
Abstract
Background
Bevacizumab, a monoclonal antibody against soluble VEGFA, is an approved and commonly administered anti-angiogenic drug in patients with metastasized colorectal cancer (mCRC). The survival benefit of anti-VEGF therapy in mCRC patients is limited to a few months, and acquired resistance mechanisms are largely unknown. Here, we employed whole-genome sequencing of plasma DNA to evaluate the tumor genome of patients undergoing treatment with bevacizumab to determine novel aberrations associated with resistance.
Methods
Using longitudinal plasma analyses, we studied the evolution of tumor genomes in a mCRC cohort (n = 150) and conducted analyses of CRC cases from The Cancer Genome Atlas (TCGA) database (n = 619) to identify associations between genomic aberrations and clinical features. We employed whole-genome sequencing to identify the most frequently occurring focal somatic copy number alterations (SCNAs). Using the TCGA data as a comparative and supporting dataset, we defined the minimally amplified overlapping region and studied the mechanistic consequences of copy number gain of the involved genes in this segment. In addition, we established an in vitro cell model and conducted downstream gene expression and cell viability assays to confirm our findings from the patient dataset.
Results
We observed a recurrent focal amplification (8.7% of cases) on chromosome 13q12.2. Analysis of CRC cases from the TCGA database suggested that this amplicon is associated with more advanced stages. We confirmed that this 13q12.2 amplicon frequently emerges later during the clinical course of disease. After defining the minimally amplified region, we observed that the amplification and expression of one gene, POLR1D, impacted cell proliferation and resulted in upregulation of VEGFA, an important regulator of angiogenesis which has been implicated in the resistance to bevacizumab treatment. In fact, in several patients, we observed the emergence of this 13q12.2 amplicon under bevacizumab treatment, which was invariably associated with therapy resistance.
Conclusions
Non-invasive analyses of cell-free DNA from patients undergoing treatment with bevacizumab enabled the tracking of evolving tumor genomes and helped identify a recurrent focal SCNA of clinical relevance. Here, we describe a novel resistance mechanism against a widely applied treatment in patients with mCRC which will impact the clinical management of patients.
Similar content being viewed by others


Background
Colorectal cancer (CRC) still remains a large global health problem, representing the third most commonly diagnosed malignancy worldwide as well as one of the major causes of morbidity and mortality across populations [1]. Its burden is projected to increase by 60% by the year 2030 with an estimated 2.2 million new cases and 1.1 million deaths [2]. Reports have shown that almost 50% of individuals with CRC who are initially diagnosed to have localized cancer will subsequently develop metastases as the disease progresses [3], and furthermore, approximately 30% of patients present with distant metastases already at the time of diagnosis [4]. Although surgical removal of early-stage or metastatic lesions represents a potential curative approach [5], therapeutic options for metastatic CRC (mCRC) are commonly limited to palliative approaches that improve quality of life and survival for a median time of about 2 to 3 years. The identification of molecular targets and pathways involved in the initiation and progression of CRC have helped to better characterize the disease and further tailor patient treatment more precisely to minimize primary resistance, or to avoid it altogether [6]. Although advances in genome sequencing technology have enabled high-resolution detection of potential molecular targets, such as somatic copy number alterations (SCNAs) or mutations, the significance of many such aberrations remains elusive in terms of guiding therapy decision-making.
Liquid biopsies, i.e., the analysis of tumor components in bodily fluids such as blood [7,8,9,10], have shown promising clinical utility in the management of CRC, ranging from applications in early detection [11,12,13,14,15,16,17], detection of relapse [18,19,20], identification of prognostic markers [21, 22], molecular characterization of metastatic disease [23], and tracking response to therapy [24,25,26,27,28].
To this end, “driver” alterations, i.e., those that actively promote cancer development, are of particular importance [29]. Of particular relevance are focal SCNA events, i.e., aberrations spanning a restricted length of a chromosome arm, as these regions harbor a limited number of genes of which one or a few may give rise to a growth advantage as a result of selection during the evolution of the cancer genome [30,31,32]. Definitions of focal events somewhat vary throughout the literature. Therefore, we analyzed the SCNAs in the TCGA pan-cancer dataset and developed a very restrictive definition of an amplicon [33], the utility of which we were able to prove using low-coverage whole-genome sequencing of plasma DNA (plasma-Seq) [34] in several studies [35,36,37,38].
In the current study, we applied plasma-Seq to our mCRC cohort (n = 150). As compared to analysis of primary tumors, the analyses of plasma DNA offer the unique opportunity to establish the sequential order of events. As expected, we identified several focal SCNAs harboring known cancer driver genes, for example, chr12p12.1 and chr8p11.23-p11.22, which include KRAS and FGFR1, respectively. However, we also found recurrent focal amplifications in which cancer driver genes have not yet been established. The most frequent amplicon was on 13q12.2, which we detected in 14 patients, and our serial plasma analyses suggested that this amplicon is a rather late event and potentially associated with resistance to administered therapies. We assessed the biological role and clinical significance of this focal event in mCRC patients and, furthermore, investigated the functional role of the genes harbored within this limited chromosomal region.
Methods
Patient cohort
The study was approved by the Ethics Committee of the Medical University of Graz (approval number 21–229 ex 09/10), conducted according to the Declaration of Helsinki, and written informed consent was obtained from all patients.
The age and sex distribution of all patients is summarized in Additional file 1. All patients had metastatic CRC and were being treated at the Department of Internal Medicine, Division of Oncology, at the Medical University of Graz. We were able to isolate DNA from pre-treatment tumor specimens in nine patients for which tumor tissue was available as a result of surgical or bioptic procedures. Imaging studies, i.e., computed tomography (CT) scans, were obtained as part of routine clinical care.
As the focus of this study was on copy number profiling of ctDNA, which requires tumor fractions of 5% and higher, we pre-screened our plasma collection using our previously published modified Fast Aneuploidy Screening Test-Sequencing System [39] to identify appropriate plasma samples with elevated tumor content. Based on these mFAST-SeqS pre-screening results, the 150 patients included in this study were selected.
TCGA data collection and analysis
TCGA data analyzed in this work originated from TCGA-COADREAD projects [40], and only cases with copy number variation data were kept. Clinical analyses and gene expression data were downloaded from Broad Institute GDAC Firehose (http://gdac.broadinstitute.org/). Absolute copy number results were downloaded from the NCI Genomic Data Commons (GDC; https://gdc.cancer.gov/about-data/publications/pancanatlas) [41]. Patient samples were categorized as “balanced,” “gain,” or “amplification” according to 13q12.2 copy number (balanced, 1 < copy number ≤ 3; gain, 3 < copy number ≤ 6; amplification, copy number > 6). Statistical analyses were performed in R. Focal event calling was performed by using an in-house script as published previously [35, 36].
Definition of the minimally 13q12 amplified region and involved genes
We first determined the minimal overlapping range of all the focal events in our patient cohort (Table 1) by calculating the median log2 ratio of chr13 against all 14 patients who harbored the 13q12.2 SCNA of each pre-defined 50 kb window. For those windows which showed a median log2 ratio over 0.55, the frequency of focal events was counted (Additional file 2: Figure S3A). Using the windows that demonstrated the lowest frequency, we calculated a p value (Fisher’s exact test) in order to identify the statistically significant minimal overlapping range. We identified a broad peak (p < 0.05; Fisher’s exact test; chr13:27,708,804-28,667,235) and a focal peak (p < 0.01; Fisher’s exact test; chr13:28,441,650-28,667,235) in our patient dataset. To confirm this finding, we applied the same method to the TCGA dataset and ended up with a broad peak at chr13:28,197,436-28,650,763 (p < 0.05; Fisher’s exact test) and a focal peak at chr13:28,382,214-28,604,579 (p < 0.01; Fisher’s exact test), which is comparable with the GISTIC analysis result (broad peak: chr13:28,192,985-28,773,237; focal peak: chr13: 28,391,954-28,558,679) (Additional file 2: Figure S3A).
Plasma-seq: whole-genome sequencing of primary tumor and plasma samples
Whole-genome sequencing libraries were prepared and sequenced for plasma and tumor samples when available by methods described previously in detail [34, 36, 42]. In brief, plasma DNA was isolated using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany) from 1 to 2 mL of plasma and primary tumor DNA was isolated from FFPE using the GeneRead DNA FFPE kit (Qiagen, Hilden, Germany). Samples were quantified with the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Vienna, Austria). Shotgun libraries were prepared using the TruSeq DNA LT Sample preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions for both primary tumor samples and for cell lines, but with several modifications for the generation of plasma libraries: 5–10 ng of input DNA was used and the fragmentation step was omitted, since plasma DNA is enriched for fragments in the range of 160 to 340 bp, and 25 PCR cycles were used for the selective amplification step of library fragments. Libraries were sequenced on either an Illumina MiSeq or NextSeq 550 instrument (Illumina, San Diego, CA, USA) for the generation of 150 bp single reads or 76 bp paired end with 5–10 million reads per sample, representing a 0.1–0.2× coverage of the whole genome. SCNA data analysis was performed as described previously [34]. Tumor fraction from plasma DNA and tumor samples were estimated with the ichorCNA algorithm, a probabilistic model for the simultaneous prediction of large-scale copy number alterations and estimation of tumor fraction, which is equivalent to tumor purity from bulk tumor analyses [43].
Digital PCR copy number assay
SCNAs of POLR1D and ERBB2 were analyzed using digital PCR (dPCR) and performed on the QuantStudio 3D platform (Life Technologies, Carlsbad, CA, USA). Pre-designed TaqMan assays specific for the detection of the copy number of POLR1D (Hs02926936_cn), ERBB2 (Hs00450668_cn), and a reference assay (TERT; 4403315) were purchased from Thermo Fisher. For dPCR, a total amount of 3–5 ng plasma DNA was used as input and samples were run in duplicate using the QuantStudio™ 3D Digital PCR 20 K Chip Kit v2 and a QuantStudio 3D instrument (Life Technologies, Carlsbad, CA, USA). Raw data were analyzed using the relative quantification module of the QuantStudio 3D Analysis Suite Software, including a Poisson correction. The confidence level was set to 95%, and the desired precision value was 10%.
Cell lines and cell culture
The human CRC cell lines OXCO-2, SW480, and HT29 were selected based on their copy number variation profiles and transfection suitability as in vitro cell models. OXCO-2 was kindly provided by Dr. Alberto Bardelli, Laboratory Molecular Oncology at the Candiolo Cancer Institute IRCCS-Candiolo (Torino). SW480 and HT29 cell lines were provided by Prof. Martin Pichler, Department of Internal Medicine, Division of Oncology, Medical University of Graz. Cell line SCNAs were profiled using whole-genome sequencing as described previously. SW480 cells harbor a complete gain of chromosome 13, whereas HT29 harbors a focal amplification of chr13q12.2. OXCO-2 cells have no changes in chromosome 13 (Additional file 2: Figure S2).
OXCO-2 cells were maintained in Iscove’s modified Dulbecco’s medium (IMDM; Gibco, Thermo Fisher Scientific, Vienna, Austria). HT29 cells were maintained in Dulbecco’s modified Eagle’s medium (DEME; Gibco, Thermo Fisher Scientific, Vienna, Austria), and SW480 cells were maintained in RPMI 1640 Medium (Gibco, Thermo Fisher Scientific, Vienna, Austria). All media were supplemented with 5% fetal bovine serum (Gibco, Thermo Fisher Scientific, Vienna, Austria) and 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific, Vienna, Austria). The CRC cell lines were authenticated at the Cell Bank of the Core Facility of the Medical University of Graz, Austria, by performing a STR profiling analysis (Kit: Promega, PowerPlex 16HS System; cat. no. DC2101, last date of testing: July 2019).
Generation of stable FLT3-overexpressing cell line
OXCO-2 cells were seeded in a 6-well plate in IMDM media at approximately 80% confluence and incubated overnight. The FLT3 (NM_004119) GFP-tagged human cDNA ORF clone (Acris, Herford, Germany) was transfected into cells 24 h post-seeding using the FuGene HD (Promega, Mannheim, Germany) transfection reagent according to the manufacturer’s recommendations. Cells were cultured in IMDM media containing 1 mg/mL Geneticin (Gibco, Thermo Fisher Scientific, Vienna, Austria) starting 24 h after transfection.
Colonies were collected via cloning discs (Sigma-Aldrich/Merck KGaA, Darmstadt, Germany) dipped in trypsin, and once detached, each colony was transferred separately into a 96-well plate containing IMDM media and Geneticin. Colonies originating from the single clones were expanded gradually into 6-well plates once they reached sufficient confluence with continued Geneticin treatment to further select for clones stably expressing FLT3-GFP (OXCO2-FLT3-GFP).
siRNA knockdown assays
Pre-designed siRNAs (Additional file 3: Table S3; Ambion, Life Technologies) were reverse transfected into SW480 and HT29 cells using Lipofectamine™ RNAiMAX transfection reagent (Invitrogen, Thermo Fisher Scientific, Vienna, Austria) as suggested by the supplier. Transfected cells were incubated for 72 h before performing proliferation assay or harvesting for expression analysis.
RNA isolation, quantitative RT-PCR, and mRNA-seq
RNA was isolated via the TRIzol (Invitrogen, Thermo Fisher Scientific, Vienna, Austria) method and reverse transcribed into cDNA using the Omniscript RT Kit (Qiagen, Hilden, Germany). Equal amounts of RNA were used in cDNA synthesis. Quantitative RT-PCR was performed on the ABI 7500 system using Biozym Blue S’Green qPCR Kit (Biozym, Hessisch Oldendorf, Germany) and pre-designed RT-PCR primers (Table 2; Microsynth AG, Switzerland) according to the manufacturer’s suggestions.
Captured coding transcriptome RNA-seq libraries were prepared using the TruSeq RNA Exome kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions with 100 ng total RNA input. Libraries were quantified using the Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina NextSeq instrument (Illumina, San Diego, CA, USA) for the generation of 75 bp paired-end reads. A pseudo-alignment approach (kallisto) was used to analyze RNA-seq data [44]. The output data of kallisto was reformed, and differential gene expression analysis was performed in R using the DESeq2 Bioconductor package [45].
Colony formation and cell viability assay
For the colony formation assay, cells were seeded in 24-well plates for 72 h and fixed in 100% methanol for 20 min. Following staining with 0.5% crystal violet, images of each well were acquired. Colonies were counted using ImageJ software.
Cell viability was assessed with the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Mannheim, Germany) according to the manufacturer’s instructions.
Gene expression prediction
Gene expression prediction analysis was performed as described previously [38]. In brief, whole-genome libraries were sequenced to obtain 200 million reads (5× coverage) and coverage values around transcription start sites (TSSs) were extracted from aligned BAM files and normalized by the mean value of the combined regions: TSS − 3000 to TSS − 1000 and TSS + 1000 and TSS + 3000. The coverage from − 1000 to + 1000 bp (2K-TSS coverage) and from − 150 to + 50 bp (NDR coverage) was used for the identification and prediction of genes as expressed or unexpressed by using support vector machines (SVMs). The SVM used for these analyses was trained on transcription start sites of genes that are constitutively expressed and constitutively unexpressed within the same sample. For every sample, a separate model is trained based on a k-fold cross-validation on those samples and the remaining genes are predicted from the trained model within each fold. Only genes that are consistently predicted across folds (the same prediction in > 95% of folds) are considered to be valid predictions. We did not use external training data to avoid inconsistencies in the models.
Results
Recurrent focal events identified by plasma-Seq
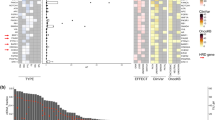
Using plasma-Seq and our aforementioned criteria for focal SCNAs [33, 35], we identified several recurrent focal events in a set of 150 mCRC patients (Additional file 1), 3 of which were found to be present in more than 5% of patients, comprising 12p12.1, 13q12.13-q12.3, and 8p11.23-p11.22 (Fig. 1a, Additional file 2: Figure S1A, Additional file 4). To compare our data with other large-scale analyses, we used publicly available SCNA data from 619 CRC patients included in The Cancer Genome Atlas (TCGA), of which a minority (n = 85) are mCRC patients. We found many similarities and few differences in focal SNCAs. Some amplifications containing known driver genes, e.g., ERBB2, EGFR, and MYC, and the deletion of tumor suppressor genes, e.g., CDKN2A and MAP2K4, were identified at a similar percentage in both cohorts (Additional file 4). The focal amplification in 8p11.23-p11.22 was also detected in both cohorts at a similar percentage (Fig. 1a, Additional file 2: Figure S1A, Additional file 4). In contrast, the focal amplification 12p12.1 harboring the KRAS oncogene was present in 6.7% of our patients, compared to only 1.6% of patient of the TCGA cohort (p = 0.002; chi-square test; Fig. 1a, Additional file 2: Figure S1A, Additional file 4), which may reflect a higher number of patients who received anti-EGFR therapy in our cohort [28, 36]. Moreover, the recurrent focal amplification of 13q12.13-q12.3 was observed with a significantly higher frequency in our cohort (8.7% vs 4.5%; p = 0.043; chi-square test; Fig. 1a, Additional file 2: Figure S1A, Additional file 4).

Identification of the 13q amplicon and establishment as a late event in CRC. a Recurrent focal events from our patient cohort with a frequency higher than 5%. Potential driver genes were identified according to a machine learning-based method for driver gene prediction [46]. The difference in these 3 recurrent focal events between our cohort and the TCGA cohort was analyzed using the chi-squared test. b The TCGA cohort was separated into 2 groups, i.e., balanced and aberrant (including gain and amplification cases). Bar charts illustrate 4 clinical features, i.e., tumor stage, distant metastasis, lymph node metastasis, and tumor location, which showed significant differences between these 2 groups. p values were calculated using the chi-squared test. c, d Plots illustrating the log2 ratio changes on chromosome 13. In C240, C118, and C79, focal amplification of chr13q12.2 was not identified in the primary tumor (PT) but found in the plasma (ctDNA) at a later stage. In C216, chr13q12.2 amplification was detected when the patient status was categorized as progressive disease. Copy number gains are shown in red and balanced regions in green. Tumor fraction (TF) of every sample was calculated using ichorCNA [43]. (SD, stable disease; PD, progressive disease)
With the exception of the 13q12 amplification, known candidate driver genes were located in all other recurrent focal amplifications. Because of the high frequency of this amplicon in our cohort, we therefore focused on a more detailed characterization of this region.
The 13q12.13-12.3 amplicon is associated with late-stage clinical features
As we only analyzed mCRC patients, whereas the TCGA data is mainly comprised of primary tumors, 55% of which are localized stages (I and II), the 13q12.13-12.3 amplicon may be more related to late-stage events of CRC. Indeed, in the TCGA dataset, patients with this 13q12 SCNA represented more late-stage disease (stage III and stage IV) (p = 1.23E−05; chi-square test), with distant (p = 1.62E−04; chi-square test) and lymph node metastases (p = 3.87E−05; chi-square test) (Fig. 1b). Furthermore, the 13q12 amplified cohort was significantly associated with primary tumors located in the rectum when compared to patients without the SCNA (p = 3.07E−04; chi-square test) (Fig. 1b).
We had access to the corresponding primary tumor in 9 of 14 patients with the amplification. Copy number analyses of the corresponding FFPE tumor tissue samples revealed that a focal amplification was already present in the primary tumor tissue in 4 patients (C123, C109, C178, and C74), whereas in 5 patients (C240, C118, C79, C206, and C166), this focal amplification was acquired at a later time point (Table 1, Fig. 1c, Additional file 2: Figure S1B and S2). In 3 patients with an undetected amplification in the primary tumor tissue (C240, C118, C79), the 13q12.13-q12.3 focal amplification appeared in 1 of the plasma DNA analyses after metastases were detected or the patient developed progressive disease (Table 1, Fig. 1c, Additional file 2: Figure S1B and S2). Furthermore, in 1 additional patient (C216), from whom an FFPE sample was not accessible, the amplification was not present in the first plasma sample but rather acquired after the patient exhibited progressive disease (Fig. 1d, Additional file 2: Figure S2). In 2 patients (C74, C123) with a detected gain in the primary tumor tissue, the copy number of the 13q12.13-q12.3 amplification increased in relation to the tumor fraction once metastases were acquired or progressive disease was exhibited (Table 1, Additional file 2: Figure S1B and S2), an observation additionally confirmed by dPCR.
The oncogene FLT3 is not associated with proliferation of CRC cells
The aforementioned data suggested that the 13q12.2 amplicon is associated with late-stage and progressive disease. In order to identify the potential driver in the amplicon, we first determined the minimal overlapping range of all the focal events in our patient cohort (Additional file 2: Figure S3A). For further confirmation, we applied the same method to the TCGA dataset and identified a broad and a focal peak, which were comparable with the GISTIC analysis result (Additional file 2: Figure S3A; further details of this region definition in the “Methods” section). In total, seven genes (POLR1D, GSX1, PDX1, ATP5EP2, CDX2, PRHOXNB, and FLT3) were completely located within the broad peak for all three analyses (Additional file 2: Figure S3A).
Since FLT3 is a well-known driver gene in hematological malignancies that can be targeted by the drug sorafenib [47], FLT3 was considered to be a reasonable driver candidate. In order to understand the potential oncogenic role of FLT3 in CRC, we investigated whether the FLT3 gene amplification correlated with gene expression in the TCGA and the CCLE CRC cell line datasets, respectively. However, consistent with a previous report, we did not observe a correlation between mRNA expression and copy number of FLT3 (Fig. 2a, b) [48]. To confirm this observation in vitro, we generated a CRC cell line stably expressing FLT3. As expected, overexpression of FLT3 in OXCO-2 cells did not lead to a significant change in cell proliferation (Fig. 2c, p > 0.05; t test).

Exclusion of FLT3 as a driver gene. a Box plot showing no significant correlation between FLT3 gene copy number and FLT3 mRNA expression (log10 (normalized RSEM value + 1)) in the TCGA cohort. Control/matched normal tissue, n = 51; Balanced, n = 196; Gain, n = 129; Amplification, n = 46. b The scatter plot illustrates no correlation in FLT3 copy number and FLT3 mRNA expression (log2 (TPM + 1)) in 58 CRC cell lines (R = − 0.41, p = 0.0016; Pearson). The red line represents the noise threshold (TPM = 1). c Colony formation assay showing significant overexpression of FLT3 in OXCO-2 cells (p = 0.03433; t test) but no significant changes in proliferation (p = 0.1866; t test)
Identification of POLR1D as a potential driver gene in 13q12.2
As the abovementioned results suggested that FLT3 may not function as a driver gene in CRC, we investigated the other five candidate genes (pseudogene ATP5EP2 was excluded) located in the overlapping broad peak regions as well as the first immediate upstream and downstream genes, LNX2 and PAN3, respectively.
In five of these genes, including LNX2, POLR1D, CDX2, PDX1, and PAN3, a positive correlation between copy number and mRNA expression could be demonstrated using the publicly available datasets from the TCGA and the CCLE databases (Fig. 3a, b; Additional file 2: Figure S3B and C). To further characterize a potential involvement of these genes in vitro, we induced a transient siRNA knockdown of these five genes in the two CRC cell lines HT29 and SW480, where 13q12.2 is overrepresented either due to a focal amplification (HT29) or gain of the entire chromosome 13 (SW480) (Additional file 2: Figure S2) and where these genes are expressed. Intriguingly, only silencing of POLR1D but not of the other genes demonstrated a significant reduction (1.3–1.6-fold) in cell viability in both the HT29 and SW480 cell line culture systems (Fig. 3c–e, Additional file 2: Figure S4A). These results suggest that POLR1D may have functional implications for CRC cell proliferation. POLR1D is a subunit of both RNA polymerases I and III. RNA polymerase I is involved in the production of 18S, 5.8S, and 28S rRNAs, while RNA polymerase III synthesizes small RNAs [50]. Despite a recent report describing the frequent overexpression of POLR1D in CRC [51], a role of POLR1D in cancer has not otherwise been thoroughly described in the literature.

Expression analyses for identification of the potential driver gene in the 13q12.2 amplicon. a Box plots showing significant positive correlation between gene copy number and mRNA expression (log10 (normalized RSEM value + 1)) in 5 genes (i.e., LNX2, POLR1D, CDX2, PDX1, and PAN3) in the TCGA cohort. Control/matched normal tissue, n = 51; Balanced, n = 196; Gain, n = 129; Amplification, n = 46. b Scatter plots illustrating positive correlation in gene copy number and mRNA expression (log2 (TPM + 1)) in 5 genes (i.e., LNX2, POLR1D, CDX2, PDX1, and PAN3) in 58 CRC cell lines. R values and p values were calculated using Pearson’s correlation test. The red line represents the noise threshold (TPM = 1). c The bar chart illustrates cell viability changes after knockdown of 5 genes (i.e., CDX2, LNX2, PAN3, PDX1, and POLR1D) in 2 CRC cell lines (i.e., HT29 and SW480). Silencing of POLR1D in both cell lines showed reduction in cell viability over 15%. p values calculated by t test are shown above the bar. d Silencing of POLR1D with 3 different siRNA constructs. RT-PCR showing that silencing provided sufficient knockdown of POLR1D expression in both cell lines. e Cell viability time curve illustrating significant reduction of cell viability after knockdown of POLR1D expression in HT29 and SW480 cells (*p < 0.1; **p < 0.05; ***p < 0.01; t test). f Box plot illustrating the different expression (normalized DESeq2 read count) of VEGFA and EREG between negative control (SCR, a scrambled siRNA) and POLR1D knockdown in SW480 (SCR, n = 6; siPOLR1D2, n = 3; siPOLR1D3, n = 3) and HT29 (SCR, n = 4; siPOLR1D2, n = 2; siPOLR1D3, n = 2) cell lines. VEGFA and EREG expression was suppressed after POLR1D silencing. Adjusted p values were calculated by DESeq2, an R package. g Violin plots of VEGFA and EREG expression (normalized RSEM value) of TCGA cases. Samples with chr13q12.2 gain (n = 129) or amplification (n = 46) showed a significant upregulation compared to balanced cases (n = 196). h Schematic presentation how nucleosome organization around promoters of repressed and active genes differ in their promoter regions. Promoters of active genes have a nucleosome-depleted region (NDR, dark blue line), whereas the nucleosome organization of promoters of repressed genes is not well-defined, resulting in different nucleosome footprints at transcription start sites. We leveraged these differences by employing our previously published nucleosome positioning [38] to determine the expression status of genes within the 13q12.2 amplicon. In addition to the genes discussed in the text, we added the gene GSX1 (light blue) as an example for a repressed gene (part of the figure adapted from [49])
POLR1D affects expression of VEGFA and EREG
We sought to elucidate the underlying oncogenic mechanisms of POLR1D using RNA-seq analysis. In addition to POLR1D, which showed an approximate 2-fold reduction in expression in the silenced cells, we detected 44 differentially expressed genes in both the HT29-POLR1D-silenced and SW480-POLR1D-silenced cell lines compared to the controls.
Moreover, a similar expression change of 8 of these 45 total genes including POLR1D (i.e. PPP1R15A, MOSPD2, FAM84B, GARS, POLR1D, KIF21B, VEGFA, and EREG) was also observed in the TCGA cases with a 13q12 SCNA (Fig. 3f, g, Additional file 2: Figure S4B, Additional file 3: Table S1). All 45 genes demonstrated an upregulation in expression in patients harboring the 13q12 SCNA compared to patients with a balanced 13q12 region (Additional file 3: Table S1).
Of particular relevance appeared to be the POLR1D-associated upregulation of VEGFA and EREG. VEGFA encodes vascular endothelial growth factor A (VEGFA), which is an important regulator of angiogenesis in the PDGF/VEGF growth factor family and which plays a role in the development and progression of CRC [52]. The anti-VEGF monoclonal antibody bevacizumab (Avastin) was approved by the US Food and Drug Administration (FDA) for the treatment of advanced CRC [53] and is a recombinant humanized IgG1 antibody against all isoforms of VEGFA. Anti-VEGF treatment may induce the expression of VEGFA, which, in turn, is involved in the resistance to anti-VEGF treatment [54, 55]. EREG encodes epiregulin, which is a member of the epidermal growth factor (EGF) family. Epiregulin is a ligand of the epidermal growth factor receptor (EGFR) and the structurally related erb-b2 receptor tyrosine kinase 4 (ERBB4) [56]. Epiregulin promotes the progression of various cancers [57, 58].
Nucleosome positioning mapping to infer POLR1D expression in plasma
Previously, we had shown that serial monitoring of tumor genomes by plasma DNA analyses may reveal focal amplifications as a mechanism of resistance to administered therapies in CRC [36] and in prostate cancer [35]. As we reasoned that due to the higher expression of VEGFA, tumors with a 13q12.2 amplification might be more resistant to anti-VEGF treatment, we implemented this serial monitoring concept here and investigated longitudinal plasma samples from two patients, i.e., C216 and C129, where we observed the emergence of the 13q12.2 amplicon under anti-VEGF treatment as described in detail below.
However, the observation of a novel amplification does not allow the conclusion that the genes located within the amplified region are actually expressed. cfDNA consists predominantly of nucleosome-protected DNA shed into the bloodstream by cells undergoing apoptosis [11, 59]. Transcription start sites (TSSs) harbor distinct nucleosome footprints, which differ for expressed and silent genes (Fig. 3h) [49]. Recently, we showed that after high-coverage whole-genome sequencing of plasma DNA, these TSS-nucleosome occupancy patterns can be leveraged to infer which cancer driver genes in regions with somatic copy number gains are expressed with high accuracy [38]. Hence, we selected for each of these two patient cases one plasma sample (C129-8 and C216-12), which both had the focal amplification on 13q12.2, and generated whole-genome libraries with 200 million reads (5× coverage) to conduct our previously described TSS profiling analyses [38].
These analyses predicted expression of POLR1D in both plasma samples along with the other four genes (LNX2, CDX2, PDX1, and PAN3) for which we had established a positive correlation between copy number and mRNA expression (Fig. 3h). In contrast, FLT3 was classified as unexpressed in C216-12 and inconclusive in C129-8 (Fig. 3h). Hence, the gene expression inference from our nucleosome positioning mapping approach suggested that our observations from the cell culture systems are applicable to these clinical cases and, furthermore, that FLT3 is indeed not the cancer driver gene within this amplicon in CRC.
The emergence of POLR1D amplification correlates with resistance to bevacizumab
In patient C216, who had been undergoing anti-VEGF treatment for 9 months, we detected the emergence of the 13q12.2 amplification which correlated with the development of progressive disease and resistance to bevacizumab, i.e., increase in blood CEA and CA19-9 tumor markers and increase in lesion size of liver metastases, as observed in the CT scan (Fig. 4; Additional file 3: Table S2; Additional file 2: Figure S5). Digital PCR was used on all serial samples to confirm the emergence of POLR1D under anti-VEGF therapy (Fig. 4b).

Emergence of the 13q12 amplicon under bevacizumab treatment in patient C216. a Genome-wide log2 ratio plots of plasma samples from C216 obtained before bevacizumab treatment (upper), after 227 days of bevacizumab treatment (middle), and after 285 days of bevacizumab treatment (bottom). The insets illustrate the respective tumor fraction (TF) for each analysis and enlarged log2 ratio plots of chromosome 13, the bottom 2 samples show gain of chromosome 13, with the highest copy number gain on chr13q12.2, the region that harbors the POLR1D gene. Copy number gains are shown in red, balanced regions in green, and copy number losses in blue. b Plot illustrating all time points of blood collection and relative marker changes. Red line: POLR1D copy number changes identified by digital PCR, showing minimum changes until day 227. Blue line: blood CEA level changes. Black line: blood CA19-9 level changes. Gray bar: tumor content identified in every sample using ichorCNA. c Four CT images obtained in 4 different time points, i.e., day 10, day 152, day 222, and day 276 after bevacizumab treatment. Compared to the first image, no significant changes were identified on day 152, during which the patient had been evaluated to have stable disease in accordance with the RECIST criteria. On day 222, the pre-present liver metastasis lesions enlarged with occurrence of new micrometastasis lesions. On day 276, all livers metastasis lesions had become larger
In patient C129, we noted a 13q12.2 amplification in the first plasma sample, which disappeared after anti-EGFR treatment. According to the blood CEA values and CT scan, the tumor acquired resistance to anti-EGFR treatment within 9 months and plasma-Seq revealed a novel focal amplification on 17q12, including ERBB2, which represents an established mechanism of resistance to anti-EGFR therapy (Fig. 5; Additional file 3: Table S2; Additional file 2: Figure S6) [36, 60,61,62]. After switching to anti-VEGF treatment for 5 months, the amplification in 13q12.2 appeared again, along with an increase in CEA level and the size of the lung metastasis region (Fig. 5; Additional file 3: Table S2; Additional file 2: Figure S6). In order to confirm this apparent clonal switch between POLR1D and ERBB2, digital PCR was performed to validate copy number amplification in all involved samples (Fig. 5b). These cases suggest that amplification of 13q12.2 along with increased expression of POLR1D and subsequent increased expression of VEGFA may contribute to anti-VEGF treatment resistance.

Alternating POLR1D and ERBB2 amplifications in serial plasma analyses of patient C129. a Genome-wide log2 ratio plots of plasma samples from C129 obtained before treatment with cetuximab (first), 160 days (second) after cetuximab, before bevacizumab (third), and 138 days (fourth) after bevacizumab. The insets illustrate the respective tumor fraction (TF) for each analysis and enlarged log2 ratio plots of chromosome 13 and 17, the first and the last sample showing gain of chromosome 13, with the highest copy number on chr13q12.2, the region harboring POLR1D. The middle 2 samples show a gain of chromosome 17 with the highest copy number on chr17q12, harboring ERBB2. The copy number color code is as in Fig. 4. b Plot illustrating blood collection time points. Red line: POLR1D copy number changes measured by dPCR. A decrease in POLR1D copy number was detected until day 274 during cetuximab treatment. After switching to bevacizumab, POLR1D copy number increased back within 138 days. Green line: ERBB2 copy number changes (dPCR). ERBB2 copy number increased until day 274 (during cetuximab treatment). After switching to bevacizumab, ERBB2 copy number decreased back within 138 days. Blue line: CEA levels decreased in the first 3 samples and slightly increased in the fourth sample. After a slight decrease in the fifth sample, CEA continuously increased up until the last sample. Black line: CA 19-9 remained at low levels across all samples. Gray bar: tumor fraction estimated with ichorCNA. c Four CT images obtained on day 6 and day 212 of cetuximab treatment, before bevacizumab treatment (day 268), and 160 days after bevacizumab treatment (day 434). No significant changes were identified on day 212, consistent with stable disease. On day 268, the pre-present lung metastasis lesion became larger in the right lung and pleural effusion appeared in the left lung, indicating progressive disease. On day 434, this pre-present lesion became larger and new metastasis lesions appeared. Pleural effusion increased, and progressive disease was designated
In summary, our results suggest that POLR1D may act as a potential driver gene in the 13q12.2 amplification and may affect cancer progression by increasing the expression of VEGFA and EREG. Because of the higher expression of VEGFA, amplification of 13q12.2 may be involved in the acquired resistance of anti-VEGF treatment.
Discussion
The identification of predictive biomarkers is necessary for the implementation of individualized tumor treatment in patients with cancer. Establishment of such biomarkers would allow the stratification of patients into appropriate treatment groups and help facilitate a transition from a one-size-fits-all approach to that of precision medicine. Although a considerable number of patients with mCRC will experience progression and eventually exhaust standard therapies approved for CRC, many of these patients remain candidates for further treatment strategies if they demonstrate an adequate performance score and lack significant comorbidities. As the instability of tumor genomes has been well-established, there have been tremendous efforts to track genetic tumor markers over time rather than solely at the point of diagnosis, which can in turn provide support for determining novel evidence-based therapies for these patients. Liquid biopsy has been at the forefront of such non-invasive profiling of the tumor at regular intervals throughout a patient’s treatment and has allowed for the monitoring of the evolution of the tumor genome [7,8,9,10].
Another application utilizing plasma DNA is the much-needed identification of novel driver genes. Although the 13q12.2 amplification is relatively frequent in CRC, a driver gene has not been identified in this amplification yet. The 13q12.2 region harbors the fms-like tyrosine kinase 3 (FLT3), a known oncogene that encodes for a tyrosine kinase which activates proliferation and differentiation pathways in hematopoietic stem cells. Although mutations in FLT3 have been well-documented in cases of acute myeloid leukemia (AML) and other hematological malignancies [63], there is currently no substantial evidence demonstrating that FLT3 plays a role in the tumorigenesis of CRC. One case report focused on a particular patient with amplification of FLT3 who demonstrated response to sorafenib, an anti-FLT3 compound [64]. However, our analysis showed no correlation between FLT3 copy number and gene expression in both the TCGA cohort and the CCLE cell line database. Furthermore, gene expression inference from TSS nucleosome positioning suggested that in our two clinical case studies C129 and C216, FLT3 was not expressed within the 13q12.2 amplicon. Moreover, our stable FLT3-overexpressing cell line did not exhibit any significant proliferation advantage, further questioning the role of FLT3 as an oncogene in mCRC. Our findings are in agreement with a previous study, which showed that FLT3 amplification does not seem to be an actionable target or a proper biomarker for FLT3 inhibitors like regorafenib or sorafenib [65].
Furthermore, two studies previously published about the potential driver gene in 13q12.2 showed varied results [66, 67]. One study suggested both LNX2 and POLR1D as potential driver genes in 13q12.2. However, in this particular in vitro model, the SW480 (whole chr13 gain) and DLD1 (whole chr13 balanced) cell lines were used, which thus excluded cell lines harboring amplification of 13q12 [66]. Another study which did use cell lines harboring 13q12.2 amplification demonstrated high expression of CDX2 and therefore concluded that CDX2 acts as a driver gene in this region [67]. According to the CCLE data, however, CDX2 copy number is poorly correlated with gene expression, and furthermore, in a real patient dataset (TCGA), mRNA expression was not significantly different between patients harboring a gain or amplification of chr13q12.2. However, as a recent study described that CDX2 loss through demethylation and HDAC inhibition is an adverse prognostic factor and linked to molecular features of the serrated pathway [68], CDX2 may act as an oncogene in tumors with a high expression of CDX2, but this is not necessarily applicable to all cases of CRC with chr13q12.2 aberration.
Our study suggests POLR1D as a potential oncogene in the 13q12.2 amplification and as a novel resistance mechanism against the anti-VEGF monoclonal antibody bevacizumab (Avastin). These conclusions are based on several observations. First, POLR1D overexpression caused proliferation of CRC cells as demonstrated by transient siRNA knockdown in the two CRC cell lines HT29 and SW480. Second, POLR1D was indeed expressed in our profoundly investigated cases C129 and C216, as inferred from plasma nucleosome positioning mapping. Third, an important consequence of POLR1D overexpression was upregulation of VEGFA, as evidenced by both our in vitro experiments and the TCGA RNA-seq data. VEGFA is an important regulator of angiogenesis, the target of bevacizumab, and plays a role in the development, progression, and metastasis of CRC [69]. Finally, given the specificity of bevacizumab to the VEGFA ligand [54] and as recent publications showed that bevacizumab treatment induces autocrine VEGF signaling [55, 70], we investigated the occurrence of POLR1D amplifications in patients receiving bevacizumab to provide in vivo data. In fact, serial plasma DNA analyses revealed that in two of our patients, the 13q12.2 amplification evolved under treatment and was in both cases linked to progressive disease (Figs. 4 and 5). It will be interesting to test the relationship between the 13q12.2 amplification and bevacizumab treatment in larger patient cohorts.
Another interesting gene is EREG, which encodes epiregulin, a member of the epidermal growth factor (EGF) family, which can bind to and activate EGFR and ERBB4 [56]. Higher EREG expression is considered to be a sign of higher activation of the EGFR pathway, which, in turn, means a better response to anti-EGFR treatment [71] but more resistance to drugs which target the other ERBB family members, e.g., ERBB2 [57, 58]. The other five genes (i.e. PPP1R15A, MOSPD2, FAM84B, GARS, KIF21B) have demonstrated involvement in the progression of various cancers. For example, FAM84B, which encodes family with sequence similarity 84, member B protein, was reported to be related to the progression of prostate cancer and esophageal squamous cell carcinoma [72, 73]. GARS encodes glycyl-tRNA synthetase and has been shown to be involved in neddylation, a post-translational modification that controls cell cycle and proliferation and thus may play a role in cancer progression [74]. KIF21B encodes a member of the kinesin superfamily and was reported to be significantly associated with poor prognosis of prostate cancer patients [75]. Mutation of PPP1R15A, which encodes protein phosphatase 1 regulatory subunit 15A, has been shown to be a valuable biomarker for mCRC patients sensitive to bevacizumab regimens [76]. MOSPD2 encodes motile sperm domain-containing protein 2 and has recently been reported to promote the metastasis of breast cancer [77].
Limitations of our study include low patient number and the need of a tumor content higher than 5–10% in plasma in order to conduct reliable copy number analyses. The two patient cases C129 and C216 demonstrate the potential of plasma-Seq for therapeutic monitoring; however, such analyses depend on increased and similar tumor fractions in serial plasma samples (Figs. 4 and 5). Reduced tumor content lowers the sensitivity of SCNA detection, and in order to avoid 13q amplicon false-negatives, we established the tumor fraction with the ichorCNA algorithm and excluded plasma DNA samples where the tumor fraction was too low. Another limitation of this study is that we only focused on one SCNA event. However, other somatic alterations may modulate therapeutic response to anti-VEGF treatment as well. For example, deletion of 18q11.2-q12.1, which co-occurred in 10 of our 14 chr13q12.2 amplified patients, was recently reported to be a predictive marker of survival for mCRC patients under undergoing treatment with bevacizumab [78].
This suggests that a variety of somatic alterations may govern response to anti-VEGF therapy so that further investigations are warranted.
Conclusions
Our results suggest that monitoring somatic focal events may allow identification of driver genes in mCRC, which has meaningful implications for the identification of novel driver genes associated with late-stage cancers. 13q12.2 is frequently amplified in CRC and may be related to tumor stage and metastasis. Here, POLR1D, a subunit of RNA polymerases I and III, was established as the most likely driver gene in this frequently amplified region, which may play a role in the oncogenesis of CRC by affecting VEGFA and EREG expression. As this may result in the acquired resistance to bevacizumab, POLR1D is a potential therapeutic target for mCRC.
Availability of data and materials
The datasets and computer code used in this study are available in the following databases:
• RNA-seq data: Gene Expression Omnibus GSE140198 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc= GSE140198)
• Low coverage WGS data: European Genome-phenome Archive EGAS00001003791 (https://www.ebi.ac.uk/ega/studies/EGAS00001003791)
• Focal SCNA identification analysis in R: GitHub (https://github.com/PeterUlz/FocalAmplifications/tree/master/Focal_amplifications_in_R.ipynb)
• TCGA-COADREAD RNA-seq: Broad GDAC Firehose [40] illuminahiseq_rnaseqv2RSEM_genes_normalized (http://gdac.broadinstitute.org/)
• TCGA-COADREAD clinic data: Broad GDAC Firehose [40] Clinical_Pick_Tier1 (http://gdac.broadinstitute.org/)
• TCGA copy number data: NCI Genomic Data Commons [41] ABSOLUTE-annotated seg file (https://gdc.cancer.gov/about-data/publications/pancanatlas)
Abbreviations
- AML:
-
Acute myeloid leukemia
- CCLE:
-
Cancer Cell Line Encyclopedia
- CRC:
-
Colorectal cancer
- CT:
-
Computerized tomography
- dPCR:
-
Digital PCR
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- ERBB4:
-
Receptor tyrosine-protein kinase erbB-4
- FDA:
-
Food and Drug Administration
- FFPE:
-
Formalin-fixed paraffin-embedded
- FLT3:
-
Fms-like tyrosine kinase 3
- GISTIC:
-
Genomic Identification of Significant Targets in Cancer
- mCRC:
-
Metastatic colorectal cancer
- rRNA:
-
Ribosomal ribonucleic acid
- SCNA:
-
Somatic copy number alteration
- SVM:
-
Support vector machine
- TCGA:
-
The Cancer Genome Atlas
- TSS:
-
Transcription start site
- VEGFA:
-
Vascular endothelial growth factor A
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–403.
DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252–71.
Saltz LB, Meropol NJ, Loehrer PJS, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8.
Reissfelder C, Timke C, Schmitz-Winnenthal H, Rahbari NN, Koch M, Klug F, et al. A randomized controlled trial to investigate the influence of low dose radiotherapy on immune stimulatory effects in liver metastases of colorectal cancer. BMC Cancer. 2011;11:419–2407.
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Bardelli A, Pantel K. Liquid biopsies, what we do not know (yet). Cancer Cell. 2017;31:172–9.
Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20:71–88.
Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–38.
Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017;14:531–48.
Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005;102:16368–73.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24.
Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017;9. https://doi.org/10.1126/scitranslmed.aan2415.
Mead R, Duku M, Bhandari P, Cree IA. Circulating tumour markers can define patients with normal colons, benign polyps, and cancers. Br J Cancer. 2011;105:239–45.
Lofton-Day C, Model F, Devos T, Tetzner R, Distler J, Schuster M, et al. DNA methylation biomarkers for blood-based colorectal cancer screening. Clin Chem. 2008;54:414–23.
Bedin C, Enzo MV, Del Bianco P, Pucciarelli S, Nitti D, Agostini M. Diagnostic and prognostic role of cell-free DNA testing for colorectal cancer patients. Int J Cancer. 2017;140:1888–98.
Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–30.
Reinert T, Henriksen TV, Christensen E, Sharma S, Salari R, Sethi H, et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol. 2019;5:1124–31.
Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra92.
Scholer LV, Reinert T, Orntoft MW, Kassentoft CG, Arnadottir SS, Vang S, et al. Clinical implications of monitoring circulating tumor DNA in patients with colorectal cancer. Clin Cancer Res. 2017;23:5437–45.
El Messaoudi S, Mouliere F, Du Manoir S, Bascoul-Mollevi C, Gillet B, Nouaille M, et al. Circulating DNA as a strong multimarker prognostic tool for metastatic colorectal cancer patient management care. Clin Cancer Res. 2016;22:3067–77.
Maierthaler M, Benner A, Hoffmeister M, Surowy H, Jansen L, Knebel P, et al. Plasma miR-122 and miR-200 family are prognostic markers in colorectal cancer. Int J Cancer. 2017;140:176–87.
Onstenk W, Sieuwerts AM, Mostert B, Lalmahomed Z, Bolt-de Vries JB, van Galen A, et al. Molecular characteristics of circulating tumor cells resemble the liver metastasis more closely than the primary tumor in metastatic colorectal cancer. Oncotarget. 2016;7:59058–69.
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90.
Tie J, Kinde I, Wang Y, Wong HL, Roebert J, Christie M, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol. 2015;26:1715–22.
Barault L, Amatu A, Siravegna G, Ponzetti A, Moran S, Cassingena A, et al. Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut. 2018;67:1995–2005.
Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40.
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6.
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905.
Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–40.
Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41–2011.
Ulz P, Heitzer E, Speicher MR. Co-occurrence of MYC amplification and TP53 mutations in human cancer. Nat Genet. 2016;48:104–6.
Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013;5:30.
Ulz P, Belic J, Graf R, Auer M, Lafer I, Fischereder K, et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat Commun. 2016;7:12008.
Mohan S, Heitzer E, Ulz P, Lafer I, Lax S, Auer M, et al. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet. 2014;10:e1004271.
Belic J, Graf R, Bauernhofer T, Cherkas Y, Ulz P, Waldispuehl-Geigl J, et al. Genomic alterations in plasma DNA from patients with metastasized prostate cancer receiving abiraterone or enzalutamide. Int J Cancer. 2018;143:1236–48.
Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet. 2016;48:1273–8.
Belic J, Koch M, Ulz P, Auer M, Gerhalter T, Mohan S, et al. Rapid identification of plasma DNA samples with increased ctDNA levels by a modified FAST-SeqS approach. Clin Chem. 2015;61:838–49.
Broad Institute TCGA Genome Data Analysis Center (2016): Firehose stddata__2016_01_28 run. Broad Institute of MIT and Harvard. doi:https://doi.org/10.7908/C11G0KM9.
Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a shared vision for cancer genomic data. N Engl J Med. 2016;375:1109–12.
Heitzer E, Auer M, Hoffmann EM, Pichler M, Gasch C, Ulz P, et al. Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int J Cancer. 2013;133:346–56.
Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8:1324–017.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550–014.
Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, Karchin R. Evaluating the evaluation of cancer driver genes. Proc Natl Acad Sci U S A. 2016;113:14330–5.
Zauli G, Voltan R, Tisato V, Secchiero P. State of the art of the therapeutic perspective of sorafenib against hematological malignancies. Curr Med Chem. 2012;19:4875–84.
Braxton DR, Zhang R, Morrissette JD, Loaiza-Bonilla A, Furth EE. Clinicopathogenomic analysis of mismatch repair proficient colorectal adenocarcinoma uncovers novel prognostic subgroups with differing patterns of genetic evolution. Int J Cancer. 2016;139:1546–56.
Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–20.
Martinez-Calvillo S, Saxena A, Green A, Leland A, Myler PJ. Characterization of the RNA polymerase II and III complexes in leishmania major. Int J Parasitol. 2007;37:491–502.
Wang M, Niu W, Hu R, Wang Y, Liu Y, Liu L, et al. POLR1D promotes colorectal cancer progression and predicts poor prognosis of patients. Mol Carcinog. 2019;58:735–48.
George ML, Tutton MG, Janssen F, Arnaout A, Abulafi AM, Eccles SA, et al. VEGF-A, VEGF-C, and VEGF-D in colorectal cancer progression. Neoplasia. 2001;3:420–7.
Kramer I, Lipp HP. Bevacizumab, a humanized anti-angiogenic monoclonal antibody for the treatment of colorectal cancer. J Clin Pharm Ther. 2007;32:1–14.
Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer: a review. Ther Adv Med Oncol. 2016;8:57–84.
Mesange P, Poindessous V, Sabbah M, Escargueil AE, de Gramont A, Larsen AK. Intrinsic bevacizumab resistance is associated with prolonged activation of autocrine VEGF signaling and hypoxia tolerance in colorectal cancer cells and can be overcome by nintedanib, a small molecule angiokinase inhibitor. Oncotarget. 2014;5:4709–21.
Shirakata Y, Komurasaki T, Toyoda H, Hanakawa Y, Yamasaki K, Tokumaru S, et al. Epiregulin, a novel member of the epidermal growth factor family, is an autocrine growth factor in normal human keratinocytes. J Biol Chem. 2000;275:5748–53.
Auf G, Jabouille A, Delugin M, Guerit S, Pineau R, North S, et al. High epiregulin expression in human U87 glioma cells relies on IRE1alpha and promotes autocrine growth through EGF receptor. BMC Cancer. 2013;13:597–2407.
Takahashi N, Furuta K, Taniguchi H, Sasaki Y, Shoji H, Honma Y, et al. Serum level of hepatocyte growth factor is a novel marker of predicting the outcome and resistance to the treatment with trastuzumab in HER2-positive patients with metastatic gastric cancer. Oncotarget. 2016;7:4925–38.
Lo YM, Chan KC, Sun H, Chen EZ, Jiang P, Lun FM, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2:61ra91.
Sartore-Bianchi A, Amatu A, Porcu L, Ghezzi S, Lonardi S, Leone F, et al. HER2 positivity predicts unresponsiveness to EGFR-targeted treatment in metastatic colorectal cancer. Oncologist. 2019;24:1395–402.
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–23.
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86.
Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev. 2012;6:e8.
Moreira RB, Peixoto RD, de Sousa Cruz MR. Clinical response to sorafenib in a patient with metastatic colorectal cancer and FLT3 amplification. Case Rep Oncol. 2015;8:83–7.
Lim SH, Kim SY, Kim K, Jang H, Ahn S, Kim KM, et al. The implication of FLT3 amplification for FLT targeted therapeutics in solid tumors. Oncotarget. 2017;8:3237–45.
Camps J, Pitt JJ, Emons G, Hummon AB, Case CM, Grade M, et al. Genetic amplification of the NOTCH modulator LNX2 upregulates the WNT/beta-catenin pathway in colorectal cancer. Cancer Res. 2013;73:2003–13.
Salari K, Spulak ME, Cuff J, Forster AD, Giacomini CP, Huang S, et al. CDX2 is an amplified lineage-survival oncogene in colorectal cancer. Proc Natl Acad Sci U S A. 2012;109:E3196–205.
Graule J, Uth K, Fischer E, Centeno I, Galvan JA, Eichmann M, et al. CDX2 in colorectal cancer is an independent prognostic factor and regulated by promoter methylation and histone deacetylation in tumors of the serrated pathway. Clin Epigenetics. 2018;10:120–018.
Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res. 2017;77:3655–65.
Fan F, Samuel S, Gaur P, Lu J, Dallas NA, Xia L, et al. Chronic exposure of colorectal cancer cells to bevacizumab promotes compensatory pathways that mediate tumour cell migration. Br J Cancer. 2011;104:1270–7.
Pentheroudakis G, Kotoula V, De Roock W, Kouvatseas G, Papakostas P, Makatsoris T, et al. Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC Cancer. 2013;13:49–2407.
Cheng C, Cui H, Zhang L, Jia Z, Song B, Wang F, et al. Genomic analyses reveal FAM84B and the NOTCH pathway are associated with the progression of esophageal squamous cell carcinoma. Gigascience. 2016;5:1–015.
Wong N, Gu Y, Kapoor A, Lin X, Ojo D, Wei F, et al. Upregulation of FAM84B during prostate cancer progression. Oncotarget. 2017;8:19218–35.
Mo Z, Zhang Q, Liu Z, Lauer J, Shi Y, Sun L, et al. Neddylation requires glycyl-tRNA synthetase to protect activated E2. Nat Struct Mol Biol. 2016;23:730–7.
Arai T, Kojima S, Yamada Y, Sugawara S, Kato M, Yamazaki K, et al. Pirin: a potential novel therapeutic target for castration-resistant prostate cancer regulated by miR-455-5p. Mol Oncol. 2019;13:322–37.
Roh SA, Park IJ, Yoon YS, Kwon YH, Chung JH, Kim TW, et al. Feasibility of novel PPP1R15A and proposed ANXA11 single nucleotide polymorphisms as predictive markers for bevacizumab regimen in metastatic colorectal cancer. J Cancer Res Clin Oncol. 2016;142:1705–14.
Salem Y, Yacov N, Propheta-Meiran O, Breitbart E, Mendel I. Newly characterized motile sperm domain-containing protein 2 promotes human breast cancer metastasis. Int J Cancer. 2019;144:125–35.
van Dijk E, Biesma HD, Cordes M, Smeets D, Neerincx M, Das S, et al. Loss of chromosome 18q11.2-q12.1 is predictive for survival in patients with metastatic colorectal cancer treated with bevacizumab. J Clin Oncol. 2018;36:2052–60.
Acknowledgements
We thank all the patients who participated in the study. We are grateful to Dr. Alberto Bardelli, Laboratory Molecular Oncology at the Candiolo Cancer Institute IRCCS-Candiolo (Torino) for providing us with the OXCO-2 cell line.
The results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding
This work was supported by CANCER-ID, a project funded by the Innovative Medicines Joint Undertaking (IMI JU; #115749-1), by the BioTechMed-Graz flagship project “EPIAge,” and by the Austrian Federal Ministry for Digital and Economic Affairs (Christian Doppler Research Fund for Liquid Biopsies for Early Detection of Cancer).
Author information
Authors and Affiliations
Contributions
QZ, SOP, SM, EH, JBG, and MRS designed the research. QZ and SOP performed the experiments. JMR, TB, MP, and AG treated the patients and provided the clinical samples and clinical information. SL, MT, and GH collected the primary tumor tissues. ET processed all the imaging data. PU supported with bioinformatics analyses. QZ, SOP, EH, and MRS analyzed the data. QZ, SOP, EH, and MRS wrote the paper. JBG, EH, and MRS supervised the study. All authors read, revised, approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the Medical University of Graz (approval number 21-229 ex 09/10), conducted according to the Declaration of Helsinki, and written informed consent was obtained from all patients.
Consent for publication
Not applicable
Competing interests
PU is employed by Freenome. The remaining authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Age spectrum and sex distribution of the CRC cohort used in this study.
Additional file 2: Figure S1.
Identification of the 13q amplicon. Figure S2. Genome-wide SCNA plots of all 13q12 focal amplification patients and three CRC cell lines used in this study. Figure S3. Definition of the minimally 13q12 amplified region and identification of the potential driver genes. Figure S4. Expression analyses for identification of the potential driver gene in the 13q12.2 amplicon. Figure S5. Emergence of the 13q12 amplicon under bevacizumab treatment in patient C216. Figure S6. Alternating POLR1D and ERBB2 amplifications in serial plasma analyses of patient C129.
Additional file 3: Table S1.
Summary of genes differently expressed after POLR1D knockdown in both HT29 and SW480 cells and their expression levels in TCGA dataset. Table S2. Summary of all patient data for C216 and C129. Table S3. Summary of all the siRNA oligos.
Additional file 4:
Summary of recurrent focal events in our cohort and TCGA dataset. Our Cohort: Summary of all recurrent focal events with a frequency over 0.01 (Upper) or 0.05 (Bottom) in our CRC cohort. Potential driver genes were identified as in Fig. 1a. TCGA: Summary of all recurrent focal events with a frequency over 0.01 (Upper) or 0.05 (Bottom) in TCGA cohort. Potential driver genes were identified as in Fig. 1a.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, Q., Perakis, S.O., Ulz, P. et al. Cell-free DNA analysis reveals POLR1D-mediated resistance to bevacizumab in colorectal cancer. Genome Med 12, 20 (2020). https://doi.org/10.1186/s13073-020-0719-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-020-0719-6