
Abstract
Background
Acral and mucosal melanomas are aggressive subtypes of melanoma, which have a significantly lower burden of somatic mutations than cutaneous melanomas, but more frequent copy number variations, focused gene amplifications, and structural alterations. The landscapes of their genomic alterations remain to be fully characterized.
Methods
We compiled sequencing data of 240 human acral and mucosal melanoma samples from 11 previously published studies and applied a uniform pipeline to call tumor cell content, ploidy, somatic and germline mutations, as well as CNVs, LOH, and SVs. We identified genes that are significantly mutated or recurrently affected by CNVs and implicated in oncogenesis. We further examined the difference in the frequency of recurrent pathogenic alterations between the two melanoma subtypes, correlation between pathogenic alterations, and their association with clinical features.
Results
We nominated PTPRJ, mutated and homozygously deleted in 3.8% (9/240) and 0.8% (2/240) of samples, respectively, as a probable tumor suppressor gene, and FER and SKP2, amplified in 3.8% and 11.7% of samples, respectively, as probable oncogenes. We further identified a long tail of infrequent pathogenic alterations, involving genes such as CIC and LZTR1. Pathogenic germline mutations were observed on MITF, PTEN, ATM, and PRKN. We found BRAF V600E mutations in acral melanomas with fewer structural variations, suggesting that they are distinct and related to cutaneous melanomas. Amplifications of PAK1 and GAB2 were more commonly observed in acral melanomas, whereas SF3B1 R625 codon mutations were unique to mucosal melanomas (12.9%). Amplifications at 11q13-14 were frequently accompanied by fusion to a region on chromosome 6q12, revealing a recurrent novel structural rearrangement whose role remains to be elucidated.
Conclusions
Our meta-analysis expands the catalog of driver mutations in acral and mucosal melanomas, sheds new light on their pathogenesis and broadens the catalog of therapeutic targets for these difficult-to-treat cancers.
Similar content being viewed by others

Background
Acral and mucosal melanomas account for approximately 3–4% and 1% of total melanoma cases in Caucasians [1,2,3,4], respectively, but are the dominant melanoma subtypes in non-Caucasian populations [5, 6]. Acral melanomas arise on the non-hair-bearing skin of palms, soles, and the nail apparatus, while mucosal melanomas primarily affect mucosal membranes of the nasopharynx and anogenital tracts. Contrary to cutaneous melanomas, which frequently originate from sun-exposed skin, the sites harboring acral and mucosal melanomas are relatively or completely shielded from environmental ultraviolet (UV) radiation. Both melanoma subtypes share similar genomic features such as low burdens of somatic mutations with an increased frequency of copy number and structural alterations instead [7,8,9]. The specific genetic alterations found in acral and mucosal melanomas only partially overlap with those in cutaneous melanomas [7]. For example, acral and mucosal melanomas have fewer BRAF mutations but more frequent KIT mutations, amplifications of CCND1, CDK4, and MDM2, and deletion of SPRED1 [7, 8, 10,11,12].
Due to their comparatively lower incidence in western countries and distinctive features, acral and mucosal melanomas were excluded from The Cancer Genome Atlas (TCGA) [13], and the number of sequencing studies of these two subtypes have comparatively smaller sample sizes [8, 10, 11, 14,15,16,17]. Consequently, the power of discovery has been lower, and the landscape of somatic and germline driver events remains incompletely characterized. In this study, we compiled a dataset of human acral and mucosal melanomas from multiple previous studies [8, 10, 11, 13,14,15,16, 18,19,20,21] and performed a meta-analysis.
Methods
Data collection
We downloaded the mapped BAM files or, where available, unprocessed FASTQ sequencing files from whole-genome or whole-exome sequencing (WGS and WXS) of 154 acral and 93 mucosal melanomas from 11 previous studies [8, 10, 11, 13,14,15,16, 18,19,20,21] (Additional file 1: Table S1). Four patients had multiple tumor samples from the same cancer sequenced, and we included only the metastasis with the highest purity for analysis for these patients. We excluded 7 acral melanomas with target coverage under 25-fold or tumor cell content under 20%, leaving 147 acral and 93 mucosal melanomas, or 165 WGS and 75 WXS samples, for downstream analyses.
Processing of sequencing data
For cases for which only BAM files were available [8, 10, 11, 13], we confirmed usage of hg19 and directly proceeded to downstream analyses. For the remaining samples, we mapped FASTQ data to the hg19 assembly, using the Burrows-Wheeler Aligner [22] and used Picard (http://broadinstitute.github.io/picard) to remove PCR duplicates and calculate insert size, followed by GATK [23] to perform base quality score recalibration and realign indel regions.
Calling of somatic mutations
We used Mutect2 [24] and Strelka2 [25] to call somatic SNVs and short indels for each melanoma DNA, using its matched germline DNA as reference. Mutations identified by both tools were merged and annotated with Annovar [26]. We removed variants corresponding to single-nucleotide polymorphisms (SNPs) found in more than 1% of any of the populations in ExAC, GnomAD, 1000 Genome, dbSNP150, or NHLBI-ESP [27,28,29,30,31] to eliminate false positive somatic calls at loci with low germline coverage. For coding mutations flagged as “PASS” by both tools, a minimum of 4 reads were required to be included in further analyses. For mutations that passed only one tool, we required a coverage of 10 reads in tumor and normal samples, a minimum of 6 mutated reads in tumor and a maximum of one mutated read or 3% mutant allele frequency (MAF) in normal. We excluded indels flagged as “clustered_events” or “clustered_events;haplotype” by Mutect2. Noncoding mutations were required to be called as “PASS” by both tools, considering the abundant repetitive elements in noncoding regions [32]. To remove potential sequencing artifacts, we applied SOBDector [33] and removed SNVs that were labeled as “artifact.” For the final set of somatic mutations, we performed principal component analysis on the matrix of 96 classes of substitutions (Additional file 2: Fig. S1A and B), and observed moderate separation of WGS and WXS samples that was probably attributable to context differences between coding and noncoding regions. Several samples with distinct mutation signatures, including UV radiation, mismatch repair defect and temozolomide treatment, were scattered as outliers. Boxplots of median MAF for samples from several major studies were shown in Additional file 2: Fig. S1C.
Identification of significantly mutated genes (SMGs)
We used 3 tools, dNdScv [34], MutPanning [35], and MutSigCV [36], to identify significantly mutated genes. For dNdScv, the R package was applied (https://github.com/im3sanger/dndscv). MutPanning was implemented through the GenePattern online server (https://cloud.genepattern.org/gp/pages/index.jsf). For MutSigCV, the GitHub version (https://github.com/getzlab/MutSig2CV) was used. Mutations located in exons or splice sites were extracted as input for the analysis.
Calling germline mutations
We used HaplotypeCaller [37] to call germline mutations in the normal DNA. Mutations with fewer than 4 reads or an allele frequency under 25% were removed and the remaining mutations annotated using Annovar. We further removed common SNPs with > 1% mutation frequency in ExAC, GnomAD, 1000 Genome, dbSNP150, or NHLBI-ESP [27,28,29,30,31]. The pathogenicity of germline mutations was estimated using annotations from OncoKB [38] and ClinVar [39]. Mutations that were annotated as “benign/likely_benign” by Clinvar, as “neutral” or “inconclusive” by OncoKB, or were located very close to protein C-terminus were not considered as possible driver events. A pathogenic JAK2 V617F mutation was identified in patient A_MELA_0061, with 45% allele frequency in the blood sample but 0 mutated reads (out of 62) in the corresponding tumor sample that retained both alleles of JAK2. The tumor and blood samples were confirmed to have originated from the same patient using the “CalculateContamination” function from GATK. This mutation was subsequently removed since it likely reflects clonal hematopoiesis [40] rather than a bona fide germline mutation.
Determining the genetic ancestry of patients
To determine the genetic ancestry of included patients, we downloaded the phase 3 autosomal variants of the 1000 Genomes Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502; PED file from: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20130606_sample_info/20130606_g1k.ped), and removed SNPs with minor allele frequency < 1%, multi-allelic variants, indels, as and those not located within exons or the 100-bp immediate flanking regions. For the remaining SNPs, we genotyped the corresponding loci in our normal samples using SAMtools and BCFtools [41], with a requirement of at least 10 reads. We further removed loci that failed genotyping in over half of the patients or had inconsistent alternative allele in any patient. We next applied PLINK v1.90b6.24 for downstream analyses. Specifically, data from our own cohort and the 1000 Genomes Project were converted to PLINK format and merged. From the merged dataset, we removed SNPs with allele frequency < 10% and further pruned to obtain the subset in approximate linkage equilibrium, with arguments “--maf 0.1 --indep 50 5 1.5”. The remaining SNPs were input for principal component analysis. Based on the closeness with 1000 genome populations, 96.7% (174/180) of patients with ancestry information available can be assigned to the correct ethnicity (Additional file 1: Table S1 and Additional file 2: Fig. S2).
Identification of pathogenic mutations
We downloaded the list of 682 annotated cancer genes from OncoKB (as of April 14, 2021) [38] and included 21 additional genes of interest (Additional file 3: Table S2), which emerged during our analyses or had been implicated in melanoma or other cancer types. Germline and somatic mutations in any of these 703 genes were considered as pathogenic, if (1) annotated as “oncogenic,” “likely oncogenic,” or “hotspot” by the OncoKB database or (2) predicted to be damaging (nonsense, stoploss, frameshift or splice site) a known tumor suppressor gene.
Mutational signature analysis of single-nucleotide substitutions
We used the R package SigProfilerExtractorR [42] (https://github.com/AlexandrovLab/SigProfilerExtractorR) with the matrix of 96 classes of substitutions in samples as input. The parameters “minimum_signatures” and “maximum_signatures” were set to be 3 and 15, respectively. The tool identified the most probable solution of de novo signatures, and subsequently decomposed them into combinations of COSMIC single base substitution (SBS) signatures (https://cancer.sanger.ac.uk/signatures/sbs/). These SBS signatures reflect different mutational processes. For instance, SBS7 is known as the “UV signature” because it is induced by exposure to UV radiation [43].
Copy number analysis
We used FACETS to assess relative and absolute copy number variation (CNV), loss of heterozygosity (LOH), and tumor purity and ploidy [44]. Overall, our purity estimates appeared comparable or better than those from the original studies (Additional file 2: Fig. S3). For a few exceptions with less accurate FACETS estimates, we recalculated purities using either the median MAF of somatic mutations, or the median VAF of germline variants at hemizygous regions [45]. We then recalculated the absolute copy number (or n) of each segment using the below formula (ρ and φ represent tumor purity and ploidy, respectively) under the assumption of diploid genome (the log2 ratios of diploid segments were near 0).
To resolve visually suspicious regions of narrow homozygous deletions or amplifications that were not called by FACETS, we used the pre-segmented log2 ratios calculated by CNVkit [46], which was run separately for the WGS and WXS datasets to account for variations in coverage across studies. Amplifications were defined as copy number increases more than twofold than the estimated ploidy of the tumor and affecting a genomic region no larger than 10 megabases.
We applied GISTIC2 [47] to the segmented and purity-adjusted copy number data derived from FACETS to identify genomic regions with recurrent copy number alterations, likely harboring genes relevant in acral and mucosal melanoma. The copy number ratios were median centered by GISTIC2 prior to the analysis.
Identifying structural variations (SV)
Delly2 [48] was used to identify somatic SVs in the WGS data.
Estimation of microsatellite instability
MSIsensor [49] was used to estimate the level of microsatellite instability within coding regions in tumor DNAs compared to their matched germline reference DNA.
Identifying driver events with distinct frequencies between the two melanoma subtypes and testing for the interdependence among driver events
We used Fisher’s exact test to compare the frequency of pathogenic alterations between the two subtypes and examine the correlations among different types of alterations. Altered tumor suppressor genes were defined as those with inactivating mutations or homozygous deletions. For oncogenes, amplification and pathogenic or likely pathogenic mutations were classified into separate groups. BRAF mutations were classified into class 1 (V600E and V600K) and other BRAF mutations. Overall, 35 groups of alterations, each observed in at least 10 samples, were included for analysis. For testing the interdependence of driver alterations, pairs of gene amplifications located on the same chromosomal arms were excluded.
Identifying driver events associated with clinical features
For acral melanomas, the frequency of pathogenic alterations were compared between those originated from sole (n = 71) and subungual area (n = 37), while for mucosal melanomas, pairwise comparison was performed among those originated from the genitourinary (n = 20), gastrointestinal (n = 15), and sinonasal/oropharyngeal (n = 37) systems, respectively. We also identified 53 primary and 85 metastatic acral melanomas, and 33 primary and 43 metastatic/recurrent mucosal melanomas, respectively, and compared primary with metastatic/recurrent samples for the merged set of both melanoma subtypes, and separately for each subtype. We further compared mucosal melanomas from patients of European and Asian ancestries (n = 48 and 42, respectively). No such comparisons were performed for acral melanomas since most were of European ancestry (n = 131). Fisher’s exact test was used for the above comparisons. Survival analysis was performed using the R package “survival.”
Results
Description of the dataset
We compiled published sequencing data from 147 acral and 93 mucosal melanomas (Additional file 1: Table S1) and applied a uniform pipeline to call tumor cell content, ploidy, somatic and germline mutations, as well as CNVs, LOH, and SVs (Additional files 4: Table S3, Additional files 5: Table S4, and Additional files 6: Table S5). Acral and mucosal melanomas had low mutation burdens (median number of mutations per megabase 1.92, range = 0.48–80.13 and 2.24, range = 0.64–20.04, respectively. Additional file 2: Figs. S4 and S5), consistent with previous estimates and considerably lower than the mutation burden in cutaneous melanomas [8]. We note that a small subset of samples showed higher mutation burdens, with 11 acral and 2 mucosal melanomas having over 10 mutations per megabase, attributable to UV radiation (SBS7; n = 8), chemotherapy with temozolomide (SBS11) and/or platinum (SBS31) (n = 4), or DNA mismatch repair defects (SBS 21 and 26) due to homozygous deletion of MLH1 in A_MELA_0271 (MSI score = 20.59) (Additional file 2: Figs. S4 and S5). Only one other melanoma, M_MELA_0581, showed evidence of mismatch repair deficiency (MSI score = 5.54. Additional file 2: Fig. S2), which was accompanied by biallelic inactivation of MSH6, but lacked an elevated mutation burden (2.24 per megabase) or mismatch repair deficiency-related signature, likely indicating that mismatch repair deficiency did not account for the majority of mutations in this melanoma.
Identification of significantly mutated genes
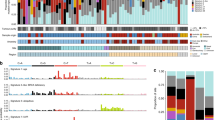
The algorithms dNdScv, MutPanning, and MutSigCV identified 16 significantly mutated genes (q < 0.1 by any tool) (Fig. 1A and Additional file 7: Table S6), 13 of which are well-documented driver genes in melanoma and known to activate the MAP kinase (MAPK) pathway (BRAF, NRAS, NF1, KIT, SPRED1, and KRAS) or other pathways (PTEN, ATRX, CTNNB1, TP53, CDKN2A, SF3B1, and B2M).

Recurring PTPRJ alterations in acral and mucosal melanomas. A PTPRJ is among the 16 SMGs identified by one or more of the three algorithms (q < 0.1) in the combined dataset of both subtypes. B Lollipop plot with somatic non-synonymous, splice site, and silent mutations in PTPRJ. C, D Focused homozygous deletions of PTPRJ in two acral melanomas
Three additional significantly mutated genes were identified that are not yet firmly associated with melanoma. Mutations in the protein tyrosine phosphatase PTPRJ, 5 truncating, 3 missense, and 1 in-frame indel, were identified in 9 melanomas, 4 acral and 5 mucosal (Fig. 1B), and PTPRJ was homozygously deleted in two additional acral melanomas (Figs. 1C and 2D). Three and 1 of the truncating and missense mutations, respectively, were coupled with LOH. Together, 5 melanomas showed genetic evidence of biallelic inactivation, with either homozygous deletion or truncating mutation coupled with LOH. One recent study observed truncating PTPRJ mutations in 23% (7/31) of canine mucosal melanomas [50], while another study reported truncating mutations in both human (1/30) and canine (2/65) mucosal melanomas [51]. PTPRJ has been nominated as a tumor suppressor in other cancers [52]. These findings implicate PTPRJ likely as a tumor suppressor gene in acral and mucosal melanomas.

Candidate genes in genomic regions recurrently affected by focused amplifications or deletions. A GISTIC scores (y-axis) per chromosome (x-axis) for combined dataset of acral and mucosal melanomas. Recurrently (q-values < 0.1) amplified regions are colored red and recurrently deleted regions are colored blue, with relevant oncogenes and tumor suppressors labeled. Putative melanoma driver genes are marked in parentheses. B, C Minimally amplified regions (indicated in vertical lines) for amplifications of chromosome 5 with candidate genes. The filled circles on the left side indicate mucosal melanomas, with unlabeled cases representing acral melanomas. For panel B, the amplified region is shown at two different resolutions
TYRP1 was mutated in 10 melanomas (Additional file 2: Fig. S6A), 8 acral and 2 mucosal melanomas. Nine mutations (90%) were frameshifting deletions, in 8 of which 4 base pairs in codons 352–353 were removed. This recurrent deletion has been reported recently in acral melanoma [11], and an identical frameshift deletion has been identified as the cause of oculocutaneous albinism type 3 [53]. The mechanisms by which this alteration contributes to melanoma pathogenesis are not understood.
SLC30A9 was mutated in 5 melanomas, 4 acral and 1 mucosal melanomas (Additional file 2: Fig. S6B). While 3 mutations were truncating, there was no evidence of biallelic inactivation in any case. SLC30A9 has not been previously implicated in melanoma or other cancer types, and additional evidence is needed to support its role as a driver gene. Summary of all non-silent mutations of the three genes can be found in Additional file 8: Table S7.
When we performed the analysis separately for acral and mucosal melanomas (Additional file 2: Fig. S7A and B), only a single additional candidate gene was flagged as significantly mutated, FYB1 in acral melanoma (Additional file 2: Fig. S7C). FYB1 is involved in coagulation and T cell signaling and its role in cancer is unclear.
Candidate cancer genes in recurrent yet previously uncharacterized amplicons
Acral and mucosal melanomas frequently have structural genomic arrangements and focal amplifications and deletions that target oncogenes and tumor suppressors, respectively [7, 8]. We applied GISTIC2 to the combined dataset and individually for each subtype to refine the boundaries of focal amplifications and deletions for genomic regions known to be recurrently altered in these melanoma subtypes (Fig. 2A, Additional file 2: Fig. S8) and to identify previously unappreciated sites affected by focal amplifications. Recurrent but previously little characterized amplifications were encountered at chromosome 5p13.2 (11.7% or 28/240 of cases; Fig. 2B) and at 5q21 (3.8%; Fig. 2C).
The amplicon at 5p13.2 (Fig. 2B) is frequently accompanied by other amplifications on chromosome 5p, including those targeting TERT at 5p15.33. It contains the SKP2 oncogene, an E3 ubiquitin ligase that degrades the cyclin-dependent kinase inhibitor p27 and thereby facilitates S-phase entry [54]. SKP2 is frequently overexpressed in melanoma and other cancer types [10, 55]. SKP2 amplification was not mutually exclusive with common alterations targeting genes that control S-phase entry such as CDKN2A or CDK4 (P > 0.05). The minimally amplified region further includes NADK2 and MIR580, genes without a direct connection to cancer and therefore less likely drivers.
The minimally amplified region at 5q21 (Fig. 2C) includes the non-transmembrane receptor tyrosine kinase FER as a candidate oncogene. Overexpression of FER has been identified in several cancer types [56, 57], and FER has been shown to activate MAP kinase signaling by phosphorylating receptor tyrosine kinases including EGFR and MET [58, 59]. FER amplifications were not mutually exclusive with other genetic alterations in this pathway. Of the 9 samples with FER amplification, 7 had genetic alterations of known MAPK drivers, including NF1 inactivation in 5 samples (P = 0.013; Fisher’s exact test for co-occurrence between the 2 genes), and KIT and SPRED1 alterations, each in 1 additional sample. FER is also involved in cell-cell adhesion through phosphorylating proteins such as CTTN and β-catenin [60] but FER amplification was not inversely correlated (P = 1) with CTTN amplification, a recurrent alteration in acral melanoma [61].
In summary, the above analyses revealed cancer genes (Fig. 3) that are significantly mutated or targeted by focused copy number alterations in acral and mucosal melanomas.

Summary of significantly altered cancer genes. Each column represents a case. For the tiling plot in the lower panel, genes are shown in rows and the type of alterations are detailed in the legend. Mutation burdens and the fraction of base changes were displayed in the upper panels
Long tails of driver events in acral and mucosal melanomas
To identify additional putative driver events with low frequencies, we cross-referenced genetic alterations with OncoKB annotations (“Methods”). We observed that a considerable number of known cancer genes were altered by mutations, focal amplifications, and homozygous deletions, each in 5% or less of cases (Fig. 4A). These less common alterations affected multiple signaling pathways. In the MAP kinase pathway MAPK1, RAC1, ERBB2, GNAQ, PTPN11, MAP2K1, MAP2K2, RAF1, SOS1, RASA1, and RASA2 alterations were observed (Additional file 2: Fig. S9). NF1 homozygous deletion was observed in 5.8% (14/240) of tumors (Additional file 2: Fig. S10). Additional drivers that likely impact this pathway were LZTR1 and CIC, which act as negative regulators of the MAPK pathway and each showed inactivating alterations in 3 tumors, with additional missense mutations of unknown significance in 1 and 3 other tumors, respectively (Fig. 4B, C, and Additional file 2: Fig. S9). LZTR1 functions as a component of the CUL3 E3-ligase complex, which mediates the degradation of RAS and is a tumor suppressor gene that can cause schwannomatosis and Noonan syndrome [62, 63]. CIC is a transcriptional repressor of ETS transcription factors and other genes downstream of the MAPK pathway and is a recurrently inactivated tumor suppressor gene in gliomas and other cancer types [64, 65].

Long tails of driver events in acral and mucosal melanomas. A Genes with at least one pathogenic somatic or germline mutation (left panel), or amplifications and homozygous deletions (right panel). B, C Lollipop diagrams of mutations in LZTR1 (B) and CIC (C). D–G Oncoprint plots of genes of SWI/SNF complex (D), WNT/β-catenin (E), Hippo pathway (F), and SOX-MITF (G) pathway with recurrent mutations
Members of the SWI/SNF complex were altered in 13.8% (33/240) of samples (Fig. 4D), with SMARCA2, SMARCA4, SMARCD1, ARID1A, ARID1B, ARID2, or PBRM1 each altered in approximately 2% of cases. Interestingly, SMARCA2 and ARID1B alterations were mainly homozygous deletions (Additional file 2: Fig. S11), possibly because they map to chromosomes 9p and 6q, respectively, which are commonly affected by arm-level losses in melanoma, whereas alterations of other genes constituted indels and SNVs.
CTNNB1, APC, and RNF43 of the WNT/β-catenin pathway were altered in 4%, 3%, and 0.8% samples, respectively (Fig. 4E).
Genes in the Hippo pathway were altered in 13.8% (33/240) of samples (Fig. 4F), most commonly by YAP1 amplification (9.6%). Inactivating mutations and mutations with unknown significance of LATS2 and MOB3B were observed in 2.1% and 1.3% of cases. WWTR1 was altered in 1.3% by amplification (n = 1) and mutations with unknown significance (n = 2).
SOX10 and MITF, genes involved in melanocyte development and differentiation, were amplified (5.8% and 2.5%, respectively), or mutated (1.5% and 0.8%) in 10.4% (25/240) of cases (Fig. 4G).
Germline mutations
We identified 52 possible pathogenic germline mutations affecting 37 genes in 44 patients (18.3%) (Additional file 2: Fig. S12). MITF E318K, a known melanoma predisposing mutation [66, 67], was observed in 2 patients. The remainders were heterogeneous loss-of-function mutations of tumor suppressor genes, mostly operative in the DNA damage response and repair pathways. Only in 9 patients were the germline mutations accompanied with loss of the wild-type allele in the tumor sample, providing evidence that the germline alterations were functionally relevant for the development of the melanoma (Additional file 2: Fig. S12). Genes with such combined biallelic inactivation were ATM (2 patients), PRKN (2 patients), PTEN, PTCH1, CMTR2, FANCG, and SMARCAL1 (1 patient each). For the remaining 41 germline mutations, the melanoma retained the corresponding wild-type allele.
Differences in the genetic alterations of acral and mucosal melanomas
We next compared the frequency of the 35 recurrent alterations, defined as at least 10 occurrences, between acral and mucosal melanomas. Inactivating mutations and homozygous deletions of a tumor suppressor gene were considered jointly as inactivation, whereas amplification and pathogenic mutations of the same oncogene were considered individually. BRAF mutations were classified as class 1 (V600E: n = 33; and V600K: n = 2) or other BRAF mutations (n = 17). Two alterations were distributed differently among the two melanoma subtypes after accounting for multiple testing (Fig. 5A). SF3B1R625 mutations were present in 12.9% of mucosal melanomas but were absent in acral melanoma (FDR-corrected P-value, Fisher’s exact test = 0.00026) (Fig. 5B). PAK1 amplifications were more frequent in acral melanoma (28.6% vs. 11.8%; FDR = 0.041). Acral melanomas also had more frequent BRAF class 1 mutations (Fig. 5C; 19.7% vs. 6.5%), and amplifications of GAB2 (30.6% vs. 16.1%), TERT (23.8% vs. 12.9%), CARD11 (12.2% vs. 3.2%), and ERBB2 (4.8% vs. 0%), whereas mucosal melanomas had more frequent ATRX (9.7% vs. 2%) and TP53 (9.7% vs. 2.7%) alterations; however, these differences did not exceed significance threshold after correction for multiple testing.

Differences of genetic alterations between acral and mucosal melanomas. A Scatter plot of mutation frequencies for the two subtypes. B Lollipop diagram of SF3B1 mutations that were exclusive to mucosal melanomas. C BRAF class 1 (V600 codon) mutations were common in acral melanoma (FDR = 0.053), with no differences for other BRAF mutations (FDR = 0.46)
GISTIC2 comparison nominated amplifications at 22q13.2 (containing EP300), which was exclusively present in acral melanoma (q = 0.0016) and 3p13 (containing MITF), exclusive to mucosal melanoma (q = 0.22), but these differences did not reach formal levels of significance (P > 0.05).
Genetic interactions among driver events
We observed the following correlations among the 35 recurring genetic alterations (Fig. 6A). ATRX inactivation correlated with TP53 mutations (FDR = 0.001), consistent with the notion that these two alterations cooperate in facilitating the alternative lengthening of telomeres [68]. This co-occurrence of ATRX and TP53 alterations was only evident in mucosal melanoma (Additional file 2: Fig. S13). While either ATRX or TP53 were altered in 7 acral melanomas, none harbored alteration of both genes. BRAF class 1 mutations, PTEN and CDKN2A inactivation, co-existed frequently, consistent with reports that BRAF V600E cooperates with PTEN and CDKN2A alterations [69, 70]. Amplification of chromosome 11q13-14, including the candidate genes CCND1, PAK1, and GAB2, frequently co-occurred with amplifications of chromosome 6q12. The WGS data revealed recurrent structural alterations joining sequences from both regions (Fig. 6B). We observed clusters of translocations between 6q12 and 11q13-14 in 46.7% (7/15) of samples that had amplifications of both regions. Thus, the co-amplification between 6q12 and 11q13-14 is likely partially driven by recurrent structural alterations between the two regions rather than functional cooperation of genes contained within these regions. The 6q12 locus harbors PTP4A1, a protein tyrosine phosphatase, previously implicated in cancer [71]. TYRP1 alterations also correlated with CCND1 amplification.

Interactions between genetic alterations. A Volcano plot with dots representing pairs of two separate genetic alterations. The log2 ratio of the observed and expected co-occurrence frequency is plotted on the x-axis against the −log10 (FDR) on the y-axis. Pairs with FDR (corrected p-value from Fisher’s exact test) < 0.05 are colored in red. B Circos plots showing clusters of translocations (gray lines) joining regions on chromosome 6q12 and 11q13-14 in 7 samples. The outer track shows the chromosome ideograms; the middle track shows the log2 ratio of tumor to normal copy number (purple dots) with the dashed black line indicating 0
The following genetic alterations were inversely correlated (Fig. 6A). CDK4 amplification and CDKN2A inactivation were mutually exclusive (FDR = 0.00069), consistent with prior reports [7]. KIT mutations were mutually exclusive with PTEN inactivation (FDR = 0.1), CDKN2A (FDR = 0.085) alterations, and BRAF class 1 mutations (FDR = 0.1). Samples with CDKN2A alterations also had fewer MDM2 amplifications, but this may be due to the frequent co-amplification of MDM2 and CDK4, which both reside on chromosome 12q, and driven by the anti-correlation of CDK4 amplification and CDKN2A inactivation. Several mutations in the MAPK pathway showed a pattern of mutual exclusivity, but only BRAF class 1 mutation and NF1 alterations reached the cutoff of FDR < 0.05, possibly reflecting the limited statistical power of our study. Melanomas with BRAF class 1 mutations harbored fewer amplifications of GAB2 (FDR = 0.099), CCND1, and PAK1 (both FDR = 0.18). However, we observed significantly fewer structural variations overall in melanomas with BRAF class 1 mutations compared to those without (Additional file 2: Fig. S14; 136 vs. 234 junctions on average; P = 0.0015), which remains significant after accounting for tumor purity and ploidy. As previously suggested, this may indicate that the BRAF V600E mutated melanomas on acral skin are likely different and more akin to cutaneous melanomas on skin with low cumulative sun-induced damage [12].
The association of genetic alterations with clinical features
Subungual melanomas (n = 37) showed significantly higher mutation burden (Fig. 7A; median mutations per megabase = 2.48 vs 1.6; P = 1.97 × 10−05 from Wilcoxon rank-sum test) and fraction of mutations that are attributable to UV signature SBS7 (P = 0.012), compared to melanomas originated from sole (n = 71). BRAF class 1 mutations occurred predominately in sole (Fig. 7C; 25.4% vs. 2.7%) compared to subungual melanoma, in contrast with CDK4 amplification (5.6% vs. 24.3%). For mucosal melanomas, significantly higher mutation burden was observed in sinonasal/oropharyngeal (n = 37) compared to genitourinary melanomas (n = 20) (Fig. 7B; median mutations per megabase = 2.98 vs. 1.71). Sinonasal/oropharyngeal melanomas also showed significantly higher fraction of SBS7 mutations compared to the other mucosal melanoma groups (P < 0.01), which is consistent with a recent study [10]. CDK4 (Fig. 7D; 32.4% vs. 5%) and MDM2 (29.7% vs. 5%) amplifications were more frequently altered in sinonasal/oropharyngeal compared to genitourinary melanomas, in contrast with SF3B1 mutations (2.7% vs. 30%) between the two subsites. We also found that BRAF class 1 mutations (20.3% vs. 5.8%) were more frequent in metastatic melanomas, while SPRED1 inactivation (3.9% vs. 19.8%), SF3B1 mutations (1.6% vs. 9.3%), and amplifications of CRKL (6.3% vs. 17.4%) and MAPK1 (1.6% vs. 11.6%) were more frequent in primary samples (Fig. 7E and Additional file 2: Fig. S15). Only SPRED1 inactivation remained significant after correction for multiple testing. We further compared mucosal melanoma from patients of European and Asian ancestry (n = 48 and 42; Additional file 2: Fig. S15E) and found that SF3B1 mutations were predominately observed in the former (22.9% vs. 2.4%), while SKP2 amplifications were more frequent in the latter (4.2% vs. 19%). However, these associations were no longer significant after correction for multiple testing.

The association of genetic alterations with clinical features. A, B The comparison of tumor mutation burden and fraction of SBS7 mutations between sole and subungual acral melanomas (A), and across mucosal melanomas from different subsites (B). The blue segments denote the medians. C–E Scatter plots of mutation frequencies between sole and subungual acral melanomas (C), between sinonasal / oropharyngeal and genitourinary mucosal melanomas (D), and between primary and metastatic samples for the merged set of both melanoma subtypes (E)
When combining both melanoma subtypes, we found that KIT (Additional file 2: Fig. S15F; P = 0.026 from log-rank test) and TP53 (P = 0.043) mutations were associated with poorer survival, while patients with CCND1 amplification had better outcome (P = 0.027). For mucosal melanomas, PTEN altered patients (n = 8) had poorer survival (Additional file 2: Fig. S15G; P = 2.33 × 10−05). Considering the limited sample size and strong variation in patient age, tumor stage and treatment condition, further study is probably needed to validate these.
Discussion
By integrating and analyzing samples from multiple previous studies, we present a wider view of the genetic landscapes of acral and mucosal melanomas (Fig. 8), provide additional support for the pathogenetic relevance of previously nominated genes, and implicate additional likely driver genes such as PTPRJ, FER, and SKP2 altered in a subset of acral and mucosal melanomas.

Major signaling pathways mutated in acral and mucosal melanoma
PTPRJ is a member of the transmembrane protein tyrosine phosphatase family. PTPRJ promotes cell adhesion and inhibits PDGFR signaling [72], and its loss may promote meningioma progression through interacting with NF2 loss [73]. The receptor tyrosine kinase FER activates MAP kinase pathway signaling and regulates cell adhesion in cancer cells [58, 59]. In our dataset, melanomas with FER amplification had frequent NF1 inactivation but lacked BRAF and NRAS alterations. The findings indicate a possible role for FER amplification in driving the MAPK pathway, similar to KIT, another receptor tyrosine kinase that can be amplified or mutated in acral and mucosal melanoma, frequently also in conjunction with NF1 mutations [74]. Functional studies have shown that inhibition of FER slows the proliferation and motility of melanoma cell lines [75]. SKP2 is involved in G1/S cell cycle transition by degrading the cyclin-dependent kinase inhibitor 1B (CDKN1B, also referred to as p27 or KIP1) [54]. It is recurrently amplified in non-small cell lung cancer. There was no inverse correlation with other common genetic alterations of the G1/S checkpoint, indicating that SKP2 amplification may not be redundant with CDKN2A inactivation. Overexpression of SKP2 has indicated worse survival in some studies [76]. Pharmacological inhibitors for FER [77] and SKP2 [78] are available, and further investigation is needed to study their roles in acral and mucosal melanomas and determine whether they constitute therapeutic targets.
Our data reveals a long list of low-frequency driver genes, which is likely to expand as more samples are being sequenced, following the experience in cutaneous melanoma [79, 80] and other cancers [81]. Among the 198 genes that had at least one pathogenic alteration, 83.8% (166/198) were present in fewer than 5% of cases, and 38.4% (76/198) were observed only once. These infrequently altered genes and pathways likely reflect the diverse trajectories that acral and mucosal melanocytes can take when evolving into melanomas and indicate that more alterations are to be discovered in these cancers.
We found germline mutations predicted to impact gene function in 18% of acral and mucosal melanoma patients, mainly functioning in the DNA damage sensing and repair pathways, similar to what has been seen in other cancer types [82]. In our analysis, only a small fraction of these germline alterations were accompanied by losses of the remaining allele in the tumors that would be required for complete inactivation. This suggests that the majority of these germline events may not contribute to tumor initiation and progression. The presence of the MITF E318K mutation in the germline of acral melanoma patients indicates that this mutation predisposes not only to cutaneous melanoma as has been previously reported [66], but may also predispose to acral melanoma. Also, none of the two MITF E318K cases contained BRAF V600 mutation. Mutations of PRKN are associated with hereditary Parkinson’s disease. The parkin protein is a component of a E3-ubiquitin ligase complex and involved in mitochondrium homeostasis and apoptosis. Recurrent loss-of-function mutations of PRKN occur in several cancer types where it acts as a tumor suppressor gene [83, 84]. Our finding of recurrent germline and somatic inactivating alterations suggests that PRKN probably also functions as a tumor suppressor in acral melanoma.
While acral and mucosal melanomas share many features such as their origin from sun-shielded or -protected sites, a precursor stage with a lentiginous growth pattern, and a high degree of chromosomal instability with frequent gene amplifications and structural rearrangements, our meta-analysis also adds to the emerging genetic differences between the two subtypes. Concordant with prior reports, pathogenic SF3B1 mutations were found only in mucosal melanoma. SF3B1 is involved in RNA splicing, and the hotspot mutation recurrent in mucosal melanoma changes the splicing of several cancer genes, including the non-canonical BAF complex member BRD9 [85]. Interestingly, similar SF3B1 mutations are found in a subset of uveal melanomas, another non-UV related melanoma subtype, which is genetically characterized by constitutive activation of the Gαq signaling pathway [86, 87]. Activating mutations of the Gαq pathway were also reported to occur in some mucosal melanomas [88] but were infrequent in our meta-analysis. The concurrent mutations of ATRX and TP53 in mucosal melanomas suggest that alternative lengthening of telomere pathway is operative in some mucosal melanomas. BRAF class 1 mutations and 11q13-14 amplifications were more common in acral melanoma and tended to be mutually exclusive of each other. In summary, the subtle but reproducible differences in mutation patterns between acral and mucosal melanoma may reflect variations among their cells of origin or their micro-environments, selecting for different alterations.
Acral melanomas with BRAF class 1 mutations, especially V600E, had fewer structural variations and focal amplifications [12], than those without and thus resemble cutaneous melanomas. This raises the possibility that melanomas on acral are of two different types, with a bona fide and unique acral type with an admixture of cutaneous melanoma of the World health Organization (WHO) low-CSD subtype, i.e., melanomas on the sun-exposed skin with low degree of cumulative sun damage [89]. A recent study on acral nevi identified BRAF V600E mutation in 86% of samples [90], which is similar to what was observed in cutaneous nevi that considered the precursors of low-CSD melanomas [45]. Another study identified transcriptome-level differences between acral and cutaneous melanocytes [91]. Further investigation is needed to determine whether acral sites contain different sublineages of melanocytes. Acral skin has a unique reservoir of melanocyte stem cells in the sweat glands, abundant in acral skin [92], where as in most of the body’s skin melanocyte stem cells are associated with hair follicles [93].
While our study expands the view of the genetic landscapes of acral and mucosal melanoma, the number of tumors sequenced is still comparatively low and additional driver mutations likely remain to be discovered. An estimated 300 tumors would be needed to identify SMGs with 5% mutation frequency or higher, based on the average mutation burden of the two subtypes [36]. To increase the sample size, we merged both acral and mucosal melanomas for most of the analyses. This helped identify driver genes that are shared by both subtypes and does not permit exhaustive characterization of driver mutations unique to either subtype. The sample size also limited the comparison of mutation patterns and interactions between genetic alterations.
Conclusions
In summary, our meta-analyses of 147 acral and 93 mucosal melanomas identified PTPRJ, FER, SKP2, LZTR1, CIC, and PRKN as part of a long tail of driver events that deserves further study and characterization. While the two melanoma subtypes have many features in common, the reproducible genetic differences support the notion that acral and mucosal melanoma are biologically distinct entities.
Availability of data and materials
Sequencing data were downloaded from European Genome-phenome (EGA) archive under accession numbers EGAD00001003388 (https://ega-archive.org/datasets/EGAD00001003388) [8], EGAD00001004409 (https://ega-archive.org/datasets/EGAD00001004409) [10], EGAD00001005500 (https://ega-archive.org/datasets/EGAD00001005500) [11], EGAD00001000944 (https://ega-archive.org/datasets/EGAD00001000944) [15] and EGAD00001000945 (https://ega-archive.org/datasets/EGAD00001000945) [16], and from Database of Genotypes and Phenotypes (dbGaP) under accession numbers phs000178 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000178.v11.p8) [13], phs001036 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001036.v1.p1) [14], and phs000452 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000452.v3.p1) [18,19,20,21]. Details about the samples can also be found in Additional file 1: Table S1. The codes used for data analysis in the article are available at https://github.com/Bioinfowangm/ac_mu_analysis [94].
Abbreviations
- UV:
-
Ultraviolet
- TCGA:
-
The Cancer Genome Atlas
- WGS:
-
Whole-genome sequencing
- WXS:
-
Whole-exome sequencing
- SNP:
-
Single-nucleotide polymorphism
- MAF:
-
Mutant allele frequency
- SMG:
-
Significantly mutated gene
- SV:
-
Structural variation
- CNV:
-
Copy number variation
- LOH:
-
Loss of heterozygosity
- SBS:
-
Single base substitution
- MSI:
-
Microsatellite instability
- MAPK:
-
Mitogen-activated protein kinase
- WHO:
-
World Health Organization
- CSD:
-
Cumulative sun damage
References
Chang AE, Karnell LH, Menck HR. The National Cancer Data Base report on cutaneous and noncutaneous melanoma. Cancer. 1998;83:1664–78.
Altieri L, Wong MK, Peng DH, Cockburn M. Mucosal melanomas in the racially diverse population of California. J Am Acad Dermatol. 2017;76:250–7.
Teramoto Y, Keim U, Gesierich A, Schuler G, Fiedler E, Tüting T, et al. Acral lentiginous melanoma: a skin cancer with unfavourable prognostic features. A study of the German central malignant melanoma registry (CMMR) in 2050 patients. Br J Dermatol. 2018;178:443–51.
Csányi I, Houshmand N, Szűcs M, Ócsai H, Kemény L, Oláh J, et al. Acral lentiginous melanoma: a single-centre retrospective review of four decades in East-Central Europe. J Eur Acad Dermatol Venereol. 2020;34:2004–10.
Chi Z, Li S, Sheng X, Si L, Cui C, Han M, et al. Clinical presentation, histology, and prognoses of malignant melanoma in ethnic Chinese: a study of 522 consecutive cases. BMC Cancer. 2011;11:85.
Huang K, Fan J, Misra S. Acral lentiginous melanoma: incidence and survival in the United States, 2006-2015, an analysis of the SEER Registry. J Surg Res. 2020;251:329–39.
Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47.
Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545:175–80.
Bastian BC, Kashani-Sabet M, Hamm H, Godfrey T, Moore DH, Bröcker E-B, et al. Gene amplifications characterize acral melanoma and permit the detection of occult tumor cells in the surrounding skin. Cancer Res. 2000;60:1968–73 American Association for Cancer Research.
Newell F, Kong Y, Wilmott JS, Johansson PA, Ferguson PM, Cui C, et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat Commun. 2019;10:3163.
Newell F, Wilmott JS, Johansson PA, Nones K, Addala V, Mukhopadhyay P, et al. Whole-genome sequencing of acral melanoma reveals genomic complexity and diversity. Nat Commun. 2020;11:5259 Nature Publishing Group.
Yeh I, Jorgenson E, Shen L, Xu M, North JP, Shain AH, et al. Targeted genomic profiling of acral melanoma. JNCI J Natl Cancer Inst. 2019;111:1068–77 Oxford Academic.
Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, et al. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–96.
Liang WS, Hendricks W, Kiefer J, Schmidt J, Sekar S, Carpten J, et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017;27:524–32.
Furney SJ, Turajlic S, Stamp G, Thomas JM, Hayes A, Strauss D, et al. The mutational burden of acral melanoma revealed by whole-genome sequencing and comparative analysis. Pigment Cell Melanoma Res. 2014;27:835–8.
Furney SJ, Turajlic S, Stamp G, Nohadani M, Carlisle A, Thomas JM, et al. Genome sequencing of mucosal melanomas reveals that they are driven by distinct mechanisms from cutaneous melanoma. J Pathol. 2013;230:261–9.
Zhou R, Shi C, Tao W, Li J, Wu J, Han Y, et al. Analysis of mucosal melanoma whole-genome landscapes reveals clinically relevant genomic aberrations. Clin Cancer Res. 2019;25:3548–60.
Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–6 Nature Publishing Group.
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63.
Allen EMV, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11 American Association for the Advancement of Science.
Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Arnon L, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med. 2019;25:1916–27 Nature Publishing Group.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–95.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018;15:591–4.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11.
The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65.
Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–9 American Association for the Advancement of Science.
Exome Aggregation Consortium, Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
de Koning APJ, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7:e1002384 Public Library of Science.
Diossy M, Sztupinszki Z, Krzystanek M, Borcsok J, Eklund AC, Csabai I, et al. Strand Orientation Bias Detector (SOBDetector) to remove FFPE sequencing artifacts. bioRxiv. 2019:386417 Available from: https://www.biorxiv.org/content/10.1101/386417v2. Cited 2022 Mar 23.
Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, et al. Universal patterns of selection in cancer and somatic tissues. Cell. 2017;171:1029–1041.e21.
Dietlein F, Weghorn D, Taylor-Weiner A, Richters A, Reardon B, Liu D, et al. Identification of cancer driver genes based on nucleotide context. Nat Genet. 2020;52:208–18 Nature Publishing Group.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501.
Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Genomics. 2017; Available from: http://biorxiv.org/lookup/doi/10.1101/201178.
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017. https://doi.org/10.1200/PO.17.00011.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–5.
Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–83.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93.
Islam SMA, Wu Y, Díaz-Gay M, Bergstrom EN, He Y, Barnes M, et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. bioRxiv. 2021. https://doi.org/10.1101/2020.12.13.422570 Cold Spring Harbor Laboratory.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng AWT, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101 Nature Publishing Group.
Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131.
Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, et al. Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell. 2018;34:45–55.e4.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873.
GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers | Genome Biology | Full Text. Available from: https://genomebiology.biomedcentral.com/articles/10.1186/gb-2011-12-4-r41. Cited 2021 Jul 2.
Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–9.
Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30:1015–6.
Hendricks WPD, Zismann V, Sivaprakasam K, Legendre C, Poorman K, Tembe W, et al. Somatic inactivating PTPRJ mutations and dysregulated pathways identified in canine malignant melanoma by integrated comparative genomic analysis. PLoS Genet. 2018;14:e1007589 Public Library of Science.
Wong K, van der Weyden L, Schott CR, Foote A, Constantino-Casas F, Smith S, et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat Commun. 2019;10:353.
Ruivenkamp CAL, van Wezel T, Zanon C, Stassen APM, Vlcek C, Csikós T, et al. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. 2002;31:295–300 Nature Publishing Group.
Chiang P-W, Spector E, Scheuerle A. A case of Asian Indian OCA3 patient. Am J Med Genet A. 2009;149A:1578–80.
Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–9.
Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, et al. Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci. 2001;98:5043–8 National Academy of Sciences.
Ivanova IA, Vermeulen JF, Ercan C, Houthuijzen JM, Saig FA, Vlug EJ, et al. FER kinase promotes breast cancer metastasis by regulating α6- and β1-integrin-dependent cell adhesion and anoikis resistance. Oncogene. 2013;32:5582–92.
Ahn J, Truesdell P, Meens J, Kadish C, Yang X, Boag AH, et al. Fer protein-tyrosine kinase promotes lung adenocarcinoma cell invasion and tumor metastasis. Mol Cancer Res. 2013;11:952–63 American Association for Cancer Research.
Guo C, Stark GR. FER tyrosine kinase (FER) overexpression mediates resistance to quinacrine through EGF-dependent activation of NF-κB. Proc Natl Acad Sci. 2011;108:7968–73 National Academy of Sciences.
Fan G, Zhang S, Gao Y, Greer PA, Tonks NK. HGF-independent regulation of MET and GAB1 by nonreceptor tyrosine kinase FER potentiates metastasis in ovarian cancer. Genes Dev. 2016;30:1542–57.
Piedra J, Miravet S, Castaño J, Pálmer HG, Heisterkamp N, García de Herreros A, et al. p120 catenin-associated Fer and Fyn tyrosine kinases regulate β-catenin Tyr-142 phosphorylation and β-catenin-α-catenin interaction. Mol Cell Biol. 2003;23:2287–97 American Society for Microbiology.
Puig-Butillé JA, Badenas C, Ogbah Z, Carrera C, Aguilera P, Malvehy J, et al. Genetic alterations in RAS-regulated pathway in acral lentiginous melanoma. Exp Dermatol. 2013;22:148–50.
Steklov M, Pandolfi S, Baietti MF, Batiuk A, Carai P, Najm P, et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science. 2018;362:1177–82 American Association for the Advancement of Science.
Motta M, Fidan M, Bellacchio E, Pantaleoni F, Schneider-Heieck K, Coppola S, et al. Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum Mol Genet. 2019;28:1007–22.
Eleveld TF, Schild L, Koster J, Zwijnenburg DA, Alles LK, Ebus ME, et al. RAS–MAPK pathway-driven tumor progression is associated with loss of CIC and other genomic aberrations in neuroblastoma. Cancer Res. 2018;78:6297–307.
Gleize V, Alentorn A, de Kérillis LC, Labussière M, Nadaradjane AA, Mundwiller E, et al. CIC inactivating mutations identify aggressive subset of 1p19q codeleted gliomas. Ann Neurol. 2015;78:355–74.
Bertolotto C, Lesueur F, Giuliano S, Strub T, de Lichy M, Bille K, et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480:94–8.
Yokoyama S, Woods SL, Boyle GM, Aoude LG, MacGregor S, Zismann V, et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011;480:99–103.
Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science. 2015;347:273–7.
Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, et al. Braf V600E cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52.
Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol Mech Dis. 2014;9:239–71.
Luo Y, Liang F, Zhang Z-Y. PRL1 promotes cell migration and invasion by increasing MMP2 and MMP9 expression through Src and ERK1/2 pathways. Biochemistry. 2009;48:1838–46 American Chemical Society.
Petermann A, Haase D, Wetzel A, Balavenkatraman KK, Tenev T, Gührs K-H, et al. Loss of the protein-tyrosine phosphatase DEP-1/PTPRJ drives meningioma cell motility. Brain Pathol. 2011;21:405–18.
Waldt N, Scharnetzki D, Kesseler C, Kirches E, Stroscher N, Böhmer F-D, et al. Loss of PTPRJ/DEP-1 enhances NF2/Merlin-dependent meningioma development. J Neurol Sci. 2020;408:116553.
Hintzsche JD, Gorden NT, Amato CM, Kim J, Wuensch KE, Robinson SE, et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017;27:189–99.
Ivanova IA, Arulanantham S, Barr K, Cepeda M, Parkins KM, Hamilton AM, et al. Targeting FER kinase inhibits melanoma growth and metastasis. Cancers. 2019;11:419 Multidisciplinary Digital Publishing Institute.
Chen G, Cheng Y, Zhang Z, Martinka M, Li G. Cytoplasmic Skp2 expression is increased in human melanoma and correlated with patient survival. PLoS One. 2011;6:e17578 Public Library of Science.
Taniguchi T, Inagaki H, Baba D, Yasumatsu I, Toyota A, Kaneta Y, et al. Discovery of novel pyrido-pyridazinone derivatives as FER tyrosine kinase inhibitors with antitumor activity. ACS Med Chem Lett. 2019;10:737–42 American Chemical Society.
Chan C-H, Morrow JK, Li C-F, Gao Y, Jin G, Moten A, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–68.
Conway JR, Dietlein F, Taylor-Weiner A, AlDubayan S, Vokes N, Keenan T, et al. Integrated molecular drivers coordinate biological and clinical states in melanoma. Nat Genet. 2020;52:1373–83.
Alkallas R, Lajoie M, Moldoveanu D, Hoang KV, Lefrançois P, Lingrand M, et al. Multi-omic analysis reveals significantly mutated genes and DDX3X as a sex-specific tumor suppressor in cutaneous melanoma. Nat Cancer. 2020;1:635–52 Nature Publishing Group.
Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018;50:645–51.
Ku GY, Kemel Y, Maron SB, Chou JF, Ravichandran V, Shameer Z, et al. Prevalence of germline alterations on targeted tumor-normal sequencing of esophagogastric cancer. JAMA Netw Open. 2021;4:e2114753.
Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, et al. Somatic mutations of the Parkinson’s disease–associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet. 2010;42:77–82.
Xiong D, Wang Y, Kupert E, Simpson C, Pinney SM, Gaba CR, et al. A recurrent mutation in PARK2 is associated with familial lung cancer. Am J Hum Genet. 2015;96:301–8.
Inoue D, Chew G-L, Liu B, Michel BC, Pangallo J, D’Avino AR, et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature. 2019;574:432–6.
Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue nevi. Nature. 2009;457:599–602.
Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in uveal melanoma: Massachusetts Medical Society; 2010. https://doi.org/10.1056/NEJMoa1000584. Available from: https://www.nejm.org/doi/10.1056/NEJMoa1000584. Cited 2021 Sep 16
Sheng X, Kong Y, Li Y, Zhang Q, Si L, Cui C, et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur J Cancer. 2016;65:156–63.
Elder DE, Bastian BC, Cree IA, Massi D, Scolyer RA. The 2018 World Health Organization classification of cutaneous, mucosal, and uveal melanoma: detailed analysis of 9 distinct subtypes defined by their evolutionary pathway. Arch Pathol Lab Med. 2020;144:500–22.
Smalley KSM, Teer JK, Chen YA, Wu J-Y, Yao J, Koomen JM, et al. A mutational survey of acral nevi. JAMA Dermatol. 2021;157:831–5.
Belote RL, Le D, Maynard A, Lang UE, Sinclair A, Lohman BK, et al. Human melanocyte development and melanoma dedifferentiation at single-cell resolution. Nat Cell Biol. 2021;23:1035–47.
Okamoto N, Aoto T, Uhara H, Yamazaki S, Akutsu H, Umezawa A, et al. A melanocyte–melanoma precursor niche in sweat glands of volar skin. Pigment Cell Melanoma Res. 2014;27:1039–50.
Nishimura EK. Melanocyte stem cells: a melanocyte reservoir in hair follicles for hair and skin pigmentation. Pigment Cell Melanoma Res. 2011;24:401–10.
Wang M, Banik I, Shain AH, Yeh I, Bastian BC. Integrated genomic analyses of acral and mucosal melanomas nominate novel driver genes: GitHub; 2022. https://github.com/Bioinfowangm/ac_mu_analysis
Acknowledgements
We acknowledge the included studies from which we obtained the sequencing data, and members in the Bastian and Yeh labs for helpful discussion. Our analyses were performed on the C4 high-performance compute cluster in the Helen Diller Family Comprehensive Cancer Center.
Funding
This study was supported by an NCI Outstanding Investigator Award (B.C.B; 1R35CA220481), a Melanoma Research Alliance Team Science Award (B.C.B, I.Y. and A.H.S.) and a Dermatology Fellows Award (M.W.; ID: 737988; DOI: https://doi.org/10.48050/pc.gr.141692).
Author information
Authors and Affiliations
Contributions
B.C.B., I.Y., and M.W. conceived the study and collected the raw data. M.W. performed bioinformatic analyses and data visualization. B.C.B., I.Y., and A.H.S. interpreted the data and contributed to data analyses. A.H.S. and I.B. contributed to manuscript review. M.W., B.C.B., and I.Y. wrote the manuscript. B.C.B. and I.Y. jointly supervised the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
A summary of all samples that were included in this study.
Additional file 2: Figure S1.
Principal component analyses of the matrix of 96 classes of substitutions and boxplots of the mutant allele frequencies (MAFs) for samples from 6 studies. Figure S2. Principal component analysis showing the ethnicities of patients. Figure S3. Tumor purities estimated through FACETS were comparable with those from the original studies with few exceptions (red dots). For the Newell et al studies we observed larger discrepancies between our and the original estimates for hyperdiploid samples compared to near diploid samples, indicating that the original estimates probably did not account for tumor ploidy. Figure S4. Features of somatic mutations in acral melanomas. Figure S5. Features of somatic mutations in mucosal melanomas. Figure S6. Lollipop plots showing somatic non-synonymous mutations on TYRP1 and SLC30A9. Figure S7. Significantly mutated genes (SMGs) in acral and mucosal melanomas. Figure S8. The GISTIC scores calculated from acral melanoma and mucosal melanoma samples. Figure S9. Oncoprint plots showing the alterations of genes that are involved in the MAPK signaling pathway. Figure S10. Copy number profiles showing homozygous deletion of NF1. Figure S11. Copy number profiles showing homozygous deletion of SMARCA2 and ARID1B. Figure S12. Tiling plot of germline mutations. Figure S13. The mutual exclusivity and co-occurrence between pairs of driver events in acral melanoma and mucosal melanoma. Figure S14. Fewer structural variation (SV) junctions for acral and mucosal melanomas with BRAF class 1 mutations compared to others. Figure S15. The association of genetic alterations with clinical features.
Additional file 3: Table S2.
The list of 703 cancer genes.
Additional file 4: Table S3.
The list of somatic mutations in exons and splice sites for all samples.
Additional file 5: Table S4.
The copy number data for all samples.
Additional file 6: Table S5.
The output of Delly2 for WGS samples.
Additional file 7: Table S6.
The outputs of dNdScv, MutPanning and MutSigCV.
Additional file 8: Table S7.
All non-silent mutations from PTPRJ, TYRP1 and SLC30A9.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, M., Banik, I., Shain, A.H. et al. Integrated genomic analyses of acral and mucosal melanomas nominate novel driver genes. Genome Med 14, 65 (2022). https://doi.org/10.1186/s13073-022-01068-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-022-01068-0