CA58860, CA92044] and the Lon V Smith Foundation [LVS39420]. The UKBGS is funded by Breast Cancer Now and the Institute of Cancer Research (ICR), London. ICR acknowledges NHS funding to the NIHR Biomedical Research Centre. The UKOPS study was funded by The Eve Appeal (The Oak Foundation) and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre with contribution to author’s salary through MRC core funding MC_UU_00004/01. The USRT Study was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. The WAABCS study was supported by grants from the National Cancer Institute of the National Institutes of Health (R01 CA89085 and P50 CA125183 and the D43 TW009112 grant), Susan G. Komen (SAC110026), the Dr. Ralph and Marian Falk Medical Research Trust, and the Avon Foundation for Women.
Author information
Authors and Affiliations
Consortia
Contributions
K.B.K. conceived the Project and edited the original and revised paper, S.H.M. conducted analysis, interpreted the results, wrote and edited the original and revised paper, U.H. led individual studies and contributed to the design of the study and data collection, edited the original, and revised paper, A.G.L. contributed to the data analyses and the interpretation of the results, edited the original, and revised paper, M.V. contributed to the data preparation, edited the original, and revised paper, H.H. edited the original and revised paper, K.Mi., M.K.B., Q.W., J.Den., M.Lus., T.U.A., I.L.A., H.A-C., N.N.A., V.A., K.J.A., A.Au., T.Bae., L.E.BF., M.W.B., S.Be., J.Ben., M.Berm., C.Bl., N.V.B., S.E.B., B.Bon., H.Bre., S.Y.B., S.S.B., J.E.C., T.L.C., J.C–C., S.J.C., J-Y.C., W.K.C., S.V.C., S.Co., F.J.C., K.Cz., M.B.D., P.D., T.D., L.Do., M.Dw., D.M.E., A.B.E., AH.E., C.En., DG.E., P.A.F., O.F., H.F., M.G-D., Y–T.G., M.G-C., J.A.G-S., A.G-M., P.Gu., M.G., L.Ha., E.Hah., C.A.H., N.Håk., P.Hall., E.F.H., P.A.H., J.M.H., M.Ha., A.Hei., W-K.H., M.J.H., R.H., J.L.Ho., R.S.H., A.How., D.J.H., D.H., H.It., M.I., A.Jak., W.Ja., E.M.J., M.E.J., A.Ju., R.Ka., D.Ka., E.K.K., S-W.K., C.M.Ki., S.Kou., P.Kr., V.N.K., K.K-S., A.W.K., A.Kw., J.V.L., D.La., L.LM., J.Li., M.Lin., W-L.L., J.Lo., A.Lo., A.Man., M.Man., S.Mar., K.Ma., D.M., U.Me., K.Mu., R.A.M., H.Ne., W.G.N., D.N., K.M.O., N.Ob., K.Of., O.L.O., A.F.O., H.O., S.K.P., A.V.P., C.M.Pe., J.Pet., P.D.P.P., D.P.K., N.Pre., B.R., P.Ra., D.Ram.,M.U.R., G.R., A.Ro., K.J.R., M.Ru., E.S., D.P.S., E.J.S., M.K.S., R.K.S., M.S., C.Sc., M.Sh., P.Sh., C-Y.S., X-O.S., J.Si., H.Sur., R.M.T., W.J.T., J.A.T., S.H.T., L.R.T., A.E.T., R.A.E.M.T., D.T., G.T-M., M.A.T., T.T.,, C.M.V, J.Vi.,C.R.W., C.We., R.Wi., A.W., A.H.W., T.Ya., X.R.Y., J-C.Y., W.Z., A.Z., E.Z., A.M.D., and D.F.E. led individual studies and contributed to the design of the study and data collection. All authors read and approved the final manuscript.
Author’s information
Twitter handle: @KKuchenbaecker (Karoline B. Kuchenbaecker).
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participants provided written informed consent and all BCAC studies have been approved by the responsible ethics review board. The ethics committees that approved the studies are listed in Additional file 1: Table S9. The research conformed to the principles of the Helsinki Declaration. This work was carried out under the approved BCAC concept form #595.
Consent for publication
Not applicable.
Competing interests
S.H.M. became an employee of Boehringer Ingelheim after completing this study. Matthias W. Beckmann conducts research funded by Amgen, Novartis, and Pfizer. P.A.F. conducts research funded by Amgen, Novartis, and Pfizer. He received Honoraria from Roche, Novartis, and Pfizer. R.A.M. is a consultant for Pharmavite. A.W.K. received research funding from Myriad Genetics for an unrelated project (funding dates 2017–2019).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Cohort Descriptives. Table S2. Included BCAC studies. Table S3. Number of genes analysed per cohort. Table S4. Genomic inflation in burden analysis. Table S5. Model covariates per cohort. Figure S1. QQ-plots of burden analysis. Figure S2. QQ-plots for meta-analysis. Figure S3. Regional plots for significantly associated genes. FAM72B, SRGAP2C, AC058822.1, ABRAXAS1, MAP3K1, MIER3, BTN2A1, FGFR2, TNNT3, LSP1, LYPD5, KCNN4, ZNF404. Table S6. All Cohort Meta-Analysis. Table S7. Suggestive Associations in Diverse Ancestries. Table S8. Prior Evidence for Suggestive Associations in Diverse Ancestries. Figure S4. Regional plot for gene ESR1. Figure S5. Regional plot for gene CBLB. Table S9. Ethics committees that provided approval for the contributing studies.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mueller, S.H., Lai, A.G., Valkovskaya, M. et al. Aggregation tests identify new gene associations with breast cancer in populations with diverse ancestry. Genome Med 15, 7 (2023). https://doi.org/10.1186/s13073-022-01152-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-022-01152-5