
Abstract
At the dawn of the twentieth century, the medical care of mothers and children was largely relegated to family members and informally trained birth attendants. As the industrial era progressed, early and key public health observations among women and children linked the persistence of adverse health outcomes to poverty and poor nutrition. In the time hence, numerous studies connecting genetics (“nature”) to public health and epidemiologic data on the role of the environment (“nurture”) have yielded insights into the importance of early life exposures in relation to the occurrence of common diseases, such as diabetes, allergic and atopic disease, cardiovascular disease, and obesity. As a result of these parallel efforts in science, medicine, and public health, the developing brain, immune system, and metabolic physiology are now recognized as being particularly vulnerable to poor nutrition and stressful environments from the start of pregnancy to 3 years of age. In particular, compelling evidence arising from a diverse array of studies across mammalian lineages suggest that modifications to our metagenome and/or microbiome occur following certain environmental exposures during pregnancy and lactation, which in turn render risk of childhood and adult diseases. In this review, we will consider the evidence suggesting that development of the offspring microbiome may be vulnerable to maternal exposures, including an analysis of the data regarding the presence or absence of a low-biomass intrauterine microbiome.
Similar content being viewed by others


Advances in culture-dependent and culture-independent molecular methodologies continue to reveal details of the human-microbe relationship, allowing for increasing identification of “who is there” (microbiota) and what the community is “capable of doing” (functional microbiome). Two working definitions of “microbiome” exist, which collectively frame an emphasis on community functionality. The first definition provided by Nature defines “microbiome” as “all of the genetic material within a microbiota (the entire collection of microorganisms in a specific niche, such as the human gut). This can also be referred to as the metagenome of the microbiota.” (https://www.nature.com/subjects/microbiome) The second definition put forward by Whipps et al. defines “microbiome” as “a characteristic microbial community occupying a reasonably well defined habitat which has distinct physio-chemical properties. The term thus not only refers to the microorganisms involved but also encompasses their theatre of activity” [1]. Although distinct, these two definitions share the commonality of emphasizing the community’s functional capacity and resultant activity, which can collectively be thought of as the microbial community functionality. Of note, the functionality need not be limited to the metabolic functions of the microbiota per se. Rather, there may be functional importance tied to niche occupancy (so-called colonization resistance). This might result from host-independent functions, such as dependence on limited substrates or production of macromolecules which exclude occupancy of other microbes. It may also result from host-dependent functions, such as antigenic exposure resulting in immune tolerance or immune activation. Of note, neither definition requires long-term establishment or high-biomass colonization. In the case of human microbiomes, this functionality may be unique to specific life stages (i.e., child to adolescent, or adult to elder adult), normal physiologic adaptations (i.e., during pregnancy and lactation), or may have a wide dynamic range which oscillates predictably within an individual but widely across the population (i.e., vaginal microbiome community dominance patterns, also called “community state types”). Throughout this review, we presume the understanding that “microbiome” infers a microbial community of live microbes with functionality, recognizing that functionality is not limited to microbial metabolic activity per se.
Much effort has been concentrated on demonstrating the association between human health and the functional high-biomass gut microbiome, with evident associations between the microbiome and human neurobehavior [2], metabolism [3], and immunity [4,5,6]. Clearly, a functional metagenome is critical for normal human physiology and neurodevelopment at all life stages. However, particular attention has been paid to the early life microbiome. “Critical developmental windows” have been identified, during which microbes and their functional pathways are necessary to promote normal metabolic, immune, and neural development [7,8,9,10]. However, the origin, timing, and route of the human microbiome acquisition, initiation, integration, and maturation during these “critical developmental windows” remains unresolved.
In this review, our goal is to provide a presentation of the evidence of a relationship between several different maternal exposures, the maternal microbiome, and the offspring’s microbiome and health outcomes. We will explore what has been learned about both the impact and lack of impact with specific maternal exposures on the development of the offspring microbiome. We will review the literature and strength of evidence both supporting and refuting the presence and functional importance of low-biomass intrauterine microbial communities in early development. In particular, we will discuss the placental microbiome and the role it may or may not play in contributing to perinatal outcomes, the offspring’s microbiome or microbiome fitness, and subsequent childhood health and disease. We will also explore the recent published evidence refuting the notion of low-biomass perinatal communities, including intrauterine communities like the placenta. Finally, we will conclude this review with our viewpoint, which challenges the long-held notion that the intrauterine low-biomass community is of little or no functional consequence [11,12,13,14,15,16]. Rather, we will argue that functional consequence remains to be determined experimentally and with an evidential basis. Solving the mystery of how and when during early development we come to tolerate commensal microorganisms is fundamental and crucial to engaging in safe and efficacious studies and trials which attempt to manipulate our microbiome [17].
Several pregnancy exposures have been associated with alterations of the offspring microbiome, which precede later onset of disease in childhood and adult life. The combination of “nature” and “nurture” has been used over the years to explain the variability in human health, including both the range of susceptibility and severity of clinical disease from one individual to the next, or among different populations. For example, following World War II, undernourished women were observed to have increased risk of miscarriage, birth malformations, and low-birthweight infants [18, 19]. Kermack et al. showed that early life exposures lead to specific and predictable effects throughout the life of offspring [20]. More formal evidence continued to accrue following a series of sentinel observations by Barker and his colleagues, which collectively supported the theory that maternal pre-conception and pregnancy exposures can result in long-term health impacts on her offspring [21,22,23,24,25,26,27]. Now formally termed the Developmental Origins of Health and Disease (DOHaD) hypothesis, we appreciate today that fetal exposures persistently alter human physiology and behavior in ways that last well into adult life [28]. We and others have described the effect of common pre-pregnancy and pregnancy exposures. We have extensively studied the effect of maternal high-fat diet consumption during pre-conception, pregnancy, and lactation and inhalation/ingestion of polycyclic aromatic hydrocarbons (found in tobacco smoke and petroleum byproducts) during pregnancy on the development of obesity, modulation of immunity, and dysfunction of metabolism in offspring via long-lasting perturbations at the level of the placenta, liver, pancreas, skeletal muscle, thyroid, and digestive tract [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78]. The mechanisms driving these long-term effects are partially explained by the developing epigenetic code [29,30,31,32,33,34,35,36,37, 39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55, 59, 60, 63,64,65,66,67,68,69, 73, 74, 76] and microbiome [56, 61, 62, 70,71,72, 77,78,79,80,81]. Specifically, the gut microbiome can contribute small molecules and nutrients to the maternal circulation that can be actively or passively transferred across the placenta (in some, but not all cases) during fetal development [82, 83], leading to varied effects in the offspring. Rodent, primate, and human studies support the observation that certain and key pregnancy exposures can lead to alterations in the maternal and/or offspring microbiomes and offspring health [57, 84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103].
In the subsequent section, we will discuss eight examples of how the offspring microbiome and health are shaped by common maternal exposures: (example 1) changes in the maternal microbiome, (example 2) women who are pregnant and living with a furry pet, (example 3) maternal exposure to herbicides, (example 4) environmental pollutants, (examples 5–7) maternal diet, and potentially (example 8) maternal stress.
Example 1
It has been established that a wide range of factors may impact the structure and function of the gut microbiome, including but not limited to sex, geography, diet, environment, genetics, and medication. However, what effect does a change in the maternal microbiome (be it transient or permanent) have on the developing fetus or its eventual microbiome? Gomez de Agüero et al. addressed this question in a rodent model by transiently colonizing pregnant germ-free mice with a specially engineered Escherichia coli strain [103]. This strain of E. coli was genetically designed to perish within the intestine; once introduced through oral gavage to the dams, its residence within the intestine was short-lived. Even though active colonization did not occur, the offspring of the E. coli-exposed dams displayed enhanced innate immune system development and intestinal epithelial maturity compared to offspring without that maternal exposure. These findings translated to the E. coli-exposed offspring having improved intestinal inflammatory response and host-microbial symbiosis to post-natal microbes due to the maternal-microbe interaction.
Example 2
The environment (both in the home and outside of the home) includes inhalation, absorption, and ingestion exposures, including exposures to domesticated animals (i.e., pets and domesticated farm animals or livestock). It has been demonstrated that children reared with domesticated animals from infancy have reduced prevalence of allergic-related illnesses [104]. Could the effect of such domesticated animal exposures extend prior to birth? Tapiainen et al. investigated the effect of common maternal exposures on the human first-pass meconium microbiome [93]. They analyzed delivery mode, perinatal antibiotics, probiotic consumption, and the number of “furry pets” living in the home with the woman during pregnancy. Multivariate analysis indicated that having “furry pets” at home impacted numerous measures of the meconium microbiome: number of operational taxonomic units, Shannon diversity, and relative abundance of Firmicutes, Bacteroidetes, Staphylococcus spp., and Faecalibaterium spp. Based on the variables that the investigators were able to control for in their analyses, the measures of the microbiome were impacted by “furry pets” in the home independently of delivery mode, perinatal antibiotics, or probiotic consumption in this cohort [93].
Example 3
The majority of crops consumed in the USA are exposed to herbicides containing chemicals such as glyphosate [105]. These environmental chemicals have demonstrable efficacy and low toxicity; however, some concerns have been raised about the effect of these herbicidal components during pregnancy. In a mouse model of maternal exposure to high-dose glyphosate during pregnancy, juvenile offspring (post-natal day 28) display abnormal behaviors, an altered gut microbiome, and increased levels of acetate in stool [102]. Although it is unlikely that humans are exposed to high-dose glyphosate through consumption of herbicide-treated crops, these data provide one example of how a prenatal environmental chemical exposure can influence the offspring microbiome composition and later gut and behavioral health down the road.
Example 4
Other ambient (inhalation) and water/soil (ingestion) environmental chemicals like polycyclic aromatic hydrocarbons (PAHs) represent a more common maternal exposure. These noxious organic substances are byproducts of carbon combustion (i.e., vehicle emissions, petroleum processing) and cigarette smoking, and they are associated with contamination of air and soil/aquatic life and perinatal complications. The adverse pregnancy outcomes associated with PAH exposure include, but are not limited to, preterm delivery, low birthweight, neonatal bronchopulmonary dysplasia, child-onset asthma, low cognitive assessment scores in children, neurodevelopmental delay, DNA adduct formation, alteration of DNA methylation patterns, and persistent fetal reprogramming [106,107,108,109,110,111,112,113,114,115,116,117,118]. PAHs are also involved in bidirectional effects on maternal physiology, as PAHs can modulate an individual’s microbiome community membership and its function, but the microbiome itself contributes to polycyclic aromatic hydrocarbon transformation and metabolism [119]. Ingested polycyclic aromatic hydrocarbons are capable of disrupting gut microbial enzymes, leading to a state of induced dysbiosis within the gut community [119]. But the relationship is not one-sided. Human commensal microbes can metabolize primary polycyclic aromatic hydrocarbon products (that are inhaled or ingested) and secondary polycyclic aromatic hydrocarbon substances (that are conjugated by the liver) [119]. For example, benzo[a]pyrene (BaP) is a polycyclic aromatic hydrocarbon compound that cannot activate human estrogen receptor; however, the human gut microbiome transforms BaP into an estrogenic compound that can activate human estrogen receptor [120]. While neither the short- nor long-term implications of exogenous estrogen receptor activation in a developing fetus are well-established [121], this is a subject of ongoing research for our laboratory and others.
Example 5
Maternal diet is an important focus of every pregnancy, leading to decades of landmark studies investigating multiple aspects and components of dietary intake in the context of perinatal (pregnancy and lactation) and offspring outcomes. A very common feature of the maternal diet during pregnancy includes efforts to reduce sugar and/or carbohydrate intake, inclusive of the use of artificial non-nutritive sweeteners. This may occur in the context of a gravidae being diagnosed with gestational diabetes, or with efforts to manage gestational weight gain. Outside of pregnancy, it has been shown that artificial non-nutritive sweeteners alter the microbiome [122] and negatively affect several measures of metabolic health [123,124,125]. Stichelen et al. investigated the effect of non-nutritive sweetener consumption during pregnancy on health outcomes in murine pups [96]. Not only were profound metabolic and hepatic detoxification changes observed, but the pups also exhibited a gut microbiome profile associated with human metabolic disease and obesity (increased Firmicutes and decreased Akkermansia muciniphila) at post-natal day 19. Further experimentation revealed that both control and sweetener-fed dams transmit A. muciniphila to their newborn pups. Interestingly, over the first 3 weeks of life, pups exposed to maternal sweetener consumption lose this beneficial microbe and acquire metabolic, hepatic, and microbiome structure disturbances.
Example 6
Another feature of maternal diet during pregnancy may be the woman’s efforts to increase fiber intake, both to avoid the constipation that accompanies physiologic adaptive smooth muscle relaxation and to optimize caloric intake. Pregnant mice fed a high-fiber diet (compared to regular-fiber and no-fiber) produce offspring that are more resistant to allergic disease [88]. Specifically, the maternal high-fiber exposed offspring are resistant to allergic airway disease (a model for human asthma) and produce fewer eosinophils and proinflammatory cytokines at 3 weeks, 6–8 weeks, and 16 weeks old. The protective effects of maternal high-fiber diet exposure were similar in maternal acetate-exposed offspring, where the dams were fed acetate in their drinking water rather than a high-fiber diet. The study’s authors propose that, since increased acetate is observed in serum and feces following high-fiber consumption, acetate may represent the causal mechanism for the anti-inflammatory effects seen in association with maternal high-fiber diet exposure [88]. Other mechanisms may be in play, since protection against atopic disease is also observed with maternal consumption of prebiotic oligosaccharides [84].
Example 7
Another dietary modification observed in gravidae is the relative increase in fat intake during pregnancy. Studies of the effect of maternal high-fat diet consumption on offspring health have been conducted in multiple animal models. For example, in non-human primates exposed to maternal high-fat diet consumption during pregnancy and lactation and then weaned onto a control diet for at least 6 months, the gut microbiome of offspring is persistently altered compared to their peers who were not exposed to maternal high-fat diet [57]. The maternal high-fat diet exposure is associated with decreases in Campylobacter spp. and Helicobacter spp. and increases in Ruminococcus spp. and Dialister spp., which occur independent of juvenile obesity, refractory to probiotic/prebiotic therapy, and observed at least 6 months and up to 2-plus years after the cessation of maternal high-fat diet consumption [57, 77].
Example 8
Exposure to “stress” during pregnancy is a common concern for many women who are pregnant, as stress can alter biological pathways, including the microbiome [90]. However, prenatal “stress” is a commonly used but poorly defined term, generally associated with both psychological and/or physical symptomology. From a pragmatic perspective, in daily life humans and animals encounter situations that demand adaptation. Stress occurs if adaptation can only be met with great difficulty or is impossible. Because it is both situational and individualized, “stress” is challenging to define and measure precisely [126]. Some studies have used quantitative measures of stress derived from numerical scores generated from standardized, self-reported questionnaires focusing on negative maternal affect, depression, anxiety, worry, perceived stress, and life satisfaction [127,128,129]. Other studies define stress by major life events [130] or with biological biomarkers like salivary cortisol and alpha amylase [129]. Although the data are divided, some studies of maternal stress during pregnancy report offspring sleep disruption [127], atopic dermatitis [128], lower cognitive development scores [129, 130], and increases in both infectious and noninfectious illnesses [131]. In a rodent model of stress during pregnancy, Jašarević et al. [90] demonstrate that maternal stress impacts the post-natal colonic microbiome beginning at post-natal day 2 and extending to post-natal day 28 (latest time point tested). A particularly disrupted microbiome structure was observed in male offspring at post-natal day 28. Interestingly, the changes observed in these males were characteristic of female-typical microbiome patterns, indicating an axis between stress, hormones, and the gut microbiome.
From these eight examples, we learn that in every mammal examined to date, a number of maternal exposures impact the offspring’s health in early life. These functional impacts are often accompanied by alterations in the microbiome community composition and function, with some intervention studies inferring causality. However, it is less clear whether there is a true causal relationship between maternal exposure-driven perturbations in the infant microbiome and the onset of disease in childhood, nor its parlance to adult disease. As one example, data from the Canadian Healthy Infant Longitudinal Development (CHILD) study demonstrated an association between the transient absence of certain gut microbes (Micrococcaceae and Veillonellaceae, genera Lachnospira, Veillonella, Faecalibacterium, and Rothia) in the infant stool microbiome at 3 months of life with a subsequent risk of a higher Asthma Predictive Index at 3 years of age [133]. However, those same gut perturbations were not observed in the interval 1 year of age cohort suggesting that the dysbiosis observed at 3 months was transient [132]. These same investigators inoculated germ-free mice with these same four bacterial taxa and observed ameliorated airway inflammation in their adult progeny [132]. However, the nature of this inflammatory suppression was not entirely consistent with the histopathology anticipated with suppression of asthmatic disease and the study was limited by its small sample size [132].
Collectively, these studies and others suggest that perinatal exposures can lead to both short-term and long-term alteration of the offspring’s gut microbes and/or health, and some of these alterations are refractory to post-natal or post-weaning correction. In addition, an emerging body of data have specifically addressed the role of the maternal microbiome during pregnancy and its impact on development of the offspring immune system [9, 52, 103, 134, 135]. However, much of this work remains correlative and not causal, and further data is necessary to understand the molecular mechanisms driving these associations. To this end, in order to further explore examples of association needing causal mechanistic links, in this next section we will review data arising from two examples commonly cited as circumstantial evidence against a role for prenatal exposures having an impact on either short- or long-term alterations to the composition or function of the offspring microbiome. These examples are (1) observations among offspring delivered via Cesarean, and (2) phenotypic analysis of gnotobiotic animal models.
Example 1: Cesarean delivery and the offspring microbiome—innocent bystander or causal driver?
In 1985, the World Health Organization called for safe delivery practices for women and stated the “ideal” Cesarean delivery rate in a population should be 10–15% [136]. Since that time, the prevalence of Cesarean delivery has increased around the world (i.e., the rate in USA is approximately 31–32% in 2016–2019 [137]). Several renowned leaders focused on maternal and neonatal safe delivery have called for a reconsideration of the WHO and other entities recommendations pertaining to “ideal Cesarean delivery rates” with more precise use of definitions and classifications of the underlying driving factors and indications for Cesarean delivery to arrive at an evidence-based recommendation of what the “right” rate of Cesarean delivery ought to be [138]. In the meantime, a number of researchers are concerned about what effect Cesarean delivery may or may not have on the offspring’s short- and long-term health and microbiome.
Numerous studies report a correlation between Cesarean delivery and long-lasting health risks in term Cesarean-born offspring, including three large cross-sectional and longitudinal studies [139,140,141]. Specifically, groups reported increased risk of childhood obesity, atopy/asthma, lower cognitive development, and Celiac disease in Cesarean-born offspring [139,140,141,142,143,144,145,146,147,148,149]. Zhang et al. performed a meta-analysis of 61 studies to assess the effects of four different delivery modes: elective versus emergent Cesarean delivery and assisted versus unassisted vaginal delivery [139]. They observed increased odds for development of autism spectrum disorder (ASD) and attention deficit hyperactivity disorder (ADHD) in offspring delivered by either Cesarean delivery mode compared to vaginal delivery. However, the authors note they were unable to account for maternal and/or infant confounders due to absent information in the included studies.
Other research groups have utilized regression analysis to include maternal and infant factors alongside Cesarean delivery as variables in offspring health prediction models. These groups did not observe robust long-term negative outcomes in offspring born by Cesarean delivery alone when accounting for maternal socioeconomic characteristics [150], geolocation [151], BMI [150, 152], parity [150], age [150, 152], smoking during pregnancy [150,151,152], education [151, 152], ethnicity [152], marital status [152], preeclampsia [152], infant sex [151, 152], gestational age (at delivery) [151, 152], and birth weight [151]. Fundamentally, the subject of Cesarean delivery can be described as follows: does the surgical procedure of Cesarean delivery itself impact health outcomes of the offspring, or do other maternal and/or infant covariates (e.g., the “company” that Cesarean delivery keeps) contribute? This question is not simple to address in an experimental model due to the wide range of variables associated with the decision to perform Cesarean delivery, which is likely responsible for the disparate conclusions observed in correlative research studies examining the effect of Cesarean delivery on offspring health. However, it is critical that this research question be sufficiently answered so that obstetricians and pediatricians can understand what exact variables are responsible for (versus covariables associated with) the increased risk of negative health outcomes in Cesarean-delivered children.
If the Cesarean surgery itself is causing these adverse short- and long-term outcomes, what would be the underlying molecular mechanism driving this association? Some have suggested that altered first microbial inoculations at birth not including vaginal microbes might be one such mechanistic culprit [153, 154]. In other words, as awareness for the importance of the microbiome and its functional metagenome in human health and development continues to grow, concerns have been raised that failure to expose the emerging neonate to the “proper” community of microbes will lead to adverse clinical outcomes throughout the offspring’s life. Dominguez-Bello and colleagues were the first to study the neonatal microbiome across body sites immediately following delivery in a small case-control study [153]. Across all body sites, vaginally born neonates harbor mostly maternal vaginal microbes, such as Lactobacillus, Prevotella, or Sneathia spp., whereas Cesarean-born neonates harbor mostly maternal skin microbes, such as Staphylococcus, Corynebacterium, and Propionibacterium spp. Subsequent experiments by this group indicate that vaginal seeding of the neonate following Cesarean delivery enriches signature vaginal taxa in the oral, skin, and gut microbiomes of 30-day-old infants [155]. Shao and colleagues further confirm the association between Cesarean delivery and an altered neonatal microbiome composition in a cohort of almost 600 term babies with repeated sampling through the first month (days 4, 7, 21) of life, where disrupted Bacteroides spp. and increased Enterococcus, Enterobacter, and Klebsiella spp. correlated with Cesarean delivery, intrapartum antibiotic use, and lack of breastfeeding [156]. Mueller et al. confirmed lower proportions of Bacteroides, Parabacteroides, and Clostridium spp. in the neonatal gut microbiome of Cesarean-delivered offspring [157]. Many additional studies have similarly reported a correlation between Cesarean delivery and altered microbiome composition profiles in the neonate (see Table 1 of the review by Moya-Pérez et al. [200] and review of colonization rates by Shaterian et al. [201]) but several questions remain. Do these changes persist? What is the biological, functional consequence of changes in the neonatal microbiome? Are these changes a result of the Cesarean delivery per se or with maternal and/or infant variables that accompany and likely drive the need for a Cesarean delivery?
Some groups have identified differences in the Cesarean-born gut microbiome beyond the neonatal period [157, 202,203,204,205,206]. Niu et al. [203] queried a cohort of 729 children under 3-years-old and discovered an association with decreased Bacteroidetes in Cesarean-born offspring. Azad et al. [207] and Coker et al. [208] analyzed term infants repeatedly up to 12 months and observed persistent changes in emergent Cesarean delivered offspring at 12 months old; however, these changes were found to be associated with the intrapartum antibiotic exposure and ameliorated by breastfeeding. These data indicate that covariables of Cesarean delivery play an important role in the microbiome changes associated with Cesarean delivery. In our own study of this issue, multivariate analysis of metagenomes from multiple body sites in our prospective, longitudinal cohort of n=81 maternal-infant dyads enabled us to parse characteristics of the neonatal (obtained at birth) and infant (4-6 weeks of age) microbiome community structure and its function [72]. Consistent with the findings of Dominguez-Bello et al. described above, we too observed that among unlabored Cesarean-born neonates, the newborn microbiota and its functional pathways were most similar to the maternal skin and relatively homogenous across all body sites, except meconium [72]. However, the neonatal microbiome from labored Cesarean deliveries more closely resembled the distribution seen in vaginally delivered neonates where the infant microbiota were more similar to the communities of the maternal vagina and skin [72]. From an obstetrician’s perspective, the underlying clinical indication for a scheduled, unlabored Cesarean would be anticipated to significantly differ from that of an intrapartum, labored Cesarean delivery. Overall, variations in microbial community structure of the oral gingiva, nares, and skin (R2=0.038) by delivery mode were observed immediately following delivery but not by 4 to 6 weeks of age, and no significant differences were ever observed in the meconium or infant stool microbiota by mode of delivery, nor any body site’s functional metabolic pathway [72]. Several antepartum, intrapartum, and/or postnatal exposures were indicated as covariates contributing to microbiome functional pathway differences in generalized linear modeling [72]. Specifically, we observed that maternal high-fat diet, intrapartum antibiotics, and any formula feeding appeared to have the greatest effect on pathway variation in the infant stool, while gestational age and pre-pregnancy BMI had little effect [72]. Of particular interest, given co-linearity with antibiotic use and exclusive breastfeeding practices, Cesarean delivery per se did not bear a differential effect on the metagenome nor its function when subject to robust linear mixed modeling controlling for covariates as fixed effects [72]. In other words, there was no discernable impact attributable to Cesarean mode of delivery on the presence or absence of specific microbes comprising the infant microbiome by 4-6 weeks [72]. These findings are further consistent with a cohort comprised of preterm born infants, were there was no observed difference in the gut microbiome community structure by 5 weeks of age by virtue of being born via vaginal or Cesarean delivery [209].
Most of these studies have analyzed the effect of Cesarean delivery on the neonatal and infant microbiome by 16S ribosomal RNA (rRNA)-based detection [153, 155, 157, 202, 203, 205, 207, 210]. 16S amplicon-based sequencing is fast, affordable, and allows taxonomic resolution to the genus level with the aid of algorithms like DADA2 [211]. However, 16S sequencing cannot reliably yield species/strain-level calls. Therefore, 16S sequencing can identify broad taxonomical differences, but is not the optimal tool to identify the functional components of the microbiome. Why should the functional microbiome matter, if we know the taxonomical structure? It is important to remember that microbial nomenclature is not synonymous with microbial functionality. Different microbial strains within a species can contain dissimilar genes and functional capacity (i.e., E. coli), and different species can contain functional overlap. For this reason, functional profiling of the microbiome is a necessary next step in understanding the microbiome in any setting [212,213,214], including that of the Cesarean-delivered offspring. Whole-genome shotgun sequencing (WGS) allows for species and strain-level calls that can specify what functions a given community has or lacks [215, 216]. A few studies have analyzed the effect of Cesarean delivery on the neonatal and infant microbiome by WGS sequencing [72, 156, 204, 206, 210], but only two [72, 210] analyzed the effect of Cesarean delivery on the functional microbiome and saw no lasting impact.
In order to consolidate the discordant conclusions of the effect of Cesarean delivery on the offspring gut microbiome and health trajectories, the causal versus correlative effect of delivery mode must be carefully examined for confounders and company of Cesarean delivery. Such variables include the underlying medical or obstetrical indication or pathology leading to the surgery, antibiotic exposure surrounding delivery, environmental exposure to the neonatal intensive care unit, human milk versus formula feeding, other maternal comorbidities, and yet-unidentified factors. For example, it has been suggested that Cesarean delivery may be associated with altered bacteria-bacteriophage interactions [217]. Bacteriophages (the viruses that infect bacteria) represent a largely unexplored component of the microbiome that may play a role in early development of the infant microbiome. As we recently commented [218], identifying intrauterine transmission of bacteriophages prior to delivery is an exciting new avenue of research. Perhaps bacteriophages represent one of multiple unexplored variables with the potential for intrauterine transmission that may play a role in affecting the gut microbiome of offspring that are born via Cesarean. The maternal dietary and medical conditions which may modulate bacteriophage transmission remain unexplored [218].
In summary, the neonatal microbiome following delivery is different in vaginal versus Cesarean-born neonates, and Cesarean delivery has been associated with a number of offspring health risks [139,140,141, 153,154,155,156,157, 200,201,202,203,204,205,206,207, 210]. Because early life is a critical window for normal human development, it stands to be concerned about these changes to the neonatal microbiome and potential lasting health impacts during early developmental windows [219].
However, outstanding speculation remains regarding the root cause of the altered neonatal microbiome and offspring health impacts associated with Cesarean delivery [72, 150,151,152, 207,208,209,210]. Namely, it is important to discern the effect of Cesarean delivery itself from the clinical confounders that accompany Cesarean delivery [72, 150,151,152, 207,208,209,210]. Resolution of this discrepancy requires incorporation of the clinical confounders that accompany Cesarean delivery with large, prospective studies naïve to eventual delivery mode. Cesarean delivery is a common and safe abdominal surgery. Ready availability to medically indicated Cesarean with surgically competent providers is crucial in the reduction of maternal and neonatal mortality and decreasing social disparities worldwide (https://www.who.int/reproductivehealth/publications/maternal_perinatal_health/cs-statement/en/). Greater understanding of the effect of birth by Cesarean in-and-of-itself on offspring health is needed without inadvertently diminishing a safe and necessary procedure. These data provoke consideration of the implications that follow if delivery mode does not meaningfully impair the long-term structure or function of the microbiome. One such implication is this: if the moment of delivery does not direct the body’s future interactions with microbes, then when does the developing host receive these instructions? What can the DOHaD hypothesis contribute to this issue? To further contemplate this latter question, we turn to observations arising from functional studies in gnotobiotic animal models.
Example 2:
Are gnotobiotic animals “normal”?
One of the most powerful tools available to study host-microbial interactions is the development of germ-free (GF) animal models, originally conceived by Louis Pasteur in 1885 and first cultivated in guinea pigs [220], rats [221], then mice [222]. Gnotobiotic animals are created and reared in environments “free” of (detectable) live bacteria. Their food, water, bedding, and any supplies are autoclaved or filter-sterilized, and sterility is continuously monitored by culture and molecular methods. While GF animals are viable and reproduce despite the lack of live microbes, they are by no means normal. Compared to conventionally colonized animals, GF animals display morphological differences, biochemical abnormalities, atypical neurobehavior, and pronounced immunological changes. GF animals are characterized by prolonged diestrus, small lymph nodes and spleen, thin intestinal villi and lamina propria, increased food/water intake, higher oxidation-reduction potential, altered mucosal enzyme patterns, and decreases in circulating leukocytes, immunoglobulin levels, Peyer’s patch size, intraepithelial T cells, inflammatory response, blood volume, regional blood flow, cardiac output, basal metabolic rate, motor activity, response to catecholamines, body fat, organ sizes, vitamin biosynthesis, enteric bile acid transformation, and intestinal-specific parameters (mass and surface area, peristalsis, epithelial cell renewal, pH levels) [223,224,225,226,227,228,229,230,231,232,233,234]. It is important to note, though, that in spite of GF and germ-depleted rodents being impressively prone to post-natal inflammation and sepsis from intestinal pathogens, gnotobiotics still carry pregnancies to term and can propagate relatively depleted or replete lineages [134, 235]. Based on these observations, we can propose that mammals have intimately coevolved with microbes and now require microbes (and the diverse functions that their metagenomes encode) for normal development.
To demonstrate the necessary role of microbes in development, GF animals are introduced to microbes through conventionalization. Studies support their proof-of-principle that microbes impact development by comparing GF controls to the conventionalized animals [135, 236]. However, a few groups have compared the conventionalized animals to conventional control animals (that have always been raised with microbes), with interesting results. For example, conventionalized mice display decreased transcription of UDP-glucuronosyltransferases 1a9 and 2a3 compared to conventional mice fed a probiotic [237]. In terms of development of immune competence, one group reports that conventionalized rats harbor altered levels of intestinal intraepithelial lymphocytes compared to both their GF and conventional control counterparts [238]. They also observe that the frequency of T cells co-expressing α4β7 in the intestinal lamina propria of conventionalized mice never normalizes to the base levels of conventional controls [239]. Another group demonstrates that conventionalized mice produce decreased IFNγ, phagocytotic ability, and reactive oxygen species production in response to fungal infection compared to GF and/or conventional controls [240]. Clearly, conventionalization does not restore all aspects of atypical development caused by a GF upbringing, particularly with respect to the immune system. Several groups identify the importance of “critical developmental windows,” where reconstitution of the microbiome after said window fails to restore normal development, even partially [7,8,9]. In an elegant study, de Agüero et al. demonstrated that transient colonization of pregnant GF dams with a mutant E. coli improved multiple aspects of GF pup health, including intestinal group 3 innate lymphoid and F4/80 + CD11c + mononuclear cell counts, epithelial antibacterial peptide transcription levels, and microbial molecular metabolism [103]. The mutant E. coli was specifically engineered to not persist in the intestine, and its transience was confirmed with negative cultures of the placenta and GF status of the neonates. Despite the absence of long-term or persistent live microbial exposure, the pups demonstrated markedly improved immune health outcomes, demonstrating the essential role of the maternal microbiome in fetal immune development.
Although the living environments of GF animals are free of live microbes, they still contain detectable dead/killed microbes and microbial particles [241,242,243]. It is possible that these nonviable microbes and microbial components can serve as pathogen/microbe-associated molecular patterns (PAMPs/MAMPs) for immune education and other aspects of host-microbe interactions. For example, germ-free mice challenged with Helicobacter pylori can reduce gastric infection levels with repeated exposure to heat-killed Lactobacillus johnsonii [244]. Similarly, heat-killed L. brevis is associated with increasing plasma serotonin concentrations in another model [245]. It is also clearly demonstrated that viable microbes are required for proper host-microbe development [246, 247].
Observations in GF animals lead to three considerations about microbes and development.
-
(1)
GF littered animals are viable without exposure to live microbes, but they display abnormal development.
-
(2)
Even early-on conventionalization of GF animals does not fully correct this abnormal development.
-
(3)
Killed microbes and microbial components present in the living environments of GF animals may potentially contribute to immune education or other aspects of normal development.
When combined with the lessons learned from Cesarean delivery, we next consider the possibility that mammalian education for “how to form a symbiotic relationship with microbes” begins in utero. We will consider the possibility of a live but low-biomass microbial community within the in utero environment. To explore this theory, we will present the historical evidence for and against the placental microbiome and how it may contribute to the development of the offspring microbiome and long-term health.
Does a low-abundance, low-biomass intrauterine microbiome exist?
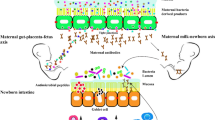
Distinct microbial communities in the meconium among preterm and healthy-term neonates are detected and described within minutes to hours of birth, and these communities expand in the first days to weeks of life to readily show discrete body niche communities long before that same infant will alter its diet or engage in meaningful contact with the outside world [204, 248,249,250]. How does this happen? Traditionally, it was assumed that the healthy intrauterine environment during pregnancy (including the uterine decidua, placenta, chorion/amnion, amniotic fluid, fetus, and meconium) was sterile until birth. Only during delivery was the neonate believed to first interact with microbes from the vagina (vaginal delivery) or skin/environment (Cesarean delivery). This dogma was further supported by the negative clinical outcomes associated with prolonged and untreated/undelivered cases of chorioamnionitis (the bacterial infection of the intra-amniotic space that leads to inflammation of fetal membranes and is associated with serious morbidity and potential mortality of both the gravidae and her fetus or neonate). Chorioamnionitis is a localized infection that is most often “cured” by delivery, plus antibiotics are used to reduce the risk of post-partum endometritis. It has been argued that if bacteria were supposed to be present in the intrauterine environment, then why does chorioamnionitis occur?
As evidence continued to accumulate challenging the sterility of the intrauterine environment during pregnancy [56, 61, 158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199], so did more evidence supporting sterility [11,12,13,14,15, 251,252,253,254,255,256,257,258,259,260,261,262,263,264,265,266,267,268,269,270,271,272] (see Tables 1 and 2, respectively). How come research efforts seemingly performing the same or similar experiments are arriving at opposing conclusions?
The search for the intrauterine microbiome has proceeded through both culture-independent and culture-dependent methods. Culture-independent methods used to identify microbial presence within the intrauterine space include histology (most commonly but not exclusively Gram staining), immunohistology, polymerase chain reaction (PCR), 16S rRNA sequencing of the hypervariable regions (HVRs), WGS sequencing, liquid or gas chromatography, rRNA in situ hybridization, and scanning electron microscopy. Of these, the most popular and common method is 16S rRNA sequencing. In order to perform 16S rRNA sequencing, it is necessary to overcome a series of challenges: (1) obtain the intrauterine sample aseptically, (2) perform DNA extraction, (3) amplify the target HVR(s), (4) perform high-fidelity sequencing, and (5) apply bioinformatic approaches that are biologically relevant. Therefore, the seemingly simple approach of 16S rRNA sequencing is actually an amalgamation of multiple steps that all have the potential to introduce error. We will discuss these technical challenges and other factors that are integral to the debate of the presence or absence of the intrauterine microbiome.
Challenge 1
Environmental microbes and contamination can potentially be construed as or obscure the signal ascribed to the placental microbiome [261]. To control for this difficulty, strict sterile procedures and rules for amount and location of starting material are required, along with inclusion of environmental swabs from every possible source of contamination (maternal body sites, surgical surfaces, DNA extraction equipment, and the sequencing site). Due to the expense of sequencing, few studies include environmental controls. Yet in the studies that do, the results are still divided in support and opposition of the intrauterine microbiome [12, 197]. However, it is important to note that the presence of the same identified microbe in both an intrauterine sample and an environment/contamination control does not automatically indicate that the microbe is a contaminant. For example, DNA signatures of Lactobacillus spp. commonly found in the vagina (a potential source of contamination) are also found in endometrial biopsies [274], suggesting that Lactobacillus spp. DNA may be present from the beginning of placental development, rather than terminally introduced during labor and delivery. In the absence of concomitantly collected samples from the same subject with deep sequencing to the species and strain level, definitive interpretations of overlapping genera as indications of contamination should exercise caution.
Challenge 2
To distinguish the low-abundance, low-biomass communities, it is essential to include extraction control samples using just the kit reagents and no input sample. DNA extraction kit and laboratory reagents contain a collection of low-abundance microbes that yield a “kit-ome,” which complicates detection of the low-abundance, low-biomass microbial signatures [208, 268, 275,276,277,278,279]. On the one hand, kit negative samples are distinguishable from intrauterine microbiome signatures [56, 61, 171, 178, 181, 183, 194]. On the other hand, however, kit negatives overlap with the intrauterine microbiome patterns [11, 12, 15, 262, 263, 273, 280]. The ability or inability to distinguish intrauterine samples from negative controls is the crux of the debate. Likely reasons for this discrepancy are differences in HVR target and sequencing technology, discussed next.
Challenge 3
The selection of primers for 16S rRNA sequencing of various HVRs is particularly influential. Some primers, such as those used by Walker et al. to cover the V1-2 HVRs, are designed as degenerate and yield short amplicons [281]. In contrast, other primers, such as those we used to span the V1-3 HVRs, are nondegenerate and yield longer amplicons [56]. Parnell et al. evaluated all nine HVRs in the context of placental samples [183]: V7-8 did not amplify at all; V1, V5, and V9 were not detected in the majority of samples; V2 and V6 generated as many reads in the negative control samples as the test samples; and V4 amplified significantly more bacterial DNA in test samples than negative control samples. This study demonstrates that, even in the same samples, the use of primers targeting different 16S rRNA HVRs can lead to drastically different conclusions.
Challenge 4
With advances in computational power come advances in computational tools and pipelines for handling large amounts of sequencing data. Bioinformatic workflows continue to be developed and enhanced to subtract artifacts, contamination, and host DNA [282,283,284,285,286]. The most popular of these tools is perhaps decontam [284]. Based on frequency and prevalence, decontam relies on multiple sequencing runs on the same sample and the inclusion of negative controls, but these practices are not always possible for all researchers and situations. A new tool called Squeegee was recently released that allows for the detection of potential contaminants at the species level of low-biomass, high-host samples without the inclusion of negative controls in the sample set [287]. Squeegee assumes that DNA extraction kits and laboratory environments will share similar contaminant features. Other approaches to handle the computational demand of identifying a low-biomass community include sequencing the genomes of contaminants to definitively subtract them post hoc [288] or computationally controlling for the low-biomass expectation [289,290,291].
Challenge 5
Alternate working definitions of “microbiota” and “microbiome” lend confusion to the field. “Microbiota” refers to the community of microbes in an ecosystem (https://www.nature.com/subjects/microbiota). As discussed earlier, “microbiome” is a more complex term with varying definitions. Whereas the Nature definition [https://www.nature.com/subjects/microbiome] emphasizes the genomics of the community, the Whipps et al. definition [1] emphasizes the ecology of the community. Together, these definitions point to a common and essential feature: functionality. Genomics, or the full content of genetic material, is a measure of capacity within a community. Ecology, or the relationship between the microorganisms, is a measure of activity within the community. Both capacity and activity are key features of function. Function is independent of the biomass of the community, be it high or low. Function is independent of the longevity of colonization. In other words, “microbiome” does not necessitate high-biomass or high abundance colonization, such as is generally implied when we consider the gut microbiome. Perhaps one of the most important questions of our time is how sparse communities remain sparse and do not increase in biomass. Furthermore, why should the functional capacity of a low-abundance, low-biomass microbiome matter? Can low numbers of microbes actually impact human physiology and development? The functional importance of these microbes still needs to be experimentally determined, but we will present the seminal research supporting and refuting a sterile intrauterine environment.
In the context of the placenta, de Goffau et al. analyzed placenta biopsies from over 500 human neonates plus microbial spike-in positive controls and DNA extraction negative controls using 16S rRNA sequencing with V1-2 degenerate HVR primers and a subset of metagenomic sequencing [273]. This study was strengthened by multiple cohort arms that include subjects with negative pregnancy outcomes (n = 318, including preeclampsia, neonates that are small for gestational age, and spontaneous preterm birth) and normal control births (n = 219). Researchers were blinded to the cohort arms. The focus of the study was to classify the reproducibility of microbial signals versus negative controls from placental biopsy samples. The researchers concluded that they reliably detected small microbial signatures from placental biopsies, but the signals overlapped between the placental samples and the negative controls. From this, the authors conclude that the taxonomic overlap between test samples and controls was attributable to contamination from lab reagents and equipment or contamination during labor/delivery. The only microbial signature they detected prior to the onset of labor was their microbial spike-in positive control (Streptococcus agalactiae). The authors note that, during sample collection, the 25 mg placental tissue was washed vigorously in sterile PBS, which is not a common methodology by others in the field. The authors also report inconsistent sequencing depth and subsequent aberrant library preparation across runs, which they assign to sequencing contamination, but could be interpreted as sequencing failure.
Subsequent work by Theis et al. analyzed samples (n = 51) from murine placental and fetal tissue via culture, quantitative real-time PCR (qPCR), and 16S sequencing, along with maternal and technical controls [12]. This study employed orthogonal methodologies to query the hypothesis of in utero colonization. The strengths of this study include (1) the use of an animal model to surgically acquire placental and fetal tissue before the onset of labor, and (2) orthogonal use of both culture-dependent and two non-culture-dependent methodologies. The researchers reported that only 5.5% of placental culture samples yielded a bacterial colony, and the bacterial loads of placental and fetal tissue were not higher than extraction negative controls. One sample of fetal brain yielded a higher bacterial load than background technical controls, and the microbe (Bacillus circulans) was traced to maternal origin. This study is limited by a lack of visualization techniques (i.e., microscopy, in situ hybridization) nor were germ-free mice included in analysis.
To summarize the stance of those refuting the intrauterine microbiome, Fricke et al. [16] asserted a “forensic approach” to detect microbiomes from bacterial DNA samples is not sufficient to infer the presence of live microbes, negating the prenatal microbiota hypothesis. However, we and other teams maintain that the functional importance of these microbes needs to be experimentally determined. It can be argued that these microbes are functionally important because maternal exposures bear an impact on the establishment of the offspring microbiome and subsequent health, as discussed in the first portion of this review.
Two studies in particular provide direct evidence for the functional significance of a low-biomass intrauterine microbiome in humans [196, 199]. In both, mid-trimester (early second trimester) human fetal tissue was examined by 16S rRNA sequencing, scanning electron microscopy, culture, and in situ hybridization. The multifaceted use of orthogonal approaches lends increased confidence to the discovery of microbial signatures and structures within placental and fetal organs. In particular, Rackaityte et al. [196] demonstrated Micrococcaceae and Lactobacillus spp. were key constituents in the fetal meconium, and Mishra et al. [199] described Staphylococcus and Lactobacillus spp. dominance patterns in fetal tissues. Both groups demonstrated with scanning electron microscopy bacteria-like cocci clustered within mucin-like structures in the fetal intestinal lumen, supporting the notion that these are nascent microbes of the fetal gut. These bacteria-like structures were not observed at 10 weeks, but were visualized at 14 weeks gestation, emphasizing the role that gestational age and temporality play in microbial detection of low-biomass communities and diminishing the likelihood of contamination. Importantly for verification of functionality from these low-biomass communities, both groups demonstrated that microbial presence can prime fetal immune education in vitro. Rackaityte et al. [196] included culture-dependent methodology in their demonstration of community functionality. Micrococcaceae isolates were cultured in media supplemented with placental steroid hormones or THP1 human monocyte cells. The fetal Micrococcus luteus isolate outperformed two reference Micrococcus strains in high-progesterone and high-β-estradiol carbon-limiting conditions, suggesting fitness specific to the in utero environment. Additionally, M. luteus alone maintained the in vitro capacity for intracellular survival in monocytes. In ex vivo experiments, M. luteus was exposed to primary fetal cells, which induced TLR6 and NKFB expression in intestinal epithelia and demonstrated tolerogenic immunomodulation in antigen presenting cells (i.e., induced granulocyte–macrophage colony-stimulating factor, granulocyte colony-stimulating factor, interleukin-10; reduced tumor necrosis factor-α). Mishra et al. [199] tested the hypothesis that microbial antigens contribute to diverse T cell responses in the fetus. Two microbes (Lactobacillus and Staphylococcus spp.) were chosen that were most consistently present across fetal tissues (as measured by 16S and culture). The microbes were heat-killed and incubated with dendritic cells isolated from fetal mesenteric lymph nodes, then co-cultured with T cells from the same lymph node in vitro. Staphylococcus antigen exposure resulted in high T cell memory expansion, increased total T cell count, increased CD69 (marker of memory activation), and production of tumor necrosis factor and interferon-γ as compared to Lactobacillus antigen and control. Memory cells would not be stimulated by spurious contaminants; therefore, these data indicate that fetal T cells displayed activated in vitro memory response toward antigens from Staphylococcus spp. and to a lesser degree Lactobacillus spp. that were observed in utero. Collectively, these two studies demonstrate the potential functional significance that a sparse, low-biomass microbiome could have on the developing fetus and potentially long-term health outcomes of the offspring. More evidence from carefully designed and well-powered studies is needed to clarify the role of live microbes in human fetal physiology and development.
In addition to these studies demonstrating functional activity within these fetal communities, both teams of investigators addressed the risk of contamination in their findings. For example, Rackaityte et al. [196] included the addition of a mock community to every PCR plate for amplification and stringent technical controls for both extraction and sequencing runs. Nonetheless, Rackaityte et al. [196] was limited by a 30% detection of microbial profiles in fetal intestine compared to controls, and microbes were detected at very low cell numbers. Additionally, a number of sequenced samples contained fewer than 1000 microbial sequence reads. When these low-sequence read number samples were included in analysis, there was an apparent batch effect in sequencing. When these low-sequence read number samples were excluded in the analysis, there was no apparent batch effect in sequencing. Mishra et al. [199] overcame some of the limitations of Rackaityte et al. [196] and were strengthened by random and well-balanced sequencing batches and inclusion of PBS controls. However, Mishra et al. were hindered by the lack of maternal vaginal swabs or cultures. Fortunately, the surgical procedures used in specimen attainment are performed in highly aseptic environments [169, 172]. Although the detected microbes are known members of the vaginal microbial community, they are also likely members of the endometrial and decidual microbial communities [292, 293]. Additionally, samples were washed with PBS, and thus could have potentially been contaminated during the wash step. However, control organs (liver, spleen, lymph nodes), and 10-week fetal intestinal samples were subject to an identical PBS wash and vaginal exposure and did not yield findings akin to the > 14-week fetal intestine.
Despite the work of Mishra et al. and Rackaityte et al., we are mindful that their work was conducted on tissues arising from the mid-gestation. We remain agnostic as to whether the placenta or other intrauterine tissues retain functional microbiomes at term gestation, and whether these communities are comprised of live microbes in a detectable abundance. Based in part on the work of Mishra et al. and Rackaityte et al, alongside others discussed further herein, we speculate that the presumptive low-biomass intrauterine live microbial communities may be a critical first step in ontogeny and education of the offspring immune system. A sparse microbial community within the intrauterine environment may participate in progressive pruning and maintenance of pregnancy, resistance to pathogenic organisms, and immune development tolerance of the soon-to-be neonate for more fulminant colonization by commensal microbiota in post-natal life. Little is currently understood about microbial interactions at the maternal-fetal interface. It is known that macrophages and natural killer cells are present, but it is possible that the constituent placental trophoblast cells also play a role. Trophoblasts can recognize and respond to PAMPs/MAMPs and promote regulatory cytokine secretion; in this way, trophoblasts may contribute to tolerogenic education [294]. It is interesting to note that the 16S rRNA signal we detected by in situ hybridization was largely localized to the synctiotrophoblast [186]. Perhaps some basal level of microbial exposure in the form of live microbes is necessary in utero to prevent a massive immune reaction to the microbial assault that accompanies entrance to the ex utero realm. Even if persistently viable microbes are absent from the intrauterine environment, it is generally understood that microbial antigenic signatures participate in offspring development. What remains to be determined with future research endeavors is the effect of live microbes versus microbial products during in utero development, including immune tolerance.
Experimental evidence supports both viewpoints of the “sterile” vs “not so sterile” intrauterine environment. As such, there is a critical need to fund high-quality research to clarify if, what, and how there is or is not a functional role of low-abundance, low-biomass communities (like the placental and the fetal microbiome) in potentially modulating fetal development. As we have stated since 2014, we remain uncertain if indeed there is a functional consequence to these low-biomass communities and are not confident which microbial members can be considered “nascent” to the intrauterine environment. However, it is a hypothesis that deserves ongoing rigorous testing to query the role for low-biomass microbial communities in modulating the fetal immune repertoire and early immune development. Furthermore, studies aimed at understanding why low-biomass communities remain sparse and how host immunity contributes to maintaining the sparsity are both intriguing and impactful. Given the data that pregnancy exposures are known to have long-lasting influences on offspring health and predisposition to adult-onset disease, the answers to these questions are vital to meaningful advances in scientific knowledge and public health. The current scientific debate pertaining to the functional role of low-biomass communities is far from settled and deserves ongoing mechanistic-based research with an objective lens.
Availability of data and materials
Not applicable
Abbreviations
- BaP:
-
Benzo[a]pyrene
- DOHaD:
-
Developmental origins of health and disease
- GF:
-
Germ-free
- HVR:
-
Hypervariable region
- PAMPs/MAMPs:
-
Pathogen/microbe-associated molecular patterns
- PCR:
-
Polymerase chain reaction
- rRNA:
-
Ribosomal RNA gene
- WGS:
-
Whole-genome shotgun
References
Whipps J, Lewis K, Cooke R. Fungi in biological control systems. Burge MN, editor. Manchester University Press; 1988.
Foster JA, Lyte M, Meyer E, Cryan JF. Gut microbiota and brain function: an evolving field in neuroscience. International Journal of Neuropsychopharmacology. 2016;19:1–7.
Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. New England Journal of Medicine. 2016;375:2369–79.
Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–73.
Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. 2016;535:65–74.
Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016;535:75–84.
Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, et al. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. Journal of Physiology. 2004;558:263–75.
Heijtz RD, Wang S, Anuar F, Qian Y, Björkholm B, Samuelsson A, et al. Normal gut microbiota modulates brain development and behavior. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3047–52.
Neufeld K-AM, Kang N, Bienenstock J, Foster JA. Effects of intestinal microbiota on anxiety-like behavior. Communicative & Integrative Biology. 2011;4:492–4.
Clarke G, Grenham S, Scully P, Fitzgerald P, Moloney RD, Shanahan F, et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Molecular Psychiatry. Nature Publishing Group. 2013;18:666–73.
Theis KR, Romero R, Winters AD, Jobe AH. Gomez-Lopez N. Lack of evidence for microbiota in the placental and fetal tissues of rhesus macaques. mSphere. 2020;5:e00210–20.
Theis KR, Romero R, Greenberg JM, Winters AD, Garcia-Flores V. Motomura K, et al. No consistent evidence for microbiota in murine placental and fetal tissues. mSphere. 2020;5:1–18.
Kuperman AA, Zimmerman A, Hamadia S, Ziv O, Gurevich V, Fichtman B, et al. Deep microbial analysis of multiple placentas shows no evidence for a placental microbiome. BJOG: An International Journal of Obstetrics and Gynaecology. 2020;127:159–69.
Burnham P, Gomez-Lopez N, Heyang M, Cheng AP, Lenz JS, Dadhania DM, et al. Separating the signal from the noise in metagenomic cell-free DNA sequencing. Microbiome. Microbiome. 2020;8:1–9.
Sterpu I, Fransson E, Hugerth LW, Du J, Pereira M, Cheng L, et al. No evidence for a placental microbiome in human pregnancies at term. American Journal of Obstetrics and Gynecology [Internet]. Elsevier Inc.; 2021;224:296.e1-296.e23. Available from. https://doi.org/10.1016/j.ajog.2020.08.103.
Fricke WF, Ravel J. Microbiome or no microbiome: are we looking at the prenatal environment through the right lens? Microbiome. 2021. p. 9.
Aagaard K, Hohmann E. Regulating microbiome manipulation. Nature medicine. United States. 2019;25:874–6.
Antonov AN. Children born during the siege of Leningrad in 1942. The Journal of Pediatrics. 1947;30:250–9.
Smith CA. Effects of maternal undernutrition upon the newborn infant in Holland (1944–1945). The Journal of Pediatrics. 1947;30:229–43.
Kermack WO, McKendrick AG, McKinlay PL. Death-rates in Great Britain and Sweden: expression of specific mortality rates as products of two factors, and some consequences thereof. Journal of Hygiene. 1934;34:433–57.
Barker DJ. Low intelligence. Its relation to length of gestation and rate of foetal growth. British journal of preventive & social medicine. 1966;20:58–66.
McCance RA, Widdowson EM. The determinants of growth and form. Proceedings of the Royal Society of London Series B, Biological sciences. England. 1974;185:1–17.
Barker DJP, Osmond C, Golding J, Kuh D, Wadsworth MEJ. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. British Medical Journal. 1989;298:564–7.
Barker DJP, Osmond C, Winter PD, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. The Lancet. 1989;334:577–80.
Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, PDW CO. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303:1019–22.
Barker DJP. Mother, babies and health in later life. Babies and Health in Later Life. Churchill Livingstone, Edinburgh: Mother; 1998.
Fleming TP, Watkins AJ, Velazquez MA, Mathers JC, Prentice AM, Stephenson J, et al. Origins of lifetime health around the time of conception: causes and consequences. Lancet. 2018;391:1842–52.
Barker DJP. The origins of the developmental origins theory. Journal of Internal Medicine. 2007;261:412–7.
Aagaard-tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, et al. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol. 2008;41:91–102.
Cox J, Williams S, Grove K, Lane RH, Aagaard-Tillery KM. A maternal high-fat diet is accompanied by alterations in the fetal primate metabolome. American Journal of Obstetrics and Gynecology. 2009;201:281.e1-281.e9.
McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. Journal of Clinical Investigation. 2009;119:323–35.
Bocock PN, Aagaard-Tillery KM. Animal models of epigenetic inheritance. Seminars in reproductive medicine. United States. 2009;27:369–79.
Sullivan EL, Grayson B, Takahashi D, Robertson N, Maier A, Bethea CL, et al. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. Journal of Neuroscience. 2010;30:3826–30.
Aagaard-Tillery KM, Suter M, Harris A, Abramovici A, Cantu J. Epigenetics and reproduction and the developmental origins of health and disease. Anim Reprod. 2010;7:103–16.
Suter M, Abramovici A, Aagaard-Tillery K. Genetic and epigenetic influences associated with intrauterine growth restriction due to in utero tobacco exposure. Pediatric Endocrinology Reviews. 2010;8:94–102.
Suter M, Abramovici A, Showalter L, Hu M. Shope C Do, Varner M, et al. in utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism - Clinical and Experimental. Elsevier. 2010;59:1481–90.
Aagaard-Tillery K, Spong CY, Thom E, Sibai B, Wendel G Jr, Wenstrom K, et al. Pharmacogenomics of maternal tobacco use: metabolic gene polymorphisms and risk of adverse pregnancy outcomes. Obstetrics and gynecology. 2010;115:568–77.
Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, et al. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology. 2011;152:2456–64.
Suter M, Bocock P, Showalter L, Hu M, Shope C, McKnight R, et al. Epigenomics: maternal high-fat diet exposure in utero disrupts peripheral circadian gene expression in nonhuman primates. FASEB Journal. 2011;25:714–26.
Sbrana E, Suter MA, Abramovici AR, Hawkins HK, Moss JE, Patterson L, et al. Maternal tobacco use is associated with increased markers of oxidative stress in the placenta. Am J Obstet Gynecol. 2011;205:246.e1–246.e7.
Suter M, Ma J, Harris A, Patterson L, Brown KA, Shope C, et al. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics. 2011;6:1284–94.
Suter MA, Chen A, Burdine MS, Choudhury M, Harris RA, Lane RH, et al. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB Journal. United States. 2012;26:5106–14.
Suter MA, Sangi-Haghpeykar H, Showalter L, Shope C, Hu M, Brown K, et al. Maternal high-fat diet modulates the fetal thyroid axis and thyroid gene expression in a nonhuman primate model. Molecular Endocrinology. United States. 2012;26:2071–80.
Roberts VJ, Rasanen JP, Novy MJ, Frias A, Louey S, Morgan TK, et al. Restriction of placental vasculature in a non-human primate: a unique model to study placental plasticity. Placenta. 2012;33:73–6.
Behl M, Rao D, Aagaard K, Davidson TL, Levin ED, Slotkin TA, et al. Evaluation of the association between maternal smoking, childhood obesity, and metabolic disorders: a national toxicology program workshop review. Environmental Health Perspectives. 2013;121:170–80.
Suter M, Takahashi D, Grove K, Aagaard K. Postweaning exposure to a high-fat diet is associated with alterations to the hepatic histone code in Japanese macaques. Pediatr Res. 2013;74:252–8.
O’Neil D, Mendez-Figueroa H, Mistretta T-A, Su C, Lane RH, Aagaard KM. Dysregulation of Npas2 leads to altered metabolic pathways in a murine knockout model. Mol Genet Metab. 2013;110:1–21.
Suter MA, Anders AM, Aagaard KM. Maternal smoking as a model for environmental epigenetic changes affecting birthweight and fetal programming. Molecular Human Reproduction. 2013;19:1–6.
Consortium TMGS and A. The common marmoset genome provides insight into primate biology and evolution. Nature genetics. United States; 2014 Aug.
Cuevas Guaman M, Sbrana E, Shope C, Showalter L, Hu M, Meloche S, et al. Administration of antenatal glucocorticoids and postnatal surfactant ameliorates respiratory distress syndrome-associated neonatal lethality in ERK3−/− pups. Pediatr Res. 2014;76:24–32.
Harris RA, Tardif SD, Vinar T, Wildman DE, Rutherford JN, Rogers J, et al. Evolutionary genetics and implications of small size and twinning in callitrichine primates. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:1467–72.
Suter MA, Ma J, Vuguin PM, Hartil K, Fiallo A, Harris RA, et al. in utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. American Journal of Obstetrics and Gynecology. 2014;210:463.e1-463.e11.
Chadderdon SM, Belcik JT, Bader L, Kirigiti MA, Peters DM, Kievit P, et al. Pro-inflammatory endothelial activation detected by molecular imaging in obese non-human primates coincides with the onset of insulin resistance and progressively increases with duration of insulin resistance. Circulation. 2014;129:471–8.
Thorn SR, Baquero KC, Newsom SA, El Kasmi KC, Bergman BC, Shulman GI, et al. Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates. Diabetes. United States. 2014;63:2702–13.
Roberts VHJ, Pound LD, Thorn SR, Gillingham MB, Thornburg KL, Friedman JE, et al. Beneficial and cautionary outcomes of resveratrol supplementation in pregnant nonhuman primates. FASEB Journal. United States. 2014;28:2466–77.
Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci T ransl Med. 2014;6:1–22.
Ma J, Prince AL, Bader D, Hu M, Ganu R, Baquero K, et al. High-fat maternal diet during pregnancy persistently alters the offspring microbiome in a primate model. Nature Communications [Internet]. Nature Publishing Group. 2014;5:1–11 Available from: https://doi.org/10.1038/ncomms4889.
Goodspeed D, Seferovic MD, Holland W, Mcknight RA, Summers SA, Branch DW, et al. Essential nutrient supplementation prevents heritable metabolic disease in multigenerational intrauterine growth-restricted rats. FASEB Journal. 2015;29:807–19.
Seferovic MD, Goodspeed DM, Chu DM, Krannich LA, Gonzalez-Rodriguez PJ, Cox JE, et al. Heritable IUGR and adult metabolic syndrome are reversible and associated with alterations in the metabolome following dietary supplementation of 1-carbon intermediates. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. United States. 2015;29:2640–52.
O’Tierney-Ginn P, Roberts V, Gillingham M, Walker J, Glazebrook PA, Thornburg KL, et al. Influence of high fat diet and resveratrol supplementation on placental fatty acid uptake in the Japanese macaque. Placenta. 2015;36:903–10.
Antony KM, Ma J, Mitchell K, Racusin DA, Versalovic J, Aagaard K. The preterm placental microbiome varies in association with excess maternal gestational weight gain. Am J Obstet Gynecol. 2015;212:653.e1-653.16.
Yasuda K, Oh K, Ren B, Tickle TL, Franzosa EA, Wachtman LM, et al. Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host Microbe. 2015;17:385–91.
Kahr MK, Suter MA, Ballas J, Ramin SM, Monga M, Lee W, et al. Geospatial analysis of food environment demonstrates associations with gestational diabetes. American Journal of Obstetrics and Gynecology. 2016;214:110.e1-110.e9.
McCurdy CE, Schenk S, Hetrick B, Houck J, Drew BG, Kaye S, et al. Maternal obesity reduces oxidative capacity in fetal skeletal muscle of Japanese macaques. JCI Insight. 2016;1:1–17.
Kahr MK, Antony KM, Delbeccaro M, Hu M, Aagaard KM, Suter MA. Increasing maternal obesity is associated with alterations in both maternal and neonatal thyroid hormone levels. Clinical Endocrinology. 2016;84:551–7.
Gonzalez-Rodriguez P, Cantu J, O’Neil D, Seferovic MD, Goodspeed DM, Suter MA, et al. Alterations in expression of imprinted genes from the H19/IGF2 loci in a multigenerational model of intrauterine growth restriction (IUGR). American Journal of Obstetrics and Gynecology. 2016;214:625.e1-625.e11.
Pew BK, Harris RA, Sbrana E, Guaman MC, Shope C, Chen R, et al. Structural and transcriptomic response to antenatal corticosteroids in an Erk3-null mouse model of respiratory distress. Am J Obstet Gynecol. 2016;215:384.e1–384.e89.
Harris RA, Alcott CE, Sullivan EL, Takahashi D, McCurdy CE, Comstock S, et al. Genomic variants associated with resistance to high fat diet induced obesity in a primate model. Scientific Reports. Nature Publishing Group. 2016;6:1–10.
Kahr MK, Suter MA, Ballas J, Ramphul R, Lubertino G, Hamilton W, et al. Preterm birth and its associations with residence and ambient vehicular traffic exposure. Am J Obstet Gynecol. 2016;215:111.e1-111.e10.
Chu DM, Aagaard KM. Microbiome: Eating for trillions. Nature. England. 2016;532:316–7.
Chu DM, Antony KM, Ma J, Prince AL, Showalter L, Moller M, et al. The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Medicine. Genome Medicine. 2016;8:1–12.
Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, Aagaard KM. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med. 2017;23:314–26.
Wesolowski SR, Mulligan CM, Janssen RC, Baker PR, Bergman BC, D’Alessandro A, et al. Switching obese mothers to a healthy diet improves fetal hypoxemia, hepatic metabolites, and lipotoxicity in non-human primates. Molecular Metabolism. Elsevier GmbH. 2018;18:25–41.
Cheung CY, Roberts VHJ, Frias AE, Brace RA. High-fat diet effects on amniotic fluid volume and amnion aquaporin expression in non-human primates. Physiological Reports. 2018;6:1–9.
Salati JA, Roberts VHJ, Schabel MC, Lo J, Kroenke CD, Lewandowski KS, et al. Maternal high fat diet reversal improves placental hemodynamics in a nonhuman primate model of diet-induced obesity. Int J Obes (Lond). 2019;43:906–16.
Seferovic MD, Beamish CA, Mosser RE, Townsend SE, Pappan K, Poitout V, et al. Increases in bioactive lipids accompany early metabolic changes associated with β-cell expansion in response to short-term high-fat diet. American Journal of Physiology - Endocrinology and Metabolism. 2018;315:e1251–63.
Pace RM, Prince AL, Ma J, Belfort BDW, Harvey AS, Hu M, et al. Modulations in the offspring gut microbiome are refractory to postnatal synbiotic supplementation among juvenile primates. BMC Microbiology. BMC Microbiology. 2018;18:1–13.
Prince AL, Pace RM, Dean T, Takahashi D, Kievit P, Friedman JE, et al. The development and ecology of the Japanese macaque gut microbiome from weaning to early adolescence in association with diet. American Journal of Primatology. 2019;81.
Ma J, Coarfa C, Qin X, Bonnen PE, Milosavljevic A, Versalovic J, et al. MtDNA haplogroup and single nucleotide polymorphisms structure human microbiome communities. BMC Genomics. BMC Genomics. 2014;15:1–14.
Barbour LA, Scifres C, Valent AM, Friedman JE, Buchanan TA, Coustan D, et al. A cautionary response to SMFM statement: pharmacological treatment of gestational diabetes. American Journal of Obstetrics and Gynecology. 2018;219:367.e1-367.e7.
Barbour LA, Farabi SS, Friedman JE, Hirsch NM, Reece MS, Van Pelt RE, et al. Postprandial triglycerides predict newborn fat more strongly than glucose in women with obesity in early pregnancy. Obesity (Silver Spring, Md). 2018;26:1347–56.
Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. 2Sci T ransl Med. 2014;6:263ra158.
Jašarević E, Morrison KE, Bale TL. Sex differences in the gut microbiome - brain axis across the lifespan. Philosophical Transactions of the Royal Society B: Biological Sciences. 2016;371:12–7.
Fujiwara R, Takemura N, Watanabe J, Sonoyama K. Maternal consumption of fructo-oligosaccharide diminishes the severity of skin inflammation in offspring of NC/Nga mice. British Journal of Nutrition. 2010;103:530–8.
Jakobsson HE, Abrahamsson TR, Jenmalm MC, Harris K, Quince C, Jernberg C, et al. Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by Caesarean section. Gut. 2014;63:559–66.
de Theije CGM, Wopereis H, Ramadan M, van Eijndthoven T, Lambert J, Knol J, et al. Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain, Behavior, and Immunity [Internet]. Elsevier Inc.; 2014;37:197–206. Available from: https://doi.org/10.1016/j.bbi.2013.12.005
Arboleya S, Sánchez B, Milani C, Duranti S, Solís G, Fernández N, et al. Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. Journal of Pediatrics. 2015;166:538–44.
Thorburn AN, McKenzie CI, Shen S, Stanley D, MacIa L, Mason LJ, et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nature Communications. 2015;6:1–13.
Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549:528–32.
Jašarević E, Howard CD, Misic AM, Beiting DP, Bale TL. Stress during pregnancy alters temporal and spatial dynamics of the maternal and offspring microbiome in a sex-specific manner. Scientific Reports. Nature Publishing Group. 2017;7:1–13.
Steegenga WT. Mischke M, Lute C, Boekschoten M v., Lendvai A, Pruis MGM, et al. Maternal exposure to a Western-style diet causes differences in intestinal microbiota composition and gene expression of suckling mouse pups. Molecular Nutrition and Food Research. 2017;61:1–17.
Dong T, Hu W, Zhou X, Lin H, Lan L, Hang B, et al. Prenatal exposure to maternal smoking during pregnancy and attention-deficit/hyperactivity disorder in offspring: a meta-analysis. Reproductive Toxicology [Internet]. Elsevier Inc.; 2018;76:63–70. Available from: https://doi.org/10.1016/j.reprotox.2017.12.010
Tapiainen T, Paalanne N, Tejesvi M v., Koivusaari P, Korpela K, Pokka T, et al. Maternal influence on the fetal microbiome in a population-based study of the first-pass meconium. Pediatric Research. 2018;84:371–9.
Lindheim L, Manti M, Fornes R, Bashir M, Czarnewski P, Diaz OE, et al. Reproductive and behavior dysfunction induced by maternal androgen exposure and obesity is likely not gut microbiome-mediated. Journal of the Endocrine Society. 2018;2:1363–80.
Dechartres J, Pawluski JL, Gueguen MM, Jablaoui A, Maguin E, Rhimi M, et al. Glyphosate and glyphosate-based herbicide exposure during the peripartum period affects maternal brain plasticity, maternal behaviour and microbiome. Journal of Neuroendocrinology. 2019;31:1–17.
Olivier-Van Stichelen S, Rother KI, Hanover JA. Maternal exposure to non-nutritive sweeteners impacts progeny’s metabolism and microbiome. Frontiers in Microbiology. 2019;10:1–13.
Luo T, Wang C, Pan Z, Jin C, Fu Z, Jin Y. Maternal polystyrene microplastic exposure during gestation and lactation altered metabolic homeostasis in the dams and their F1 and F2 offspring. Environmental Science and Technology. 2019;53:10978–92.
Li H, Xiao B, Zhang Y, Xiao S, Luo J, Huang W. Impact of maternal intrapartum antibiotics on the initial oral microbiome of neonates. Pediatrics and Neonatology [Internet]. Elsevier Taiwan LLC; 2019;60:654–61. Available from: https://doi.org/10.1016/j.pedneo.2019.03.011
Dong T, Guan Q, Hu W, Zhang M, Zhang Y, Chen M, et al. Prenatal exposure to glufosinate ammonium disturbs gut microbiome and induces behavioral abnormalities in mice. Journal of Hazardous Materials [Internet]. Elsevier; 2020;389:1–10. Available from: https://doi.org/10.1016/j.jhazmat.2020.122152
Zhou P, Zhou Y, Liu B, Jin Z, Zhuang X, Dai W, et al. Perinatal antibiotic exposure affects the transmission between maternal and neonatal microbiota and is associated with early-onset sepsis. mSphere. 2020;5:1–6.
Johanson SM, Swann JR, Umu ÖCO, Aleksandersen M, Müller MHB, Berntsen HF, et al. Maternal exposure to a human relevant mixture of persistent organic pollutants reduces colorectal carcinogenesis in A/J Min/+ mice. Chemosphere. 2020;252:1–14.
Pu Y, Yang J, Chang L, Qu Y, Wang S, Zhang K, et al. Maternal glyphosate exposure causes autism-like behaviors in offspring through increased expression of soluble epoxide hydrolase. Proceedings of the National Academy of Sciences of the United States of America. 2021;118:11753–9.
de Agüero MG, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The maternal microbiota drives early postnatal innate immune development. Science [Internet]. 2016;351:1296 LP – 1302. Available from: http://science.sciencemag.org/content/351/6279/1296.abstract
Hesselmar B, Hicke-Roberts A, Lundell AC, Adlerberth I, Rudin A, Saalman R, et al. Pet-keeping in early life reduces the risk of allergy in a dose-dependent fashion. PLoS ONE. 2018;13:1–13.
Bradberry SM, Proudfoot AT, Vale JA. Glyphosate poisoning. Toxicological reviews. New Zealand. 2004;23:159–67.
Duarte-Salles T, Mendez MA, Meltzer HM, Alexander J, Haugen M. Dietary benzo(a)pyrene intake during pregnancy and birth weight: associations modified by vitamin C intakes in the Norwegian Mother and Child Cohort Study (MoBa). Environment International [Internet]. Elsevier Ltd. 2013;60:217–23 Available from: https://doi.org/10.1016/j.envint.2013.08.016.
Langlois P, Hoyt A, Desrosiers T, Lupo P, Lawson C, Waters M, et al. Maternal occupational exposure to polycyclic aromatic hydrocarbons and small for gestational age offspring. Occup Environ Med. 2014;71:529–35.
Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, et al. Relation of DNA methylation of 5′-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS ONE. 2009;4:e4488.
Miller R, Yan Z, Maher C, Zhang H, Gudsnuk K, McDonald J, et al. Impact of prenatal polycyclic aromatic hydrocarbon exposure on behavior, cortical gene expression and DNA methylation of the Bdnf gene. Neuroepigenetics. 2016;5:11–8.
Suter MA, Aagaard KM, Coarfa C, Robertson M, Zhou G, Jackson BP, et al. Association between elevated placental polycyclic aromatic hydrocarbons (PAHs) and PAH-DNA adducts from Superfund sites in Harris County, and increased risk of preterm birth (PTB). Biochemical and Biophysical Research Communications [Internet]. Elsevier Ltd; 2019;516:344–9. Available from: https://doi.org/10.1016/j.bbrc.2019.06.049
Polanska K, Dettbarn G, Jurewicz J, Sobala W, Magnus P, Seidel A, et al. Effect of prenatal polycyclic aromatic hydrocarbons exposure on birth outcomes: The Polish mother and child cohort study. BioMed Research International. Hindawi Publishing Corporation. 2014;2014.
Thakur V, Liang Y, Lingappan K, Jiang W, Wang L, Barrios R, et al. Increased susceptibility to hyperoxic lung injury and alveolar simplification in newborn rats by prenatal administration of benzo[a]pyrene. Toxicol Lett. 2014;230:322–32.
Jedrychowski WA, Perera FP, Camann D, Spengler J, Butscher M, Mroz E, et al. Prenatal exposure to polycyclic aromatic hydrocarbons and cognitive dysfunction in children. Environmental Science and Pollution Research. 2015;22:3631–9.
Lovasi GS, Eldred-Skemp N, Quinn JW, Wen CH, Rauh VA, Rundle A, et al. Neighborhood social context and individual polycyclic aromatic hydrocarbon exposures sssociated with child cognitive test scores. Journal of Child and Family Studies. 2014;23:785–99.
Jedrychowski W, Perera F, Majewska R, Camman D, Spengler J, Mroz E, et al. Separate and joint effects of tranplacental and aromatic hydrocarbons: prospective birth cohort study on wheezing events. Pediatr Pulmonol. 2014;49:162–72.
Zhang X, Li X, Jing Y, Fang X, Zhang X, Lei B, et al. Transplacental transfer of polycyclic aromatic hydrocarbons in paired samples of maternal serum, umbilical cord serum, and placenta in Shanghai, China. Environmental Pollution [Internet]. Elsevier Ltd. 2017;222:267–75 Available from: https://doi.org/10.1016/j.envpol.2016.12.046.
Henkler F, Stolpmann K, Luch A. Exposure to polycyclic aromatic hydrocarbons: bulky DNA adducts and cellular responses. Experientia supplementum (2012). Switzerland. 2012;101:107–31.
Herbstman JB, Tang D, Zhu D, Qu L, Sjödin A, Li Z, et al. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[a]pyrene-DNA adducts, and genomic DNA methylation in cord blood. Environmental Health Perspectives. 2012;120:733–8.
Claus SP, Guillou H, Ellero-Simatos S. The gut microbiota: a major player in the toxicity of environmental pollutants? npj Biofilms and Microbiomes. Nature Publishing Group. 2016;2:1–12.
Van de Wiele T, Vanhaecke L, Boeckaert C, Peru K, Headley J, Verstraete W, et al. Human colon microbiota transform polycyclic aromatic hydrocarbons to estrogenic metabolites. Environmental Health Perspectives. 2005;113:6–10.
Sahay D, Lloyd SE, Rivera JA, Jezioro J, McDonald JD, Pitiranggon M, Yan B, Szabolcs M, Terry MB, Miller RL. Prenatal polycyclic aromatic hydrocarbons, altered ERα pathway-related methylation and expression, and mammary epithelial cell proliferation in offspring and grandoffspring adult mice. Environ Res. 2021;196:110961. Available from: https://doi.org/10.1016/j.envres.2021.110961.
Abou-Donia MB, El-Masry EM, Abdel-Rahman AA, McLendon RE, Schiffman SS. Splenda alters gut microflora and increases intestinal P-Glycoprotein and Cytochrome P-450 in male rats. Journal of Toxicology and Environmental Health, Part A [Internet]. Taylor & Francis; 2008;71:1415–29. Available from: https://doi.org/10.1080/15287390802328630
Simon BR, Parlee SD, Learman BS, Mori H, Scheller EL, Cawthorn WP, et al. Artificial sweeteners stimulate adipogenesis and suppress lipolysis independently of sweet taste receptors. Journal of Biological Chemistry. 2013;288:32475–89.
Jang H-J, Kokrashvili Z, Theodorakis MJ, Carlson OD, Kim B-J, Zhou J, et al. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proceedings of the National Academy of Sciences [Internet]. 2007;104:15069 LP – 15074. Available from: http://www.pnas.org/content/104/38/15069.abstract
Tellez LA, Han W, Zhang X, Ferreira TL, Perez IO, Shammah-Lagnado SJ, et al. Separate circuitries encode the hedonic and nutritional values of sugar. Nature Neuroscience [Internet]. 2016;19:465–70. Available from. https://doi.org/10.1038/nn.4224.
Tarabulsy GM, Pearson J, Vaillancourt-Morel M-P, Bussières E-L, Madigan S, Lemelin J-P, et al. Meta-analytic findings of the relation between maternal prenatal stress and anxiety and child cognitive outcome. Journal of Developmental & Behavioral Pediatrics [Internet]. 2014;35:38–43 Available from: https://journals.lww.com/jrnldbp/Fulltext/2014/01000/Meta_Analytic_Findings_of_the_Relation_Between.5.aspx.
van den Heuvel MI, Hect JL, Smarr BL, Qawasmeh T, Kriegsfeld LJ, Barcelona J, et al. Maternal stress during pregnancy alters fetal cortico-cerebellar connectivity in utero and increases child sleep problems after birth. Scientific Reports [Internet]. Nature Publishing Group UK; 2021;11:1–12. Available from: https://doi.org/10.1038/s41598-021-81681-y
Shen Q, Zhang Q, Zhao J, Huang Z, Wang X, Ni M, et al. Association between maternal perceived stress in all trimesters of pregnancy and infant atopic dermatitis: a prospective birth cohort study. Frontiers in Pediatrics. 2020;8:1–7.
Nazzari S, Fearon P, Rice F, Ciceri F, Molteni M, Frigerio A. Neuroendocrine and immune markers of maternal stress during pregnancy and infant cognitive development. Developmental Psychobiology. 2020;62:1100–10.
King S, Laplante DP. The effects of prenatal maternal stress on children’s cognitive development: Project Ice Storm. Stress (Amsterdam, Netherlands). England. 2005;8:35–45.
Bush NR, Savitz J, Coccia M, Jones-Mason K, Adler N, Boyce WT, et al. Maternal stress during pregnancy predicts infant infectious and noninfectious illness. Journal of Pediatrics [Internet]. Elsevier Inc; 2021;228:117-125.e2. Available from: https://doi.org/10.1016/j.jpeds.2020.08.041
Arrieta M-C, Sadarangani M, Brown EM, Russell SL, Nimmo M, Dean J, et al. A humanized microbiota mouse model of ovalbumin-induced lung inflammation. Gut microbes. 2016;7:342–52.
Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Science Translational Medicine. 2015;7.
Deshmukh H, Liu Y, Menkiti O, Mei J, Dai N, O’Leary C, et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med. 2014;20:524–30.
Gensollen T, Iyer SS, Kasper DL, Blumberg RS, Medical H. How colonization by microbiota in early life shapes the immune system. Science (New York, NY) [Internet]. 2016;352:539–44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27126036%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid = PMC5050524
Appropriate technology for birth. Lancet (London, England). England; 1985;2:436–437.
Martin JA, Hamilton BE, Osterman MJ, Driscoll AK. Births: Final Data for 2019. National Vital Statistics Reports. 2021;70:1–51.
Betran AP. WHO statement on caesarean section rates. BJOG. 2016;123(5):667-70. Available from: https://doi.org/10.1111/1471-0528.13526.
Zhang T, Sidorchuk A, Sevilla-Cermeño L, Vilaplana-Pérez A, Chang Z, Larsson H, et al. Association of Cesarean delivery with risk of neurodevelopmental and psychiatric disorders in the offspring: a systematic review and meta-analysis. JAMA network open. 2019;2:e1910236.
Polidano C, Zhu A, Bornstein JC. The relation between cesarean birth and child cognitive development. Scientific reports. 2017;7:11483.
Thavagnanam S, Fleming J, Bromley A, Shields MD, Cardwell CR. A meta-analysis of the association between Caesarean section and childhood asthma. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. England. 2008;38:629–33.
Huh SY, Rifas-Shiman SL, Zera CA, Rich Edwards JW, Oken E, Weiss ST, et al. Delivery by caesarean section and risk of obesity in preschool age children: a prospective cohort study. Arch Dis Child. 2012;97:610–6.
Yuan C, Gaskins AJ, Blaine AI, Zhang C, Gillman MW, Missmer SA, et al. Cesarean birth and risk of offspring obesity in childhood, adolescence and early adulthood. JAMA Pediatr. 2016;170:e162385.
Bager P, Wohlfahrt J, Westergaard T. Caesarean delivery and risk of atopy and allergic disease: meta-analyses. Clinical and Experimental Allergy. 2008;38:634–42.
Kolokotroni O, Middleton N, Gavatha M, Lamnisos D, Priftis KN, Yiallouros PK. Asthma and atopy in children born by caesarean section: effect modification by family history of allergies - a population based cross-sectional study. BMC Pediatrics [Internet]. BMC Pediatrics; 2012;12:1. Available from: BMC Pediatrics
Sevelsted A, Stokholm J, Bisgaard H. Risk of asthma from Cesarean delivery depends on membrane rupture. Journal of Pediatrics [Internet]. Elsevier Inc.; 2016;171:38-42.e4. Available from: https://doi.org/10.1016/j.jpeds.2015.12.066
Decker E, Engelmann G, Findeisen A, Gerner P, Laaß M, Ney D, et al. Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Pediatrics. 2010;125:e1433–40.
Cardwell CR, Stene LC, Joner G, Cinek O, Svensson J, Goldacre MJ, et al. Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: a meta-analysis of observational studies. Diabetologia. 2008;51:726–35.
Phillips J, Gill N, Sikdar K, Penney S, Newhook LA. History of cesarean section associated with childhood onset of T1DM in Newfoundland and Labrador. Canada. Journal of Environmental and Public Health. 2012;2012:1–6.
Barros AJD, Santos LP, Wehrmeister F, Motta JVDS, Matijasevich A, Santos IS, et al. Caesarean section and adiposity at 6, 18 and 30 years of age: results from three Pelotas (Brazil) birth cohorts. BMC Public Health. BMC Public Health. 2017;17:1–9.
Rifas-Shiman SL, Huh SY, Martin RM, Kramer M, Patel R, Bogdanovich N, et al. Delivery by caesarean section and offspring adiposity and cardio-metabolic health at ages 6.5, 11.5 and 16 years: results from the PROBIT cohort in Belarus. Pediatric obesity. England; 2021;e12783.
Masukume G, McCarthy FP, Baker PN, Kenny LC, Morton SMB, Murray DM, et al. Association between caesarean section delivery and obesity in childhood: a longitudinal cohort study in Ireland. BMJ Open. 2019;9:1–8.
Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11971–5.
Mueller NT, Bakacs E, Combellick J, Grigoryan Z, Dominguez-Bello MG. The infant microbiome development: mom matters. Trends in molecular medicine. 2015;21:109–17.
Dominguez-Bello MG, de Jesus-Laboy KM, Shen N, Cox LM, Amir A, Gonzalez A, et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med. 2016;22:250–3.
Shao Y, Forster SC, Tsaliki E, Vervier K, Strang A, Simpson N, et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature [Internet]. 2019;574:117–21. Available from: https://doi.org/10.1038/s41586-019-1560-1
Mueller NT, Shin H, Pizoni A, Werlang IC, Matte U, Goldani MZ, et al. Delivery mode and the transition of pioneering gut-microbiota structure, composition and predicted metabolic function. Genes. 2017;8:364.
Burrage S. Bacteria in the supposedly sterile meconium. Journal of Bacteriology: Twenty-eighth Annual Meeting of the Society of American Bacteriologists; 1926. p. 1–71.
Hall IC, O’Toole E. Bacterial flora of first specimens of meconium passed by fifty newborn infants. Am J Dis Child. 1934;47:1279–85.
Snyder ML. The bacterial flora of meconium specimenscollected from sixty-four infants within four hours after delivery. Journal of Pediatrics. 1936;9:624–32.
Kovalovszki L. Isolation of aerobic bacteria from the placenta. Acta paediatrica Academiae Scientiarum Hungaricae. 1982;23:357–60.
Hitti J, Riley D, Krohn MA, Hillier SL, Agnew K, Krieger J, et al. Broad-spectrum bacterial ribosomal rna polymerase chain reaction for the detection of amniotic fluid infection among women in preterm labor. Infectious Diseases in Obstetrics and Gynecology. 1997;4:48–9.
Gardella C, Riley DE, Hitti J, Agnew K, Krieger JN, Eschenbach D. Identification and sequencing of bacterial rDNAs in culture-negative amniotic fluid from women in premature labor. American Journal of Perinatology. 2004;21:319–23.
León R, Silva N, Ovalle A, Chaparro A, Ahumada A, Gajardo M, et al. Detection of Porphyromonas gingivalis in the amniotic fluid in pregnant women with a diagnosis of threatened premature labor. Journal of Periodontology. 2007;78:1249–55.
Pettker CM, Buhimschi IA, Magloire LK, Sfakianaki AK, Hamar BD, Buhimschi CS. Value of placental microbial evaluation in diagnosing intra-amniotic infection. Obstetrics and Gynecology. 2007;109:739–49.
Onderdonk AB, Delaney ML, DuBois AM, Allred EN, Leviton A. Detection of bacteria in placental tissues obtained from extremely low gestational age neonates. American Journal of Obstetrics and Gynecology. 2008;198:110.e1-110.e7.
DiGiulio DB, Romero R, Amogan HP, Kusanovic JP, Bik EM, Gotsch F, et al. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PLoS ONE. 2008;3:1–10.
Jiménez E, Marín ML, Martín R, Odriozola JM, Olivares M, Xaus J, et al. Is meconium from healthy newborns actually sterile? Research in Microbiology. 2008;159:187–93.
Satokari R, Grönroos T, Laitinen K, Salminen S, Isolauri E. Bifidobacterium and Lactobacillus DNA in the human placenta. Letters in Applied Microbiology. 2009;48:8–12.
Fardini Y, Chung P, Dumm R, Joshi N, Han YW. Transmission of diverse oral bacteria to murine placenta: Evidence for the oral microbiome as a potential source of intrauterine infection. Infection and Immunity. 2010;78:1789–96.
DiGiulio D, Romero R, Kusanovic J, Gómez R, Kim C, Seok K, et al. Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am J Reprod Immunol. 2010;64:38–57.
Stout MJ, Conlon B, Landeau M, Lee I, Bower C, Zhao Q, et al. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. American Journal of Obstetrics and Gynecology. 2013;208:226.e1-226.e7.
Moles L, Gómez M, Heilig H, Bustos G, Fuentes S, de Vos W, et al. Bacterial diversity in meconium of preterm neonates and evolution of their fecal microbiota during the first month of life. PLoS ONE. 2013;8.
Doyle RM, Alber DG, Jones HE, Harris K, Fitzgerald F, Peebles D, et al. Term and preterm labour are associated with distinct microbial community structures in placental membranes which are independent of mode of delivery. Placenta. Elsevier Ltd. 2014;35:1099–101.
Cao B, Mysorekar IU. Intracellular bacteria in placental basal plate localize to extravillous trophoblasts. Placenta. Elsevier Ltd. 2014;35:139–42.
Zheng J, Xiao X, Zhang Q, Mao L, Yu M, Xu J. The placental microbiome varies in association with low birth weight in full-term neonates. Nutrients. 2015;7:6924–37.
Collado MC, Rautava S, Aakko J, Isolauri E, Salminen S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Scientific Reports [Internet]. Nature Publishing Group. 2016;6:1–13 Available from: https://doi.org/10.1038/srep23129.
Prince AL, Ma J, Kannan PS, Alvarez M, Gisslen T, Harris RA, et al. The placental membrane microbiome is altered among subjects with spontaneous preterm birth with and without chorioamnionitis. Am J Obstet Gynecol. 2016;214:627.e1-627.e16.
Bassols J, Serino M, Carreras-Badosa G, Burcelin R, Blasco-Baque V, Lopez-Bermejo A, et al. Gestational diabetes is associated with changes in placental microbiota and microbiome. Pediatric Research. 2016;80:777–84.
Gomez-Arango LF, Barrett HL, McIntyre HD, Callaway LK, Morrison M, Nitert MD. Contributions of the maternal oral and gut microbiome to placental microbial colonization in overweight and obese pregnant women. Scientific Reports. Springer US. 2017;7:1–10.
Doyle RM, Harris K, Kamiza S, Harjunmaa U, Ashorn U, Nkhoma M, et al. Bacterial communities found in placental tissues are associated with severe chorioamnionitis and adverse birth outcomes. PLoS ONE. 2017;12:1–23.
Zheng J, Xiao XH, Zhang Q, Mao LL, Yu M, Xu JP, et al. Correlation of placental microbiota with fetal macrosomia and clinical characteristics in mothers and newborns. Oncotarget. 2017;8:82314–25.
Parnell LA, Briggs CM, Cao B, Delannoy-Bruno O, Schrieffer AE, Mysorekar IU. Microbial communities in placentas from term normal pregnancy exhibit spatially variable profiles. Scientific Reports [Internet]. Springer US. 2017;7:1–11. Available from: https://doi.org/10.1038/s41598-017-11514-4.
Zhu L, Luo F, Hu W, Han Y, Wang Y, Zheng H, et al. Bacterial communities in the womb during healthy pregnancy. Frontiers in Microbiology. 2018;9:1–6.
Tuominen H, Rautava S, Syrjänen S, Collado MC, Rautava J. HPV infection and bacterial microbiota in the placenta, uterine cervix and oral mucosa. Scientific Reports. 2018;8:1–11.
Seferovic MD, Pace RM, Carroll M, Belfort B, Major AM, Chu DM, et al. Visualization of microbes by 16S in situ hybridization in term and preterm placentas without intraamniotic infection. American Journal of Obstetrics and Gynecology. Elsevier Inc.; 2019;221:146.e1-146.e23.
Stinson LF, Boyce MC, Payne MS, Keelan JA. The not-so-sterile womb: Evidence that the human fetus is exposed to bacteria prior to birth. Frontiers in Microbiology. 2019;10:1–15.
Yu K, Rodriguez MD, Paul Z, Gordon E, Rice K, Triplett EW, et al. Proof of principle: Physiological transfer of small numbers of bacteria from mother to fetus in late-gestation pregnant sheep. PLoS ONE. 2019;14:4–15.
Leoni C, Ceci O, Manzari C, Fosso B, Volpicella M, Ferrari A, et al. Human endometrial microbiota at term of normal pregnancies. Genes. 2019;10:1–11.
Liu CJ, Liang X, Niu ZY, Jin Q, Zeng XQ, Wang WX, et al. Is the delivery mode a critical factor for the microbial communities in the meconium. EBioMedicine. Elsevier B.V. 2019;49:354–63.
Younge N, Brandon D, Seed PC, Younge N, Mccann JR, Ballard J, et al. Fetal exposure to the maternal microbiota in humans and mice. JCI Insight. 2019;4:e127806.
Tuominen H, Collado MC, Rautava J, Syrjänen S, Rautava S. Composition and maternal origin of the neonatal oral cavity microbiota. Journal of Oral Microbiology. Taylor & Francis; 2019;11.
Zakošek Pipan M, Kajdič L, Kalin A, Plavec T, Zdovc I. Do newborn puppies have their own microbiota at birth? Influence of type of birth on newborn puppy microbiota. Theriogenology. 2020;152:18–28.
Hockney R, Waring GJ, Taylor G, Cummings SP, Robson SC, Orr CH, et al. Fetal membrane bacterial load is increased in histologically confirmed inflammatory chorioamnionitis: A retrospective cohort study: Fetal membrane bacterial load is increased in HCA. Placenta [Internet]. Elsevier Ltd; 2020;91:43–51. Available from: https://doi.org/10.1016/j.placenta.2020.01.006
Chen X, Li P, Liu M, Zheng H, He Y, Chen MX, et al. Gut dysbiosis induces the development of pre-eclampsia through bacterial translocation. Gut. 2020;69:513–22.
Rackaityte E, Halkias J, Fukui EM, Mendoza VF, Hayzelden C, Crawford ED, et al. Viable bacterial colonization is highly limited in the human intestine in utero. Nature Medicine [Internet]. Springer US. 2020;26:599–607 Available from: https://doi.org/10.1038/s41591-020-0761-3.
Benny PA, Al-Akwaa FM, Dirkx C, Schlueter RJ, Wolfgruber TK, Chern IY, et al. Placentas delivered by pre-pregnant obese women have reduced abundance and diversity in the microbiome. The FASEB Journal. 2021;35:1–8.
Bi Y, Tu Y, Zhang N, Wang S, Zhang F, Suen G, et al. Multiomics analysis reveals the presence of a microbiome in the gut of fetal lambs. Gut. 2021;70:853–64.
Mishra A, Lai GC, Yao LJ, Aung TT, Shental N, Rotter-Maskowitz A, et al. Microbial exposure during early human development primes fetal immune cells. Cell [Internet]. 2021;184:1–16 Available from: https://www.sciencedirect.com/science/article/pii/S0092867421005742.
Moya-Pérez A, Luczynski P, Renes IB, Wang S, Borre Y, Ryan CA, et al. Intervention strategies for cesarean section- induced alterations in the microbiota-gut-brain axis. Nutrition Reviews. 2017;75:225–40.
Shaterian N, Abdi F, Ghavidel N, Alidost F. Role of cesarean section in the development of neonatal gut microbiota: a systematic review. Open Medicine (Poland). 2021;16:624–39.
Martinez KA, Devlin JC, Lacher CR, Yin Y, Cai Y, Wang J, et al. Increased weight gain by C-section: functional significance of the primordial microbiome. Science Advances. 2017;3:1–5.
Niu J, Xu L, Qian Y, Sun Z, Yu D, Huang J, et al. Evolution of the gut microbiome in rarly childhood: a cross-sectional study of Chinese children. Frontiers in Microbiology. 2020;11:1–16.
Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host and Microbe. 2015;17:690–703.
Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. United States. 2006;118:511–21.
Vatanen T, Kostic AD, D’Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165:842–53.
Azad MB, Konya T, Persaud RR, Guttman DS, Chari RS, Field CJ, et al. Impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: a prospective cohort study. BJOG : an international journal of obstetrics and gynaecology. England. 2016;123:983–93.
Kim D, Hofstaedter CE, Zhao C, Mattei L, Tanes C, Clarke E, et al. Optimizing methods and dodging pitfalls in microbiome research. Microbiome. Microbiome. 2017;5:1–14.
Stewart CJ, Embleton ND, Clements E, Luna PN, Smith DP, Fofanova TY, et al. Cesarean or vaginal birth does not impact the longitudinal development of the gut microbiome in a cohort of exclusively preterm infants. Frontiers in Microbiology. 2017;8:1–9.
Coker MO, Laue HE, Hoen AG, Hilliard M, Dade E, Li Z, et al. Infant feeding alters the longitudinal impact of birth mode on the development of the gut microbiota in the first year of life. Frontiers in microbiology. 2021;12:642197.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods. 2016;13:581–3.
Hugenholtz P, Skarshewski A, Parks DH. Genome-based microbial taxonomy coming of age. Cold Spring Harbor perspectives in biology [Internet]. Cold Spring Harbor Laboratory Press; 2016;8:a018085. Available from: https://pubmed.ncbi.nlm.nih.gov/26988968
Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nature methods. 2018;15:962–8.
Yan Y, Nguyen LH, Franzosa EA, Huttenhower C. Strain-level epidemiology of microbial communities and the human microbiome. Genome medicine. 2020;12:71.
Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL. Analysis of the microbiome: advantages of whole genome shotgun versus 16S amplicon sequencing. Biochemical and biophysical research communications. 2016;469:967–77.
Ma J, Prince A, Aagaard KM. Use of whole genome shotgun metagenomics: a practical guide for the microbiome-minded physician scientist. Seminars in reproductive medicine. United States. 2014;32:5–13.
Maqsood R, Rodgers R, Rodriguez C, Handley SA, Ndao IM, Tarr PI, et al. Discordant transmission of bacteria and viruses from mothers to babies at birth. Microbiome. Microbiome. 2019;7:1–13.
Bolte EE, Aagaard KM. Turning the “phage” on malnutrition and stunting. Cell Host and Microbe [Internet]. Elsevier Inc.; 2020;27:159–61. Available from: https://doi.org/10.1016/j.chom.2020.01.020
Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705–21.
Nuttall G, Thierfelder H. No Title. Physiological Chem. 1896;21:109–21.
Reyniers J, Trexler P, Ervin R. Rearing germ-free albino rats. Lobund reports. United States. 1946:1–84.
Pleasants JR. Rearing germfree Cesarean-born rats, mice, and rabbits through weaning. Ann N Y Acad Sci. 1959;78:116–26.
Yoshida T, Pleasants J, Reddy B, Wostmann B. Efficiency of digestion in germ -free and conventional rabbits. Br J Nutr. 1968;22:723–37.
Reddy BS, Pleasants JR, Wostmann BS. Effect of dietary carbohydrates on intestinal disaccharidases in germfree and conventional rats. The Journal of nutrition. United States. 1968;95:413–9.
Coates M, Fuller R. The gnotobiotic animal in the study of gut microbiology. In: Clark R, Bauchop T, editors. Microbial ecology of the gut. London: Academic Press; 1977. p. 311–46.
Berg RD. The indigenous gastrointestinal microflora. Trends in Microbiology. 1996;4:430–5.
Wostmann BS, Bruckner-Kardoss E, Knight PLJ. Cecal enlargement, cardiac output, and O2 consumption in germfree rats. Proceedings of the Society for Experimental Biology and Medicine Society for Experimental Biology and Medicine (New York, NY). United States. 1968;128:137–41.
Pleasants J. Animal production and rearing. I. Small laboratory animals. In: Coates M, editor. The Germfree Animal in Research. London: Academic Press; 1968. p. 113–125.
Kellogg TF, Wostmann BS. Fecal neutral steroids and bile acids from germfree rats. Journal of Lipid Research. 1969;10:495–503.
Wostmann BS, Olson GB. Persistence of primary antibody formation caused by the absence of antigen of microbial origin in the germ-free chicken. Immunology. 1969;17:199–206.
Wostmann BS, Pleasants JR, Bealmear P, Kincade PW. Serum proteins and lymphoid tissues in germ-free mice fed a chemically defined, water soluble, low molecular weight diet. Immunology. 1970;19:443–8.
Wostmann BS. Intestinal bile acids and cholesterol absorption in the germfree rat. The Journal of Nutrition [Internet]. 1973;103:982–90. Available from: https://doi.org/10.1093/jn/103.7.982
Reddy B, Pleasants J. Wostmann B. Metabolic enzymes in liver and kidney of the germfree rat. 1973;320:1–8.
Wostmann BS. Nutrition and metabolism of the germfree mammal. World review of nutrition and dietetics. Switzerland. 1975;22:40–92.
Fox AC, McConnell KW, Yoseph BP, Breed E, Liang Z, Clark AT, et al. The endogenous bacteria alter gut epithelial apoptosis and decrease mortality following pseudomonas aeruginosa pneumonia. Shock. 2012;38:508–14.
Luczynski P, Neufeld KAMV, Oriach CS, Clarke G, Dinan TG, Cryan JF. Growing up in a bubble: using germ-free animals to assess the influence of the gut microbiota on brain and behavior. International Journal of Neuropsychopharmacology. 2016;19:1–17.
Selwyn FP, Cheng SL, Klaassen CD, Cui JY. Regulation of hepatic drug-metabolizing enzymes in germ-free mice by conventionalization and probiotics. Drug Metabolism and Disposition. 2016;44:262–74.
Stepankova R, Šinkora J, Hudcovic T, Kozáková H, Tlaskalová-Hogenová H. Differences in development of lymphocyte subpopulations from gut-associated lymphatic tissue (GALT) of germfree and conventional rats: effect of aging. Folia Microbiologica. 1998;43:531–4.
Williams AM, Probert CSJ, Stepankova R, Tlaskalova-Hogenova H, Phillips A, Bland PW. Effects of microflora on the neonatal development of gut mucosal T cells and myeloid cells in the mouse. Immunology. 2006;119:470–8.
Costa MC, Santos JRA, Ribeiro MJA, Freitas GJC de, Bastos RW, Ferreira GF, et al. The absence of microbiota delays the inflammatory response to Cryptococcus gattii. International Journal of Medical Microbiology. Elsevier GmbH.; 2016;306:187–95.
Midtvedt T, Gustafsson BE. Digestion of dead bacteria by germ-free rats. Current Microbiology. 1981;6:13–5.
Taylor DM, Read L, Neal DL. Determining the viability of faecal bacteria present in germ-free mice. Laboratory Animals. 1986;20:22–6.
Fontaine CA, Skorupski AM, Vowles CJ, Anderson NE, Poe SA, Eaton KA. How free of germs is germ-free? Detection of bacterial contamination in a germ free mouse unit. Gut Microbes. 2015;6:225–33.
Aiba Y, Ishikawa H, Tokunaga M, Komatsu Y. Anti-Helicobacter pylori activity of non-living, heat-killed form of lactobacilli including Lactobacillus johnsonii No.1088. FEMS microbiology letters. England; 2017;364.
Nakaita Y, Kaneda H, Shigyo T. Heat-Killed Lactobacillus brevis SBC8803 induces serotonin release from intestinal cells. Food and Nutrition Sciences. 2013;04:767–71.
Engevik MA, Luk B, Chang-Graham AL, Hall A, Herrmann B, Ruan W, et al. Bifidobacterium dentium fortifies the intestinal mucus layer via autophagy and calcium signaling pathways. mBio. 2019;10.
Engevik MA, Luck B, Visuthranukul C, Ihekweazu FD, Engevik AC, Shi Z, et al. Human-derived Bifidobacterium dentium modulates the mammalian serotonergic system and gut–brain axis. Cellular and Molecular Gastroenterology and Hepatology [Internet]. Elsevier Inc; 2020;11:221–48. Available from: https://doi.org/10.1016/j.jcmgh.2020.08.002
Mackie RI, Sghir A, Gaskins HR. Developmental microbial ecology of the neonatal gastrointestinal tract. American Journal of Clinical Nutrition. 1999;69.
Chu DM, Meyer KM, Prince AL, Aagaard KM. Impact of maternal nutrition in pregnancy and lactation on offspring gut microbial composition and function. Gut Microbes. Taylor & Francis. 2016;7:459–70.
Brooks B, Firek BA, Miller CS, Sharon I, Thomas BC, Baker R, et al. Microbes in the neonatal intensive care unit resemble those found in the gut of premature infants. Microbiome. 2014;2:1–16.
Butler B. Value of endometrial cultures in sterility investigation. Fertility and sterility [Internet]. Elsevier Masson SAS. 1958;9:269–73 Available from: https://doi.org/10.1016/S0015-0282(16)33070-9.
Stroup P. Amniotic fluid infection and the intact fetal membrane. Obstetrics & Gynecology. 1962;19:736–9.
Mishell DR, Bell JH, Good RG, Moyer DL. The intrauterine device: a bacteriologic study of the endometrial cavity. American journal of obstetrics and gynecology [Internet]. Elsevier Masson SAS. 1966;96:119–26 Available from: https://doi.org/10.1016/S0002-9378(16)34650-6.
Harwick HJ, Iuppa JB, Fekety FR. Microorganisms and amniotic fluid. Obstetrics & Gynecology. 1969;33:256–9.
Prevedourakis C, Papadimitriou G, Ioannidou A. Isolation of pathogenic bacteria in the amniotic fluid during pregnancy and labor. American Journal of Obstetrics & Gynecology1. 1970;106:400–2.
Prevedourakis C, Strigou-Charalabis E, Kaskarelis D. Bacterial Invasion of Amniotic Cavity During Pregnancy and Labor. Obstetrics & Gynecology Survey. 1971;26:577–8.
Lewis JF, Johnson P, Miller P. Evaluation of amniotic fluid for aerobic and anaerobic bacteria. American Journal of Clinical Pathology. 1976;65:58–63.
Aquino TI, Zhang J, Kraus FT, Knefel R, Taff T. Subchorionic fibrin cultures for bacteriologic study of the placenta. Obstetrical and Gynecological Survey. 1984;39:689–91.
Teisala K. Endometrial microbial flora of hysterectomy specimens. European Journal of Obstetrics and Gynecology and Reproductive Biology. 1987;26:151–5.
Romero R, Miranda J, Chaiworapongsa T, Chaemsaithong P, Gotsch F, Dong Z, et al. Sterile intra-amniotic inflammation in asymptomatic patients with a sonographic short cervix: prevalence and clinical significance. Journal of Maternal-Fetal and Neonatal Medicine. 2015;28:1343–59.
Lauder AP, Roche AM, Sherrill-Mix S, Bailey A, Laughlin AL, Bittinger K, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome [Internet]. Microbiome. 2016;4:1–11 Available from: https://doi.org/10.1186/s40168-016-0172-3.
Leiby JS, McCormick K, Sherrill-Mix S, Clarke EL, Kessler LR, Taylor LJ, et al. Lack of detection of a human placenta microbiome in samples from preterm and term deliveries. Microbiome. Microbiome. 2018;6:1–11.
Leon LJ, Doyle R, Diez-Benavente E, Clark TG, Klein N, Stanier P, et al. Enrichment of clinically relevant organisms in spontaneous preterm-delivered placentas and reagent contamination across all clinical groups in a large pregnancy cohort in the United Kingdom. Applied and Environmental Microbiology. 2018;84:1–13.
Rehbinder EM, Lødrup Carlsen KC, Staff AC, Angell IL, Landrø L, Hilde K, et al. Is amniotic fluid of women with uncomplicated term pregnancies free of bacteria? American Journal of Obstetrics and Gynecology [Internet]. Elsevier Inc.; 2018;219:289.e1-289.e12. Available from: https://doi.org/10.1016/j.ajog.2018.05.028
Lim ES, Rodriguez C, Holtz LR. Amniotic fluid from healthy term pregnancies does not harbor a detectable microbial community. Microbiome. Microbiome. 2018;6:4–11.
Lager S, de Goffau MC, Sovio U, Peacock SJ, Parkhill J, Charnock-Jones DS, et al. Detecting eukaryotic microbiota with single-cell sensitivity in human tissue. Microbiome. Microbiome. 2018;6:1–11.
Theis KR, Romero R, Winters AD, Greenberg JM, Gomez-Lopez N, Alhousseini A, et al. Does the human placenta delivered at term have a microbiota? Results of cultivation, quantitative real-time PCR, 16S rRNA gene sequencing, and metagenomics. American Journal of Obstetrics and Gynecology [Internet]. Elsevier Inc.; 2019;220:267.e1-267.e39. Available from: https://doi.org/10.1016/j.ajog.2018.10.018
Glassing A, Dowd SE, Galandiuk S, Davis B, Chiodini RJ. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathogens. BioMed Central. 2016;8:1–12.
Perez-Muñoz ME, Arrieta MC, Ramer-Tait AE, Walter J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome. Microbiome. 2017;5:1–19.
Bushman FD. De-discovery of the placenta microbiome. American Journal of Obstetrics and Gynecology [Internet]. Elsevier Inc.; 2019;220:213–4. Available from: https://doi.org/10.1016/j.ajog.2018.11.1093
Ansbacher R, Boyson WA, Morris JA. Sterility of the uterine cavity. American Journal of Obstetrics and Gynecology [Internet]. 1967;99:394–6 Available from: https://www.sciencedirect.com/science/article/pii/S0002937816345495.
Kennedy KM, Gerlach MJ, Adam T, Heimesaat MM, Rossi L, Surette MG, et al. Fetal meconium does not have a detectable microbiota before birth. Nature microbiology. England. 2021;6:865–73.
de Goffau MC, Lager S, Sovio U, Gaccioli F, Cook E, Peacock SJ, et al. Human placenta has no microbiome but can contain potential pathogens. Nature [Internet]. Springer US. 2019;572:329–34 Available from: https://doi.org/10.1038/s41586-019-1451-5.
Franasiak JM, Werner MD, Juneau CR, Tao X, Landis J, Zhan Y, et al. Endometrial microbiome at the time of embryo transfer: next-generation sequencing of the 16S ribosomal subunit. Journal of Assisted Reproduction and Genetics. 2016;33:129–36.
Tanner MA, Goebel BM, Dojka MA, Pace NR. Specific ribosomal DNA sequences from diverse environmental settings correlate with experimental contaminants. Applied and environmental microbiology. 1998;64:3110–3.
van der Zee A, Peeters M, de Jong C, Verbakel H, Crielaard JW, Claas ECJ, et al. Qiagen DNA Extraction Kits for Sample Preparation for <em>Legionella</em> PCR Are Not Suitable for Diagnostic Purposes. Journal of Clinical Microbiology [Internet]. 2002;40:1126 LP – 1126. Available from: http://jcm.asm.org/content/40/3/1126.abstract
Grahn N, Olofsson M, Ellnebo-Svedlund K, Monstein H-J, Jonasson J. Identification of mixed bacterial DNA contamination in broad-range PCR amplification of 16S rDNA V1 and V3 variable regions by pyrosequencing of cloned amplicons. FEMS Microbiology Letters [Internet]. 2003;219:87–91. Available from: https://doi.org/10.1016/S0378-1097(02)01190-4
Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biology [Internet]. 2014;12:87. Available from: https://doi.org/10.1186/s12915-014-0087-z
Stinson LF, Keelan JA, Payne MS. Identification and removal of contaminating microbial DNA from PCR reagents: impact on low-biomass microbiome analyses. Letters in applied microbiology. England. 2019;68:2–8.
Olomu IN, Pena-cortes LC, Long RA, Vyas A, Krichevskiy O, Luellwitz R, et al. Elimination of “ kitome ” and “ splashome ” contamination results in lack of detection of a unique placental microbiome. Microbiome. BMC Microbiology; 2020;20:157.
Walker AW, Martin JC, Scott P, Parkhill J, Flint HJ, Scott KP. 16S rRNA gene-based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice. Microbiome. Microbiome. 2015;3:1–11.
Jervis-Bardy J, Leong LEX, Marri S, Smith RJ, Choo JM, Smith-Vaughan HC, et al. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome [Internet]. 2015;3:19. Available from: https://doi.org/10.1186/s40168-015-0083-8
Lazarevic V, Gaïa N, Girard M, Schrenzel J. Decontamination of 16S rRNA gene amplicon sequence datasets based on bacterial load assessment by qPCR. BMC microbiology [Internet]. BioMed Central; 2016;16:73. Available from: https://pubmed.ncbi.nlm.nih.gov/27107811
Davis NM, Proctor DiM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. bioRxiv [Internet]. Microbiome; 2018;6:221499. Available from: http://biorxiv.org/content/early/2018/07/25/221499.abstract
Lu J, Salzberg SL. Removing contaminants from metagenomic databases. bioRxiv [Internet]. 2018;261859. Available from: http://biorxiv.org/content/early/2018/02/08/261859.abstract
Karstens L, Asquith M, Davin S, Fair D, Gregory WT, Wolfe AJ, et al. Controlling for contaminants in low-biomass 16S rRNA gene sequencing experiments. mSystems [Internet]. American Society for Microbiology; 2019;4:e00290-19. Available from: https://pubmed.ncbi.nlm.nih.gov/31164452
Liu Y, Elworth RAL, Jochum MD, Aagaard KM, Treangen TJ. Squeegee: de-novo identification of reagent and laboratory induced microbial contaminants in low biomass microbiomes. bioRxiv [Internet]. 2021;2021.05.06.442815. Available from: http://biorxiv.org/content/early/2021/05/07/2021.05.06.442815.abstract
Laurence M, Hatzis C, Brash DE. Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLOS ONE [Internet]. Public Library of Science; 2014;9:e97876. Available from. https://doi.org/10.1371/journal.pone.0097876.
Minich JJ, Zhu Q, Janssen S, Hendrickson R, Amir A, Vetter R, et al. KatharoSeq Enables High-Throughput Microbiome Analysis from Low-Biomass Samples. McFall-Ngai MJ, editor. mSystems [Internet]. 2018;3:e00218-17. Available from: http://msystems.asm.org/content/3/3/e00218-17.abstract
Moossavi S, Fehr K, Khafipour E, Azad MB. Repeatability and reproducibility assessment in a large-scale population-based microbiota study: case study on human milk microbiota. Microbiome [Internet]. BioMed Central; 2021;9:41. Available from: https://pubmed.ncbi.nlm.nih.gov/33568231
Bender JM, Li F, Adisetiyo H, Lee D, Zabih S, Hung L, et al. Quantification of variation and the impact of biomass in targeted 16S rRNA gene sequencing studies. Microbiome. 2018;6:155.
Chen C, Song X, Wei W, Zhong H, Dai J, Lan Z, et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nature Communications [Internet]. Springer US; 2017;8:875. Available from: https://doi.org/10.1038/s41467-017-00901-0
Moreno I, Franasiak JM. Endometrial microbiota—new player in town. Fertility and Sterility [Internet]. Elsevier Inc.; 2017;108:32–9. Available from: https://doi.org/10.1016/j.fertnstert.2017.05.034
Mor G, Kwon J-Y. Trophoblast-microbiome iInteraction: a new paradigm on immune regulation. Am J Obstet Gynecol. 2015;213:S131–7.
Funding
This study was supported in part by NIH grants R01-HD091731 (KMA), R21-ES029462 (KMA), R01-DK089201 (KMA), and F30-HD104278 (EEB), and the Robert and Janice McNair Foundation (EEB). KMA holds the Henry & Emma Meyer Endowed Chair at Baylor College of Medicine. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
EB, DM, and KA equally contributed to the literature review and manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bolte, E.E., Moorshead, D. & Aagaard, K.M. Maternal and early life exposures and their potential to influence development of the microbiome. Genome Med 14, 4 (2022). https://doi.org/10.1186/s13073-021-01005-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-021-01005-7