
Abstract
HIV/AIDS is a chronic and debilitating disease that cannot be cured with current antiretroviral drugs. While combinatorial antiretroviral therapy (cART) can potently suppress HIV-1 replication and delay the onset of AIDS, viral mutagenesis often leads to viral escape from multiple drugs. In addition to the pharmacological agents that comprise cART drug cocktails, new biological therapeutics are reaching the clinic. These include gene-based therapies that utilize RNA interference (RNAi) to silence the expression of viral or host mRNA targets that are required for HIV-1 infection and/or replication. RNAi allows sequence-specific design to compensate for viral mutants and natural variants, thereby drastically expanding the number of therapeutic targets beyond the capabilities of cART. Recent advances in clinical and preclinical studies have demonstrated the promise of RNAi therapeutics, reinforcing the concept that RNAi-based agents might offer a safe, effective, and more durable approach for the treatment of HIV/AIDS. Nevertheless, there are challenges that must be overcome in order for RNAi therapeutics to reach their clinical potential. These include the refinement of strategies for delivery and to reduce the risk of mutational escape. In this review, we provide an overview of RNAi-based therapies for HIV-1, examine a variety of combinatorial RNAi strategies, and discuss approaches for ex vivo delivery and in vivo delivery.
Similar content being viewed by others

Introduction
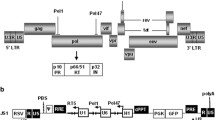
The genome of the human immunodeficiency virus-1 (HIV-1) is composed of nine viral genes (gag, pol, vif, vpr, tat, rev, vpu, env, and nef) that are required for all processes in the viral replicative cycle, including viral assembly, viral entry and receptor binding, membrane fusion, reverse transcription, integration, and proteolytic protein processing (Fig. 1). HIV-1 preferentially infects human CD4+ T-lymphocytes, eventually leading to a depletion of CD4+ cells and the clinical progression to acquired immunodeficiency syndrome (AIDS). Since the discovery of the antiretroviral activity of azidothymidine (AZT) in 1985 [1], a variety of antiretroviral drugs have been developed that target multiple steps in the viral replication cycle (Table 1). Typically, a cocktail of three antiretroviral drugs is used as a combinatorial antiretroviral therapy (cART) that can effectively suppress viral replication, reduce rates of transmission, and improve patients’ life expectancy by prolonging the onset of AIDS [2]. Nevertheless, cART does not fully restore health, as patients on cART still experience chronic inflammation and increased rates of non-AIDS morbidity and mortality [3]. Moreover, current cART regimens are unable to cure HIV-1 patients, as these drugs cannot eradicate latent viral reservoirs and may also fall short in completely suppressing viral replication despite drug intensification [4–7]. Thus, nearly all HIV-infected individuals on cART will need to maintain their medications for the entirety of their lives, resulting in considerable expense, development of resistance, and toxic side effects. Owing to improvements in the efficacy and availability of antiretroviral drugs and efforts in HIV/AIDS prevention, recent years have seen a decrease in the incidence of HIV-1 infection. Nevertheless, there are still 35.3 million people living with HIV-1, with 2.3 million new cases and 1.6 million deaths in 2012 [8]. Because of the limitations and adverse effects associated with current cART regimens, it is necessary to develop alternative therapeutic strategies that are safer, more efficacious, and more resistant to viral escape. Such emerging therapeutic strategies include gene-based and nucleic-acid-based therapies that are based on gene editing, ribozymes, and RNA interference (RNAi).

The HIV-1 genome and strategies for antiviral targeting. a Structure of the HIV-1 genome. The genome contains nine genes and two long terminal repeats (LTRs) that can be targeted by RNA interference (RNAi). Certain genomic regions are more conserved than others, making them better targets. In addition, many of the genes are alternatively spliced, requiring careful target design. b HIV-1 targeting. Several steps of the HIV-1 viral replication cycle can be targeted by RNAi. Current drug targets are in parentheses. (1) The first step is receptor binding and membrane fusion by the HIV envelope glycoproteins gp120 and gp41 to host receptors CD4 and either CCR5 or CXCR4. This step can be inhibited by knocking down the HIV-1 co-receptors, CCR5 or CXCR4. (2) Next, the viral genome must be reverse transcribed by the viral reverse transcriptase (RT) and (3) integrated into the cellular genome which is mediated by the viral integrase protein and host factors LEDGF, Importin, and Chaperonin. After integration, (4) the virus is transcribed, which is mediated by viral (TAR and tat) and host (pTEFb, tat-SF1, SPT5, cyclin T1) factors, (5) exported to the cytoplasm (dependent on DDX3 and Rev) and then translated and (6) subjected to post-translational processing by the viral protease. (7) Finally, the proteins are processed and (8) packaged into new viral particles
Like cART, gene-therapy strategies against HIV-1 are often designed to target multiple viral mechanisms, which reduces the possibility of viral-escape-induced drug resistance and improves therapeutic efficacy. This is particularly true for RNAi therapeutics, as small interfering RNAs (siRNAs) and short hairpin RNAs (shRNAs) can be designed to target conserved 21-nucleotide sequences within the 9.2-kb HIV-1 genomic RNA, thereby expanding the possible targets far beyond those of current drugs. Moreover, multiplexing siRNAs or shRNAs as a single therapy can increase the overall potency and reduce the dosage of each component required to minimize off-target toxicity.
This review highlights recent advances in gene-therapy technologies for the treatment and eradication of HIV-1. Many new therapies are currently in development with the hope of preventing drug resistance, toxicity, and latent infection. Owing to their design flexibility and target specificity, we pay particular attention to RNAi-based therapies that target viral or host targets, including some that have reached human clinical testing. Delivery of the RNAi agents to HIV-infected or HIV-susceptible cells remains a challenging problem that is currently being addressed by ex vivo delivery, targeted in vivo delivery, and non-targeted in vivo delivery strategies.
HIV-1 infection and therapeutic targets
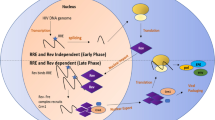
HIV-1 gains entry into CD4-expressing T-lymphocytes, macrophages, and monocytes by fusion of the viral envelope with the host cell membrane (reviewed in [9]). The HIV-1 envelope is characterized by trimeric spikes, and each spike contains three glycoprotein 120 (gp120) surface subunits and three gp41 trans-membrane subunits. The process of viral binding and entry into the cell generally requires the CD4 receptor and either the CCR5 or the CXCR4 co-receptor. Upon receptor-mediated binding and internalization, the viral RNA genome is converted to double-stranded DNA, using reverse transcriptase that is provided by the virus. The viral genomic DNA is then imported into the nucleus and integrated into the host genome, where it is transcribed into the full-length 9.2-kb mRNA transcript. This transcript can undergo double splicing into 1.8-kb mRNAs encoding Tat, Rev, and Nef proteins. Tat and Rev travel to the nucleus where Tat activates transcription and Rev mediates the export of mRNAs in the late phase. Singly-spliced 4.0-kb transcripts encoding Env, Vpu, Vpr, and Vif proteins [10] and unspliced 9.2-kb mRNA transcripts encoding Gag and Gag-Pol are exported from the nucleus in a Rev-dependent manner. These unspliced mRNAs also function as the genomic RNA for progeny virus. Several Gag molecules bind to the viral genome and are trafficked to lipid rafts on the plasma membrane of the host cell [11, 12]. The viral genome is assembled as a dimer with viral proteins and exits the cell via budding. Final maturation occurs once the viral particles are released from the host cell. These steps provide many opportunities for therapeutic intervention.
Owing to the limitations of current cART approaches, alternative gene- and nucleic-acid-based therapies have been proposed to control HIV-1 infection, including gene editing, ribozymes, and utilization of RNA decoys. In addition, gene- and nucleic-acid-based agents have been developed as HIV-1 prophylactics, including RNA aptamers and siRNAs that are used as microbicides and viral genes used as vaccines (Table 1).
Gene editing, which requires a sequence-specific nuclease that modifies a target gene, is a particularly attractive approach that aims to develop cells that are permanently resistant to HIV-1 infection or to eliminate the HIV-1 provirus from infected cells [13]. HIV-resistant cells can be established by genomic editing of the CCR5 or CXCR4 co-receptors using zinc finger nucleases (ZFNs) [14–21], transcription activator-like effector nucleases (TALENs) [22], designer endonucleases [23], peptide nucleic acids (PNAs) [24], or CRISPR/Cas9 [22, 25, 26]. Alternatively, the integrated HIV-1 genome in infected cells can be targeted and destroyed using CRISPR/Cas9 [27, 28], homing endonucleases [29], and HIV-1 long terminal repeat (LTR)-specific recombinases [30–32]. Catalytic RNAs, known as ribozymes, can complete a similar task, but the effects are not permanent because their target molecule is mRNA [33]. RNA aptamers and decoys can bind viral proteins to act as steric or competitive inhibitors [34–36]. By combining an anti-HIV-1 microRNA (miRNA) with a TAR decoy (TARmiR), a dual-functioning RNA molecule can suppress HIV-1 expression by RNAi and also bind to the HIV-1 transactivating protein Tat, reducing the numbers of Tat molecules that can bind to its cognate target TAR to transactivate viral transcription [37]. Several of these technologies have reached clinical testing as single or combinatorial HIV-1 therapies [2], including CCR5 ZFN [20], CCR5 ribozymes [38, 39], HIV-1 ribozymes [40–43], and TAR decoys [38, 39].
Endogenous RNA silencing pathway
RNAi is a natural cellular mechanism first identified by Craig Mello and Andrew Fire in 1998, which involves the pairing of a short miRNA sequence to an endogenous mRNA target [44]. Endogenous miRNAs regulate many cellular mechanisms and are processed in a very specific manner. First, double-stranded miRNAs are loaded into the RNA-induced silencing complex (RISC), where the non-targeting (passenger) strand is removed. The RISC complex then finds the complementary mRNA sequence for the guide strand of the miRNA. The binding of the miRNA to the mRNA target results in gene silencing through degradation of the mRNA (Fig. 2). While the canonical RNAi pathway occurs in the cytoplasm, RNAi can also function in the nucleus to induce gene-specific transcriptional gene silencing or transcriptional gene activation by directing epigenetic changes in gene promoters [45–47]. Teasing out these mechanisms has resulted in a better understanding of how mammalian cells use this system to regulate endogenous gene expression.

RNA silencing mechanism. For therapeutic RNAi applications, a lentiviral vector or plasmid is transcribed by RNA pol III into shRNA. This shRNA is then exported from the nucleus to the cytoplasm by Exportin 5. After export, shRNA or miRNA are processed by Dicer and loaded into RISC. For exogenously delivered siRNA or Dicer substrate RNA (dsiRNA), the processed siRNA is loaded directly into RISC. The passenger strand is removed from the guide strand and RISC guides the remaining strand to its complementary mRNA target. RISC then cleaves the mRNA for degradation. Abbreviations: pol III, RNA polymerase III; pol II, RNA polymerase II; RISC, RNA-induced silencing complex; shRNA, short hairpin RNA; siRNA, short interfering RNA
While the role of the RNAi system in mammalian cells is well documented (reviewed in [48]), the role of mammalian RNAi in response to viral infections is still the subject of much debate [49]. It is well established that viral RNAs trigger innate interferon (IFN) responses in mammalian cells as a first line of defense. While viral small RNAs (vsRNAs) have been detected in infected mammalian cells [50], it was previously uncertain whether vsRNAs could function via an antiviral RNAi pathway. One reason for this uncertainty is that many viruses encode viral suppressors of RNAi (VSRs), which are viral proteins that inhibit the host RNAi pathway [51, 52]. Two important studies from 2013, which examined conditions that are unfavorable for VSRs, reported that vsRNAs act via an RNAi pathway [53, 54]. Encephalomyocarditis virus infection of undifferentiated mouse embryonic stem cells, which have low or no innate IFN response, resulted in the accumulation of virus-derived siRNAs (vsiRNAs) that associated with Argonaute-2 (Ago2) [53]. vsiRNAs were not, however, observed in differentiated cells, which have high innate IFN activity. Thus, antiviral RNAi appears to be most prevalent in conditions that are unfavorable for innate IFN viral clearance. Similarly, infection by a Nodamura virus strain that contained a mutant VSR protein was attenuated because host cells produced vsRNAs in response to infection with these mutant viruses [54]. Despite these compelling reports, seemingly contradictory observations have recently been reported that indicate that viral RNAi does not occur in Dicer-knockout cells or in cells with disabled IFN function [55, 56]. Thus, the potential role of antiviral RNAi in mammalians remains controversial and appears to be highly dependent on the host cells and the particular virus.
The RNA silencing pathway provides a tool for gene therapy by the delivery of artificial RNAi in the form of a small RNA duplex, such as a miRNA, siRNA, Dicer substrate RNA (DsiRNA), or shRNA [57]. Although siRNA, DsiRNA, and shRNA all function through the same RNAi pathway, shRNA requires additional processing in the nucleus (Fig. 2). shRNAs differ from siRNAs because they comprise a single strand of RNA with two complementary tails connected by a hairpin loop, and are commonly transcribed from viral vectors or plasmid DNA in the nucleus. Following transcription, the shRNA is exported to the cytoplasm, where it is processed in the same manner as siRNA by Dicer in complex with TRBP, a member of the RISC. Alternatively, Dicer-independent Ago2-shRNAs are shRNAs that have a short stem length and are incorporated into RISC without Dicer processing [58]. After incorporation into RISC, the processed siRNA is loaded onto an mRNA target to induce either mRNA cleavage by Ago2 or translational repression and subsequent mRNA degradation.
Toxicity of RNAi can occur through improper target recognition resulting from suboptimal annealing of the antisense guide strand to the target strand, improper selection of an unintended passenger strand by the RISC complex, or overloading of the silencing machinery. Biasing the strand-selection of the guide strand, rather than of the passenger strand, can enhance potency while also reducing off-target effects of the passenger strand. Strand selection can be biased by designing asymmetric siRNAs with less stable thermodynamic stability at the 5′ end of the guide strand [59, 60], by chemical modifications at one or both ends of the siRNA [61, 62], or by the design of structurally asymmetric shRNAs or siRNAs that have a single 2-nucleotide 3′-overhang on the guide strand [63–66]. One of the most reliable design strategies for generating strand-biased siRNAs is to use asymmetric 25/27-nucleotide DsiRNAs [64, 65]. After processing by Dicer, the guide strand from the asymmetric 25/27mer DsiRNA is preferentially incorporated into the RISC, although the siRNA potency can still be influenced by thermodynamic stabilities at both ends [67, 68].
In addition, overloading can be avoided by limiting the amounts of exogenous RNA added or by using single stranded siRNA [69]. Toxicity in vivo often results from the immunogenic effects of introducing foreign RNA into cells. Most issues arise as a result of IFN-α production following Toll-like receptor 7/8 activation. These issues can be abrogated by replacing uridines with 2′-O-methyluridine in siRNAs [70]. Additionally, delivery agents may cause harmful side effects because the introduced RNA is trafficked through pathways that expose them to the innate immune system. For example, liposome-mediated delivery of siRNA exposes the RNA to endosomal Toll-like receptors, cytoplasmic RIG-I and protein kinase R (PKR), and other inflammatory pathways [71, 72]. Other delivery agents expose the RNA to a completely different set of pattern recognition receptors, resulting in an interferon response and inflammation [73].
Therapeutic RNA interference strategies
Many researchers have used RNAi to develop therapeutics against acute and persistent viral infections, and several clinical trials have been carried out. For acute viral infections, siRNA treatments for respiratory syncytial virus (NCT01065935) and influenza (NCT01747148) are currently in clinical trials. These viruses present good targets for RNAi therapeutics because they cause acute infections with pathogenic effects that can be prevented using this approach. In addition, an acute infection is often localized to a single organ or region, requiring only a relatively straightforward delivery approach for RNAi. To date, Alnylam (Cambridge, MA, USA) has successfully conducted phase II clinical trials using the RNAi therapeutic ALN-RSV01, which targets RSV nucleocapsid RNA [74]. Safety and tolerability have been reported, paving the way for future clinical trials to investigate efficacy.
RNAi approaches have also been investigated to treat persistent viral infections such as HIV-1, hepatitis B, and hepatitis C. These are targeted in a different manner to acute infections because the RNA therapy must be present for a longer period of time [75, 76]. The pre-clinical efficacy is promising; nevertheless, a different set of problems are associated with using RNAi for long-term infections and these issues must be further explored before moving to the clinic [77, 78]. The viral proteins and cellular machinery necessary for viral replication are obvious targets for preventing initial infection, but viral targets are susceptible to mutational escape resulting from the relatively low fidelity of the viral RNA polymerase enzymes. Additionally, in the case of HIV-1, cells may be infected with latent virus that can persist for years before viral production, with no or only low levels of viral mRNA being produced during the latent period. Nevertheless, in vitro targeting of initial HIV-1 infection has been very successful at inhibiting viral replication and the infection of new cells [79, 80]. Translating the success of RNAi against HIV-1 in vitro to an in vivo or clinical setting has been hampered by problems with delivery and sustained expression [81, 82]. An optimal delivery agent would efficiently target only those cells with the potential to be infected by HIV-1, or already-infected cells. Current delivery methods do not offer this advantage, so non-specific or ex vivo methods are used. Enhancing RNAi expression is also necessary to advance therapeutics to a clinical setting; this can be achieved by increasing the molar ratio of RNAi to delivery reagent or by improving the potency of the RNAi [83]. Alternatively, the HIV-1 promoter can be targeted using transcriptional gene silencing to prevent reactivation of viral production [47, 84]. Peripheral blood mononuclear cells transduced with a lentivirus expressing shRNA against the U3 region of HIV-1 successfully repopulated CD4+ T-cell lineages in mice after HIV-1 infection, suggesting that this approach might be a feasible functional cure [85]. These approaches are very promising, but must be carefully assessed because increasing the concentration of RNAi also increases the possibility of off-target effects and/or the mutation rate [86, 87].
RNA interference approaches for HIV-1 treatment
RNA-silencing approaches have been designed to control viral replication by blocking key steps within the viral replication cycle [88]. Several considerations must be made when choosing targets for HIV-1 treatment with RNAi (Table 2), including design strategies to target highly conserved regions within viral RNAs or cellular mRNAs that are necessary for viral production [89].
Viral targets
The viral genome (Fig. 1a) consists of several highly conserved regions that have proven to be useful targets as viral escape via mutagenesis is less likely in these regions than in other parts of the genome. To identify highly conserved regions, it is useful to analyze HIV-1 sequences from global clades and primary isolates that are available online from the Los Alamos National Laboratory [90].
Selection of targets for RNAi-based therapies against HIV-1 is a crucial step in obtaining a long-term therapeutic effect without the emergence of resistant strains. Various steps throughout the HIV-1 replication cycle have been targeted, although it can be particularly beneficial to inhibit the early stages of infection (class I targets), such as viral entry, reverse transcription, and integration [2, 91]. Owing to the versatility of RNAi-based therapeutics against any HIV-1 mRNA sequence, it is also possible to target steps of the HIV-1 replication cycle that are not targeted by clinical cART agents (Table 2).
As the unspliced HIV-1 genomic RNA transcripts contain the full sequences for all nine viral genes, it is possible to design anti-HIV-1 siRNAs against any particular conserved coding region. Owing to alternative splicing patterns, however, it can be advantageous to design siRNAs against the common sequences that occur on all spliced and unspliced HIV-1 transcripts, including the early spliced viral mRNAs that contain tat, rev, and nef reading frames. These sequences are among the earliest spliced transcripts of the integrated provirus. Thus, targeting with siRNA can inhibit production of the associated protein and downstream proteins necessary for viral maturation, because the early proteins are required for production of late transcripts. Target sites present in the HIV-1 subgenomic RNAs, including nef and the untranslated LTR regions, can be targeted in all spliced and unspliced transcripts. Alterations in the RNA folding and structure of these regions can, however, lead to RNAi-resistance [92, 93]. Alternatively, the targeting of genes that encode the structural and enzymatic proteins required for viral assembly and infection (including gag, pol, vif, and env) is effective, despite the fact that these genes are not found in all transcripts [94–97]. While the integrase and reverse transcriptase proteins are targeted by different classes of antiretroviral drugs, the coding regions of these proteins can also be targeted by RNAi. Most approaches have relied on using RNAi to target viral mRNA transcripts in the cytoplasm, but the HIV-1 promoter may also be targeted by transcriptional gene silencing, which induces epigenetic silencing of the integrated provirus [47, 85, 98, 99].
HIV-1 rapidly escapes drug inhibition and RNAi because of its high rate of mutation, so finding highly conserved regions of the HIV-1 genome to target with RNAi is a common strategy. Escape from targeting by mutagenesis at conserved regions may be restricted by a consequent loss of viral fitness. A major goal of developing anti-HIV-1 RNAi therapy is to target HIV-1 in a manner that prevents viral escape; such strategies may include targeting multiple viral sites and endogenous host factors that are required for HIV-1 infection and replication.
Owing to the low fidelity of HIV-1 reverse transcriptase, with an estimated error rate of 3 × 10−5 bases per replication [100, 101], the virus can evolve to escape most monotherapies. A variety of mechanisms are responsible for RNAi escape, including single-point mutations (which may disrupt the specific recognition of and binding to the siRNA-binding site), deletion of the target sequence, and transcriptional upregulation [102]. Single-point mutations within the target site of a specific siRNA and mutations that cause structural changes to the mRNA can abate guide-strand recognition of the target mRNA, thereby preventing Ago2-mediated silencing [92, 103–105]. Indirect resistance, such as mutation of non-targeted sites within the viral promoter, can lead to upregulation of viral transcription, thereby allowing viral escape by overwhelming the RNAi-mediated selective pressure [106, 107]. Mutational escape can be avoided by using combination siRNAs that target multiple sites, which may act synergistically to lower the overall required siRNA concentration, or by combining siRNAs with other cART drugs [78, 108]. Like cART, such siRNA-based cocktail approaches increase the threshold for mutational escape, and any HIV-1 escape variants that do occur may also result in a loss of viral fitness [109–111].
Cellular targets
Targeting the host cellular co-factors that are required for HIV-1 infection is an attractive option for RNAi therapies because endogenous genes are not as prone to mutational escape as viral genes, which must undergo reverse transcription by an error-prone polymerase [75, 112]. The concept of generating an HIV-resistant immune system has been supported by a report on the ‘Berlin Patient’ [113], an individual with HIV-1 infection who required an allogeneic bone marrow transplant for acute myeloid leukemia. The donor hematopoietic progenitor cells (HPCs) were homozygous for a CCR5 partial gene deletion (CCR5∆32). In the absence of the CCR5 co-receptor on the surface of the HPC-derived CD4+ T cells, macrophages, and monocytes from the donor, the CCR5-tropic HIV-1 virus was unable to infect new cells. After the transplant, HIV-1 was no longer present at detectable levels, suggesting that the patient has been functionally cured of HIV/AIDS [114, 115]. This approach is not feasible for the majority of HIV/AIDS patients who do not require bone marrow transplants for pre-existing cancer, but other modes of cellular targeting can be considered. Moreover, a second attempt at an allogeneic transplant of CCR5∆32 HPCs was not successful because CXCR4-tropic virus became dominant after the interruption of cART [116]. Therefore, it may be necessary to target multiple cellular co-factors, including endogenous genes that are required for cell entry, integration, transcription, and maturation of the virus.
Cellular co-factors that are required for infection include the CD4 receptor as well as the CCR5 and CXCR4 co-receptors. Targeting these receptors can protect the cell from viral entry and infection, but this strategy has limitations depending on the receptor targeted [117, 118]. CCR5 is an attractive target because it is required for CCR5-tropic viral infection and, because it shares functional redundancy with CCR2, it is not required for the normal development and function of T-lymphocytes [119]. By contrast, CXCR4 and CD4 receptors are less favored as targets as they are involved in important cellular processes. CXCR4 is required for T-cell maturation, so disruption of CXCR4 must be targeted only in mature T cells [118]. Targeting of CD4 is also problematic because of its significant immunological function [120].
Host factors play a role in the integration of the HIV-1 viral genome into cells. Therefore, the HIV-1 replication cycle may be disrupted by using these host factors as targets [75, 121]. Co-factors of integrase, including LEDGF/p75, Importin-7, and Chaperonin, are potential targets [78]. Elongation factors P-TEFb, Tat-SF1, and SPT5 regulate HIV-1 transcription by interaction with the Tat protein [87]. Cyclin T1 interacts with P-TEFb and Tat to initiate HIV-1 transcription [122]. The human RNA helicase DDX3 is responsible for the export of spliced HIV-1 transcripts, and human SOCS1 is responsible for trafficking of HIV-1 gag [123]. Additionally, RNAi has been used to inhibit several autophagy inhibitory factors that are responsible for HIV-1 replication and prevention of HIV-1 degradation [121]. As HIV-1 is capable of modulating the immune system through TRBP-TAR-mediated inhibition of PKR- or SOCS1-dysregulation of immune activation, silencing these genes results in an antiviral response [124]. RNAi silencing of TRBP can also inhibit HIV-1 replication [75, 125, 126], but this could potentially disrupt the RNAi pathway because of the role of TRBP in Dicer processing and RISC assembly [127]. In general, cellular targets must be considered with caution because these targets affect cell growth and function, and because they may be important for cell signaling not observed in vitro. Another approach is to use a combination of RNAi or other biological agents against cellular and viral targets using gene therapy [128]. There are two clinical trials using this approach [38, 129], which will be discussed in the ex vivo delivery section below.
Delivery
One of the largest hurdles in developing effective gene therapy is delivery. Considerations for the delivery of RNAi include immunogenicity, cell type uptake, and specificity. RNAi-based therapeutics can be delivered as RNA molecules (such as siRNAs or shRNAs), as DNA (plasmids or minicircles), or as therapeutic transgenes in viral vectors [130]. Delivery reagents can be targeted to specific cell types or can be non-specific. The advantage of targeted delivery is that side effects can be reduced, while increasing the efficiency of delivery to target cells. To date, the RNAi-based agents that have reached clinical testing for HIV/AIDS have relied on ex vivo delivery of lentiviral vectors into specific cell types, such as CD4+ T cells or CD34+ HPCs, which are then transfused back into the patient [2]. Alternatively, in vivo delivery approaches may utilize non-targeted or targeted delivery agents, which include non-viral carriers such as liposomes, dendrimers, and aptamers.
Ex vivo delivery
Ex vivo delivery approaches for RNAi-based therapeutics include viral vectors, nanoparticles, and electroporation [131]. The most common of these is ex vivo transduction with virus, including adenovirus, adeno-associated virus (AAV), and lentivirus. shRNAs can be encoded in viral vectors to produce stable cell lines and to transduce cells that are refractory to transfection, such as hematopoietic stem cells and T cells [132]. In viral vectors derived from adenovirus, AAV, and lentivirus, shRNA transcripts driven by polymerase III promoters synthesize a single-strand hairpin loop that can be incorporated into the RNAi pathway. Lentivirus is integrative, so the RNAi therapy delivered is incorporated into the cellular genomic DNA, and although the site of insertion is semi-random, lentiviral vectors do not share the same tendency for insertional oncogenesis as gammaretroviral vectors [133]. As lentiviral vectors are constructed using a limited HIV-1 genome, vector production and transduction of the lentiviral vector may be poor when expressing vector-encoded shRNAs that target regions that are common to HIV-1 transcripts and lentiviral vector transcripts, such as the LTR region, or regions that are encoded by helper plasmids, such as gag, pol, and rev [134, 135]. Different strategies can be employed to overcome this restriction by increasing the amount of helper plasmids [135] or by knocking down Ago2 with a separate shRNA [136]. As a ssDNA virus, AAV is mostly non-integrative, although limited integration occurs at a specific locus within cellular genomic DNA. Several non-viral approaches have been taken, including plasmid delivery in liposomes and other nanoparticles, but this results in very inefficient delivery of transcripts to T cells; transcripts that are delivered in this way are also non-integrative, limiting the longevity of their therapeutic value. Other non-specific strategies are non-integrative but also have reduced efficiency compared to lentivirus [137].
The first clinical demonstration of the use of RNAi against HIV-1 was a phase 0 safety and feasibility clinical trial conducted by the City of Hope Hospital (Duarte, CA, USA) and Benitec, Inc. (Sydney, Australia). This study involved transducing human CD34+ HPCs with a self-inactivating and non-replicating lentivirus containing a tat/rev shRNA, a TAR decoy, and a CCR5 ribozyme [38]. Four patients with AIDS-related lymphoma received autologous bone marrow transplants with lentiviral-transduced CD34+ HPCs in an effort to treat both lymphoma and HIV/AIDS. The lentiviral-based therapy was well tolerated and no drug-related adverse events were observed in the four patients. Expression of the shRNA and ribozyme components was detected in the peripheral blood and bone marrow of all four patients for at least 8 months, and for more than 3 years in one patient [38, 39]. More recently, we have developed a next-generation lentiviral construct that expresses three small anti-HIV-1 RNAs from the MCM7 intron and a fourth CCR5 shRNA from the tRNASer promoter [136, 138].
Calimmune, Inc. (Los Angeles, CA, USA) has developed a similar strategy using a dual-therapeutic anti-HIV-1 lentiviral vector that expresses a CCR5 shRNA and a C46 peptide, which is derived from the HIV-1 gp41 envelope protein that inhibits viral fusion [139]. In a current phase I/II trial (NCT01734850), the anti-HIV-1 vector is transduced ex vivo into autologous CD34+ HPCs and CD4+ T-lymphocytes, which are then transplanted back into the HIV-infected patient [129]. The CCR5 shRNA component is capable of inhibiting CCR5-tropic (R5-tropic) but not CXCR4-tropic (X4-tropic) strains, whereas the C46 peptide inhibits both tropisms of HIV-1 and is thus an essential factor in the dual-therapeutic design of the vector.
Non-specific in vivo delivery
A variety of nanoparticles have been used for encapsulating and delivering HIV-1 siRNAs, including dendrimers, polymers, and liposomes. Using the G5 PAMAM (generation 5 polyamidoamine) dendrimer with three anti-HIV-1 DsiRNAs, HIV-1 replication was potently suppressed in a humanized mouse model [140]. This cocktail strategy combined one DsiRNA against the HIV-1 tat/rev overlapping reading frame, another DsiRNA against the host CD4 receptor, and a third DsiRNA against Transportin-3 (TNPO3), which is a host factor required for nuclear entry of the HIV-1 pre-integration complex. Biodegradable polymers can protect siRNAs from ribonuclease degradation but release the siRNAs upon cell entry. This design was used to encapsulate CCR5 siRNAs in a polymer nanocapsule to knockdown levels of CCR5 with significantly greater efficiency than liposome-based delivery systems [141].
Advances in electroporation are creating opportunity for siRNA delivery in vivo. RNAi has been delivered to the brain, muscle, and skin [142, 143]. Most recently, Inovio Pharmaceuticals, Inc. (Blue Bell, PA, USA) has developed ELGEN in vivo surface electroporation for delivery of siRNA [142] or DNA vaccines to CD4+ memory T cells [144, 145], which is promising because memory T cells are typically thought to be reservoirs for HIV-1. This strategy has yet to be tested with siRNA against HIV-1 targets, and in vivo electroporation will need to target a wider range of locations that include other regions of high T-cell concentration, such as the spleen and blood.
Targeted delivery
Targeted delivery approaches include the use of tissue-specific serotypes of AAV, nucleic acid aptamers, antibodies, and modified nanoparticles. These approaches are non-integrative, but each has its own set of challenges that need to be considered in order to obtain an effective delivery method.
AAV is a DNA virus that is used for gene therapy and that undergoes limited integration into host genomes. AAV serotypes are selected for specificity to certain regions of an organism. For example, AAV2 targets heparan sulfate proteoglycan, αVβ5 integrin and fibroblast growth receptor 1, which is found on hepatocytes, skeletal muscle cells, smooth muscle cells, and neurons [146]. AAV is a very effective vector for gene therapy, but the vector has limited space for exogenous gene insertion and can typically only be delivered once because of its immunogenic properties [147].
Cell-specific nanoparticles comprise another class of targeted delivery molecules that include aptamers, antibodies, and targeted lipid nanoparticles. These particles are very small and often must overcome multiple barriers, such as the cell membrane and endosome, for the siRNA to be loaded into the RISC. Targeted lipid nanoparticles are widely used to deliver siRNA in vitro, but can also be applied in vivo by conjugating a variety of functionalities to liposomes [71, 148, 149]. These functionalities include proteins, chemical conjugates, and beads. RNAi molecules are encapsulated in a lipid bilayer with targeting molecules facing outwards to bind to receptors of interest.
Aptamers and antibodies selectively deliver siRNAs to cells of interest by binding to surface receptors and then entering the cells by receptor-mediated endocytosis. Unlike some siRNA-nanoparticle designs, aptamers or antibodies do not bestow a protective layer around the siRNAs, so the siRNAs are susceptible to nucleases if not modified. Aptamers are selected through the systematic evolution of ligands by exponential enrichment, and are similar to antibodies in binding affinity and specificity to the target receptor or protein. Several delivery targets for these molecules include the HIV-1 gp120 protein and the CD4 receptor [150–152]. These particles are typically not immunogenic, but are rapidly cleared from the circulation and are non-integrative, so multiple doses of anti-HIV-1 siRNAs must be given [149, 153]. Dimeric bacteriophage pRNA complexes have been engineered with an HIV-1 gp120 aptamer and a tat/rev siRNA for targeted delivery to HIV-infected cells [154]. Antibodies have also been used to deliver siRNAs in cell-specific fashion, including gag siRNAs complexed with a protamine-gp160 antibody fusion protein [155] and siRNAs against the CCR5 co-receptor and viral genes tat and vif conjugated with poly-(D)-arginine and a CD7 antibody [156].
Microbicides loaded with siRNAs or aptamer-siRNAs are designed to prevent the transmission of the virus, and their efficacy is increased by targeting localized areas. CD4 aptamer-siRNA chimeras can be administered topically as microbicides for prophylaxis against HIV-1 [150, 157]. In other systems, the siRNA is either naked or conjugated to cholesterol and applied topically to the skin, and results in prevention of viral transmission [158]. For complete and effective HIV-1 prophylaxis, however, siRNA microbicides must achieve durable protection from HIV-1, broad target specificity against potential viral variants, and favorable kinetics for cellular uptake and virus neutralization.
Conclusions
The potential for RNAi to be used against HIV-1 in the clinic is feasible with improvements in current strategies. Several points to consider when using RNAi to treat HIV-1 include delivery, immunogenicity, toxicity, and viral mutagenesis. In this review, we have discussed strategies that are used to overcome problems in each of these areas. Modified base incorporation and improved siRNA specificity have led the way in reducing the immunogenicity and improving efficacy. In addition, target identification has produced a wide array of viral and cellular targets that inhibit HIV-1 replication upon silencing. As a therapeutic, siRNAs have great promise due to their target specificity, potency, and ability to be chemically synthesized for manufacturing. They are widely adaptable, which is advantageous in the case of HIV-1 mutagenesis. By developing a platform for clinical use of RNAi against HIV, targets can be changed rapidly as the virus mutates.
The mode of delivery remains one of the highest hurdles in bringing RNAi to the clinic to treat HIV. To have an effective siRNA therapeutic, delivery must efficiently reach the intended cells and tissues. To date, the most efficient delivery methods are viral vectors, including the ex vivo transduction of cells with lentivirus and in vivo delivery with AAV. Unfortunately, these delivery types are only moderately specific, and AAV is prone to inducing an immunogenic response in vivo. More targeted delivery methods are applicable in an in vivo setting, where they offer specificity and reduced immunogenicity; nevertheless, these methods often lack the pharmacokinetics and scalability of non-specific delivery carriers. Therefore, continued progress to improve each of these shortcomings is required to fulfill the promise of RNAi therapeutics for HIV.
Abbreviations
- AAV:
-
adeno-associated virus
- Ago2:
-
Argonaute-2
- AIDS:
-
acquired immunodeficiency syndrome
- cART:
-
combinatorial antiretroviral therapy
- DsiRNA:
-
Dicer substrate RNA
- gp120:
-
glycoprotein 120
- HIV-1:
-
human immunodeficiency virus-1
- HPC:
-
human progenitor cells
- IFN:
-
interferon
- LTR:
-
long terminal repeat
- miRNA:
-
microRNA
- PKR:
-
protein kinase R
- RISC:
-
RNA-induced silencing complex
- RNAi:
-
RNA interference
- shRNA:
-
short hairpin RNA
- siRNA:
-
short interfering RNA
- vsiRNA:
-
viral suppressors of RNAi
- VSR:
-
viral suppressors of RNAi
- vsRNA:
-
viral small RNA
- ZFN:
-
zinc finger nucleases
References
Mitsuya H, Weinhold KJ, Furman PA, St Clair MH, Lehrman SN, Gallo RC, et al. 3′-Azido-3″-deoxythymidine (BW A509U). an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc Natl Acad Sci U S A. 1985;82:7096–100.
Burnett JC, Zaia JA, Rossi JJ. Creating genetic resistance to HIV. Curr Opin Immunol. 2012;24:625–32.
Deeks SG. HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med. 2011;62:141–55.
Buzon MJ, Codoner FM, Frost SD, Pou C, Puertas MC, Massanella M, et al. Deep molecular characterization of HIV-1 dynamics under suppressive HAART. PLoS Pathog. 2011;7, e1002314.
Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–5.
Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–300.
Yukl SA, Shergill AK, McQuaid K, Gianella S, Lampiris H, Hare CB, et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS. 2010;24:2451–60.
UNAIDS. Global Report: UNAIDS report on the global AIDS epidemic 2013. http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf. Accessed 15 Apr 2015.
Gorry PR, Ancuta P. Coreceptors and HIV-1 pathogenesis. Curr HIV/AIDS Rep. 2011;8:45–53.
Purcell DF, Martin MA. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J Virol. 1993;67:6365–78.
Hogue IB, Grover JR, Soheilian F, Nagashima K, Ono A. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J Virol. 2011;85:9749–66.
Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci U S A. 2001;98:13925–30.
Stone D, Kiem HP, Jerome KR. Targeted gene disruption to cure HIV. Curr Opin HIV AIDS. 2013;8:217–23.
Maier DA, Brennan AL, Jiang S, Binder-Scholl GK, Lee G, Plesa G, et al. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Hum Gene Ther. 2013;24:245–58.
Yuan J, Wang J, Crain K, Fearns C, Kim KA, Hua KL, et al. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4(+) T cell resistance and enrichment. Mol Ther. 2012;20:849–59.
Didigu CA, Wilen CB, Wang J, Duong J, Secreto AJ, Danet-Desnoyers GA, et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood. 2014;123:61–9.
Wilen CB, Wang J, Tilton JC, Miller JC, Kim KA, Rebar EJ, et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 2011;7, e1002020.
Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–47.
Li L, Krymskaya L, Wang J, Henley J, Rao A, Cao LF, et al. Genomic editing of the HIV-1 coreceptor CCR5 in adult hematopoietic stem and progenitor cells using zinc finger nucleases. Mol Ther. 2013;21:1259–69.
Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–10.
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–16.
Ye L, Wang J, Beyer AI, Teque F, Cradick TJ, Qi Z, et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Delta32 mutation confers resistance to HIV infection. Proc Natl Acad Sci U S A. 2014;111:9591–6.
Certo MT, Gwiazda KS, Kuhar R, Sather B, Curinga G, Mandt T, et al. Coupling endonucleases with DNA end-processing enzymes to drive gene disruption. Nat Methods. 2012;9:973–5.
Schleifman EB, Bindra R, Leif J, del Campo J, Rogers FA, Uchil P, et al. Targeted disruption of the CCR5 gene in human hematopoietic stem cells stimulated by peptide nucleic acids. Chem Biol. 2011;18:1189–98.
Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–92.
Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–2.
Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A. 2014;111:11461–6.
Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:2510.
Aubert M, Ryu BY, Banks L, Rawlings DJ, Scharenberg AM, Jerome KR. Successful targeting and disruption of an integrated reporter lentivirus using the engineered homing endonuclease Y2 I-AniI. PLoS One. 2011;6, e16825.
Mariyanna L, Priyadarshini P, Hofmann-Sieber H, Krepstakies M, Walz N, Grundhoff A, et al. Excision of HIV-1 proviral DNA by recombinant cell permeable tre-recombinase. PLoS One. 2012;7, e31576.
Sarkar I, Hauber I, Hauber J, Buchholz F. HIV-1 proviral DNA excision using an evolved recombinase. Science. 2007;316:1912–5.
Buchholz F, Hauber J. In vitro evolution and analysis of HIV-1 LTR-specific recombinases. Methods. 2011;53:102–9.
Nagawa T, Habu Y, Matsumoto N, Miyano-Kurosaki N, Takaku H. Long-term transgene expression and inhibition of HIV-1 replication by a Cre/loxP-EBNA-1/oriP HIV-1-dependent ribozyme vector: applications for HIV-1 gene therapy. J RNAi Gene Silencing. 2006;2:146–53.
Symensma TL, Giver L, Zapp M, Takle GB, Ellington AD. RNA aptamers selected to bind human immunodeficiency virus type 1 Rev in vitro are Rev responsive in vivo. J Virol. 1996;70:179–87.
Konopka K, Lee NS, Rossi J, Duzgunes N. Rev-binding aptamer and CMV promoter act as decoys to inhibit HIV replication. Gene. 2000;255:235–44.
Michienzi A, Li S, Zaia JA, Rossi JJ. A nucleolar TAR decoy inhibitor of HIV-1 replication. Proc Natl Acad Sci U S A. 2002;99:14047–52.
Unwalla HJ, Rossi JJ. A dual function TAR decoy serves as an anti-HIV siRNA delivery vehicle. Virol J. 2010;7:33.
DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A, et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med. 2010;2:36ra43.
DiGiusto DL, Stan R, Krishnan A, Li H, Rossi JJ, Zaia JA. Development of hematopoietic stem cell based gene therapy for HIV-1 infection: considerations for proof of concept studies and translation to standard medical practice. Viruses. 2013;5:2898–919.
Mitsuyasu RT, Zack JA, Macpherson JL, Symonds GP. Phase I/II clinical trials using gene-modified adult hematopoietic stem cells for HIV: lessons learnt. Stem Cells Int. 2011;2011:393698.
Mitsuyasu RT, Merigan TC, Carr A, Zack JA, Winters MA, Workman C, et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat Med. 2009;15:285–92.
Wong-Staal F, Poeschla EM, Looney DJ. A controlled, phase 1 clinical trial to evaluate the safety and effects in HIV-1 infected humans of autologous lymphocytes transduced with a ribozyme that cleaves HIV-1 RNA. Hum Gene Ther. 1998;9:2407–25.
Macpherson JL, Boyd MP, Arndt AJ, Todd AV, Fanning GC, Ely JA, et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J Gene Med. 2005;7:552–64.
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11.
Gagnon KT, Li L, Chu Y, Janowski BA, Corey DR. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014;6:211–21.
Zhang X, Li H, Burnett JC, Rossi JJ. The role of antisense long noncoding RNA in small RNA-triggered gene activation. RNA. 2014;20:1916–28.
Weinberg MS, Villeneuve LM, Ehsani A, Amarzguioui M, Aagaard L, Chen ZX, et al. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA. 2006;12:256–62.
Siomi H, Siomi MC. Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell. 2010;38:323–32.
Umbach JL, Cullen BR. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009;23:1151–64.
Parameswaran P, Sklan E, Wilkins C, Burgon T, Samuel MA, Lu R, et al. Six RNA viruses and forty-one hosts: viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog. 2010;6, e1000764.
Bivalkar-Mehla S, Vakharia J, Mehla R, Abreha M, Kanwar JR, Tikoo A, et al. Viral RNA silencing suppressors (RSS): novel strategy of viruses to ablate the host RNA interference (RNAi) defense system. Virus Res. 2011;155:1–9.
Backes S, Shapiro JS, Sabin LR, Pham AM, Reyes I, Moss B, et al. Degradation of host microRNAs by poxvirus poly(A) polymerase reveals terminal RNA methylation as a protective antiviral mechanism. Cell Host Microbe. 2012;12:200–10.
Maillard PV, Ciaudo C, Marchais A, Li Y, Jay F, Ding SW, et al. Antiviral RNA interference in mammalian cells. Science. 2013;342:235–8.
Li Y, Lu J, Han Y, Fan X, Ding SW. RNA interference functions as an antiviral immunity mechanism in mammals. Science. 2013;342:231–4.
Backes S, Langlois RA, Schmid S, Varble A, Shim JV, Sachs D, et al. The mammalian response to virus infection is independent of small RNA silencing. Cell Rep. 2014;8:114–25.
Bogerd HP, Skalsky RL, Kennedy EM, Furuse Y, Whisnant AW, Flores O, et al. Replication of many human viruses is refractory to inhibition by endogenous cellular microRNAs. J Virol. 2014;88:8065–76.
Rettig GR, Behlke MA. Progress toward in vivo use of siRNAs-II. Mol Ther. 2012;20:483–512.
Liu YP, Schopman NC, Berkhout B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res. 2013;41:3723–33.
Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–16.
Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208.
Snead NM, Escamilla-Powers JR, Rossi JJ, McCaffrey AP. 5′ Unlocked nucleic acid modification improves siRNA targeting. Mol Ther Nucleic Acids. 2013;2, e103.
Kubo T, Zhelev Z, Ohba H, Bakalova R. Chemically modified symmetric and asymmetric duplex RNAs: an enhanced stability to nuclease degradation and gene silencing effect. Biochem Biophys Res Commun. 2008;365:54–61.
Sano M, Sierant M, Miyagishi M, Nakanishi M, Takagi Y, Sutou S. Effect of asymmetric terminal structures of short RNA duplexes on the RNA interference activity and strand selection. Nucleic Acids Res. 2008;36:5812–21.
Rose SD, Kim DH, Amarzguioui M, Heidel JD, Collingwood MA, Davis ME, et al. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33:4140–56.
Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23:222–6.
Siolas D, Lerner C, Burchard J, Ge W, Linsley PS, Paddison PJ, et al. Synthetic shRNAs as potent RNAi triggers. Nat Biotechnol. 2005;23:227–31.
Snead NM, Wu X, Li A, Cui Q, Sakurai K, Burnett JC, et al. Molecular basis for improved gene silencing by Dicer substrate interfering RNA compared to other siRNA variants. Nucleic Acids Res. 2013. doi:10.1093/nar/gkt200.
Zhou J, Song MS, Jacobi AM, Behlke MA, Wu X, Rossi JJ. Deep sequencing analyses of DsiRNAs reveal the influence of 3′ terminal overhangs on Dicing polarity, strand selectivity, and RNA editing of siRNAs. Mol Ther Nucleic Acids. 2012;1, e17.
Kanasty RL, Whitehead KA, Vegas AJ, Anderson DG. Action and reaction: the biological response to siRNA and its delivery vehicles. Mol Ther. 2012;20:513–24.
Sioud M. Targeted delivery of antisense oligonucleotides and siRNAs into mammalian cells. Methods Mol Biol. 2009;487:61–82.
Karlsen TA, Brinchmann JE. Liposome delivery of microRNA-145 to mesenchymal stem cells leads to immunological off-target effects mediated by RIG-I. Mol Ther. 2013;21:1169–81.
Sui H, Zhou M, Chen Q, Lane HC, Imamichi T. siRNA enhances DNA-mediated interferon lambda-1 response through crosstalk between RIG-I and IFI16 signalling pathway. Nucleic Acids Res. 2014;42:583–98.
Nejepinska J, Flemr M, Svoboda P. Control of the interferon response in RNAi experiments. Methods Mol Biol. 2012;820:133–61.
Zamora MR, Budev M, Rolfe M, Gottlieb J, Humar A, Devincenzo J, et al. RNA interference therapy in lung transplant patients infected with respiratory syncytial virus. Am J Respir Crit Care Med. 2011;183:531–8.
Eekels JJ, Geerts D, Jeeninga RE, Berkhout B. Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antiviral Res. 2011;89:43–53.
Singh A, Palanichamy JK, Ramalingam P, Kassab MA, Bhagat M, Andrabi R, et al. Long-term suppression of HIV-1C virus production in human peripheral blood mononuclear cells by LTR heterochromatization with a short double-stranded RNA. J Antimicrob Chemother. 2014;69:404–15.
Pan JS, Wang XZ, Ren JL. Long-term RNA interference and its application to hepatitis B virus. J Dig Dis. 2009;10:165–71.
Boutimah F, Eekels JJ, Liu YP, Berkhout B. Antiviral strategies combining antiretroviral drugs with RNAi-mediated attack on HIV-1 and cellular co-factors. Antiviral Res. 2013;98:121–9.
Rossi JJ, June CH, Kohn DB. Genetic therapies against HIV. Nat Biotechnol. 2007;25:1444–54.
Vlachakis D, Tsiliki G, Pavlopoulou A, Roubelakis MG, Tsaniras SC, Kossida S. Antiviral stratagems against HIV-1 using RNA interference (RNAi) technology. Evol Bioinform Online. 2013;9:203–13.
Singh SK, Gaur RK. Progress towards therapeutic application of RNA interference for HIV infection. BioDrugs. 2009;23:269–76.
Soejitno A, Wihandani DM, Kuswardhani T. The therapeutic potential of RNA interference in controlling HIV-1 replication. Acta Med Indones. 2009;41:215–21.
Liu YP, von Eije KJ, Schopman NC, Westerink JT, ter Brake O, Haasnoot J, et al. Combinatorial RNAi against HIV-1 using extended short hairpin RNAs. Mol Ther. 2009;17:1712–3.
Suzuki K, Juelich T, Lim H, Ishida T, Watanebe T, Cooper DA, et al. Closed chromatin architecture is induced by an RNA duplex targeting the HIV-1 promoter region. J Biol Chem. 2008;283:23353–63.
Suzuki K, Hattori S, Marks K, Ahlenstiel C, Maeda Y, Ishida T, et al. Promoter targeting shRNA suppresses HIV-1 infection in vivo through transcriptional gene silencing. Mol Ther Nucleic Acids. 2013;2, e137.
Caffrey DR, Zhao J, Song Z, Schaffer ME, Haney SA, Subramanian RR, et al. siRNA off-target effects can be reduced at concentrations that match their individual potency. PLoS One. 2011;6:e21503.
Green VA, Arbuthnot P, Weinberg MS. Impact of sustained RNAi-mediated suppression of cellular cofactor Tat-SF1 on HIV-1 replication in CD4+ T cells. Virol J. 2012;9:272.
Li M, Li H, Rossi JJ. RNAi in combination with a ribozyme and TAR decoy for treatment of HIV infection in hematopoietic cell gene therapy. Ann N Y Acad Sci. 2006;1082:172–9.
Eekels JJ, Berkhout B. Toward a durable treatment of HIV-1 infection using RNA interference. Prog Mol Biol Transl Sci. 2011;102:141–63.
HIV Databases. Los Alamos National Laboratory. http://www.hiv.lanl.gov. Accessed 15 Apr 2015.
Lau TS, Li Y, Kameoka M, Ng TB, Wan DC. Suppression of HIV replication using RNA interference against HIV-1 integrase. FEBS Lett. 2007;581:3253–9.
Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005;33:796–804.
Puerta-Fernandez E, Barroso-del Jesus A, Romero-Lopez C, Tapia N, Martinez MA, Berzal-Herranz A. Inhibition of HIV-1 replication by RNA targeted against the LTR region. AIDS. 2005;19:863–70.
Cave E, Weinberg MS, Cilliers T, Carmona S, Morris L, Arbuthnot P. Silencing of HIV-1 subtype C primary isolates by expressed small hairpin RNAs targeted to gag. AIDS Res Hum Retroviruses. 2006;22:401–10.
Morris KV, Chung CH, Witke W, Looney DJ. Inhibition of HIV-1 replication by siRNA targeting conserved regions of gag/pol. RNA Biol. 2005;2:17–20.
Lee SK, Dykxhoorn DM, Kumar P, Ranjbar S, Song E, Maliszewski LE, et al. Lentiviral delivery of short hairpin RNAs protects CD4 T cells from multiple clades and primary isolates of HIV. Blood. 2005;106:818–26.
McIntyre GJ, Groneman JL, Yu YH, Tran A, Applegate TL. Multiple shRNA combinations for near-complete coverage of all HIV-1 strains. AIDS Res Ther. 2011;8:1.
Suzuki K, Ishida T, Yamagishi M, Ahlenstiel C, Swaminathan S, Marks K, et al. Transcriptional gene silencing of HIV-1 through promoter targeted RNA is highly specific. RNA Biol. 2011;8:1035–46.
Knowling S, Stapleton K, Turner AM, Uhlmann E, Lehmann T, Vollmer J, et al. Chemically modified oligonucleotides modulate an epigenetically varied and transient form of transcription silencing of HIV-1 in human cells. Mol Ther Nucleic Acids. 2012;1, e16.
Bebenek K, Abbotts J, Wilson SH, Kunkel TA. Error-prone polymerization by HIV-1 reverse transcriptase. Contribution of template-primer misalignment, miscoding, and termination probability to mutational hot spots. J Biol Chem. 1993;268:10324–34.
Takeuchi Y, Nagumo T, Hoshino H. Low fidelity of cell-free DNA synthesis by reverse transcriptase of human immunodeficiency virus. J Virol. 1988;62:3900–2.
Berkhout B, Sanders RW. Molecular strategies to design an escape-proof antiviral therapy. Antiviral Res. 2011;92:7–14.
Schopman NC, Braun A, Berkhout B. Directed HIV-1 evolution of protease inhibitor resistance by second-generation short hairpin RNAs. Antimicrob Agents Chemother. 2012;56:479–86.
Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77:11531–5.
Das AT, Brummelkamp TR, Westerhout EM, Vink M, Madiredjo M, Bernards R, et al. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol. 2004;78:2601–5.
Leonard JN, Shah PS, Burnett JC, Schaffer DV. HIV evades RNA interference directed at TAR by an indirect compensatory mechanism. Cell Host Microbe. 2008;4:484–94.
Shah PS, Pham NP, Schaffer DV. HIV develops indirect cross-resistance to combinatorial RNAi targeting two distinct and spatially distant sites. Mol Ther. 2012;20:840–8.
Zhang T, Cheng T, Wei L, Cai Y, Yeo AE, Han J, et al. Efficient inhibition of HIV-1 replication by an artificial polycistronic miRNA construct. Virol J. 2012;9:118.
Schopman NC, ter Brake O, Berkhout B. Anticipating and blocking HIV-1 escape by second generation antiviral shRNAs. Retrovirology. 2010;7:52.
von Eije KJ, ter Brake O, Berkhout B. Human immunodeficiency virus type 1 escape is restricted when conserved genome sequences are targeted by RNA interference. J Virol. 2008;82:2895–903.
Centlivre M, Legrand N, Klamer S, Liu YP, Jasmijn von Eije K, Bohne M, et al. Preclinical in vivo evaluation of the safety of a multi-shRNA-based gene therapy against HIV-1. Mol Ther Nucleic Acids. 2013;2:e120.
Prussia A, Thepchatri P, Snyder JP, Plemper RK. Systematic approaches towards the development of host-directed antiviral therapeutics. Int J Mol Sci. 2011;12:4027–52.
Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–8.
Hutter G, Ganepola S. Eradication of HIV by transplantation of CCR5-deficient hematopoietic stem cells. Scientific World Journal. 2011;11:1068–76.
Allers K, Hutter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood. 2011;117:2791–9.
Kordelas L, Verheyen J, Beelen DW, Horn PA, Heinold A, Kaiser R, et al. Shift of HIV tropism in stem-cell transplantation with CCR5 Delta32 mutation. N Engl J Med. 2014;371:880–2.
Shimizu S, Kamata M, Kittipongdaja P, Chen KN, Kim S, Pang S, et al. Characterization of a potent non-cytotoxic shRNA directed to the HIV-1 co-receptor CCR5. Genet Vaccines Ther. 2009;7:8.
Tamhane M, Akkina R. Stable gene transfer of CCR5 and CXCR4 siRNAs by sleeping beauty transposon system to confer HIV-1 resistance. AIDS Res Ther. 2008;5:16.
Vazquez-Salat N, Yuhki N, Beck T, O’Brien SJ, Murphy WJ. Gene conversion between mammalian CCR2 and CCR5 chemokine receptor genes: a potential mechanism for receptor dimerization. Genomics. 2007;90:213–24.
Anderson J, Banerjea A, Akkina R. Bispecific short hairpin siRNA constructs targeted to CD4, CXCR4, and CCR5 confer HIV-1 resistance. Oligonucleotides. 2003;13:303–12.
Eekels JJ, Sagnier S, Geerts D, Jeeninga RE, Biard-Piechaczyk M, Berkhout B. Inhibition of HIV-1 replication with stable RNAi-mediated knockdown of autophagy factors. Virol J. 2012;9:69.
Li MJ, Kim J, Li S, Zaia J, Yee JK, Anderson J, et al. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol Ther. 2005;12:900–9.
Ishaq M, Hu J, Wu X, Fu Q, Yang Y, Liu Q, et al. Knockdown of cellular RNA helicase DDX3 by short hairpin RNAs suppresses HIV-1 viral replication without inducing apoptosis. Mol Biotechnol. 2008;39:231–8.
Subramanya S, Armant M, Salkowitz JR, Nyakeriga AM, Haridas V, Hasan M, et al. Enhanced induction of HIV-specific cytotoxic T lymphocytes by dendritic cell-targeted delivery of SOCS-1 siRNA. Mol Ther. 2010;18:2028–37.
Christensen HS, Daher A, Soye KJ, Frankel LB, Alexander MR, Laine S, et al. Small interfering RNAs against the TAR RNA binding protein, TRBP, a Dicer cofactor, inhibit human immunodeficiency virus type 1 long terminal repeat expression and viral production. J Virol. 2007;81:5121–31.
Sanghvi VR, Steel LF. The cellular TAR RNA binding protein, TRBP, promotes HIV-1 replication primarily by inhibiting the activation of double-stranded RNA-dependent kinase PKR. J Virol. 2011;85:12614–21.
Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–4.
McIntyre GJ, Yu YH, Lomas M, Fanning GC. The effects of stem length and core placement on shRNA activity. BMC Mol Biol. 2011;12:34.
Burke BP, Boyd MP, Impey H, Breton LR, Bartlett JS, Symonds GP, et al. CCR5 as a natural and modulated target for inhibition of HIV. Viruses. 2014;6:54–68.
Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet. 2011;12:316–28.
Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6:1130–46.
Grimm D, Wang L, Lee JS, Schurmann N, Gu S, Borner K, et al. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. JClin Invest. 2010;120:3106–19.
Persons DA. Lentiviral vector gene therapy: effective and safe? Mol Ther. 2010;18:861–2.
Morris KV, Rossi JJ. Lentivirus-mediated RNA interference therapy for human immunodeficiency virus type 1 infection. Hum Gene Ther. 2006;17:479–86.
Liu YP, Vink MA, Westerink JT, Ramirez de Arellano E, Konstantinova P, Ter Brake O, et al. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA. 2010;16:1328–39.
Chung J, Zhang J, Li H, Ouellet DL, DiGiusto DL, Rossi JJ. Endogenous MCM7 microRNA cluster as a novel platform to multiplex small interfering and nucleolar RNAs for combinational HIV-1 gene therapy. Hum Gene Ther. 2012;23:1200–8.
Subramanya S, Kim SS, Manjunath N, Shankar P. RNA interference-based therapeutics for human immunodeficiency virus HIV-1 treatment: synthetic siRNA or vector-based shRNA? Expert Opin Biol Ther. 2010;10:201–13.
Chung J, DiGiusto DL, Rossi JJ. Combinatorial RNA-based gene therapy for the treatment of HIV/AIDS. Expert Opin Biol Ther. 2013;13:437–45.
Wolstein O, Boyd M, Millington M, Impey H, Boyer J, Howe A, et al. Preclinical safety and efficacy of an anti–HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol Ther. 2014;1. doi:10.1038/mtm.2013.11.
Zhou J, Neff CP, Liu X, Zhang J, Li H, Smith DD, et al. Systemic administration of combinatorial dsiRNAs via nanoparticles efficiently suppresses HIV-1 infection in humanized mice. Mol Ther. 2011;19:2228–38.
Yan M, Liang M, Wen J, Liu Y, Lu Y, Chen IS. Single siRNA nanocapsules for enhanced RNAi delivery. J Am Chem Soc. 2012;134:13542–5.
Broderick KE, Chan A, Lin F, Shen X, Kichaev G, Khan AS, et al. Optimized in vivo transfer of small interfering RNA targeting dermal tissue using in vivo surface electroporation. Mol Ther Nucleic Acids. 2012;1, e11.
Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest. 2007;117:3623–32.
Patel V, Jalah R, Kulkarni V, Valentin A, Rosati M, Alicea C, et al. DNA and virus particle vaccination protects against acquisition and confers control of viremia upon heterologous simian immunodeficiency virus challenge. Proc Natl Acad Sci U S A. 2013;110:2975–80.
Kulkarni V, Rosati M, Valentin A, Ganneru B, Singh AK, Yan J, et al. HIV-1 p24(gag) derived conserved element DNA vaccine increases the breadth of immune response in mice. PLoS One. 2013;8, e60245.
Kashiwakura Y, Tamayose K, Iwabuchi K, Hirai Y, Shimada T, Matsumoto K, et al. Hepatocyte growth factor receptor is a coreceptor for adeno-associated virus type 2 infection. J Virol. 2005;79:609–14.
Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet. 2014;15:445–51.
Kim SS, Peer D, Kumar P, Subramanya S, Wu H, Asthana D, et al. RNAi-mediated CCR5 silencing by LFA-1-targeted nanoparticles prevents HIV infection in BLT mice. Mol Ther. 2010;18:370–6.
Krebs MD, Alsberg E. Localized, targeted, and sustained siRNA delivery. Chemistry. 2011;17:3054–62.
Wheeler LA, Vrbanac V, Trifonova R, Brehm MA, Gilboa-Geffen A, Tanno S, et al. Durable knockdown and protection from HIV transmission in humanized mice treated with gel-formulated CD4 aptamer-siRNA chimeras. Mol Ther. 2013;21:1378–89.
Zhou J, Neff CP, Swiderski P, Li H, Smith DD, Aboellail T, et al. Functional in vivo delivery of multiplexed anti-HIV-1 siRNAs via a chemically synthesized aptamer with a sticky bridge. Mol Ther. 2013;21:192–200.
Neff CP, Zhou J, Remling L, Kuruvilla J, Zhang J, Li H, et al. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci Transl Med. 2011;3:66ra66.
Pan Y, Zhang Y, Jia T, Zhang K, Li J, Wang L. Development of a microRNA delivery system based on bacteriophage MS2 virus-like particles. FEBS J. 2012;279:1198–208.
Zhou J, Shu Y, Guo P, Smith DD, Rossi JJ. Dual functional RNA nanoparticles containing phi29 motor pRNA and anti-gp120 aptamer for cell-type specific delivery and HIV-1 inhibition. Methods. 2011;54:284–94.
Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–17.
Kumar P, Ban HS, Kim SS, Wu H, Pearson T, Greiner DL, et al. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell. 2008;134:577–86.
Yang S, Chen Y, Ahmadie R, Ho EA. Advancements in the field of intravaginal siRNA delivery. J Control Release. 2013;167:29–39.
Katakowski JA, Palliser D. siRNA-based topical microbicides targeting sexually transmitted infections. Curr Opin Mol Ther. 2010;12:192–202.
Arts EJ, Hazuda DJ. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med. 2012;2:a007161.
Vaccari M, Poonam P, Franchini G. Phase III HIV vaccine trial in Thailand: a step toward a protective vaccine for HIV. Expert Rev Vaccines. 2010;9:997–1005.
de Souza MS, Ratto-Kim S, Chuenarom W, Schuetz A, Chantakulkij S, Nuntapinit B, et al. The Thai phase III trial (RV144) vaccine regimen induces T cell responses that preferentially target epitopes within the V2 region of HIV-1 envelope. J Immunol. 2012;188:5166–76.
Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–20.
van Lunzen J, Glaunsinger T, Stahmer I, von Baehr V, Baum C, Schilz A, et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol Ther. 2007;15:1024–33.
Rossi JJ. Ribozyme therapy for HIV infection. Adv Drug Deliv Rev. 2000;44:71–8.
Levine BL, Humeau LM, Boyer J, MacGregor RR, Rebello T, Lu X, et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci U S A. 2006;103:17372–7.
Liu D, Donegan J, Nuovo G, Mitra D, Laurence J. Stable human immunodeficiency virus type 1 (HIV-1) resistance in transformed CD4+ monocytic cells treated with multitargeting HIV-1 antisense sequences incorporated into U1 snRNA. J Virol. 1997;71:4079–85.
Kohn DB, Bauer G, Rice CR, Rothschild JC, Carbonaro DA, Valdez P, et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34(+) cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood. 1999;94:368–71.
Scarborough RJ, Levesque MV, Boudrias-Dalle E, Chute IC, Daniels SM, Ouellette RJ, et al. A conserved target site in HIV-1 Gag RNA is accessible to inhibition by both an HDV ribozyme and a short hairpin RNA. Mol Ther Nucleic Acids. 2014;3, e178.
Park WS, Hayafune M, Miyano-Kurosaki N, Takaku H. Specific HIV-1 env gene silencing by small interfering RNAs in human peripheral blood mononuclear cells. Gene Ther. 2003;10:2046–50.
Flynn NM, Forthal DN, Harro CD, Judson FN, Mayer KH, Para MF. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J Infect Dis. 2005;191:654–65.
Omoto S, Ito M, Tsutsumi Y, Ichikawa Y, Okuyama H, Brisibe EA, et al. HIV-1 nef suppression by virally encoded microRNA. Retrovirology. 2004;1:44.
ter Brake O, Konstantinova P, Ceylan M, Berkhout B. Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Mol Ther. 2006;14:883–92.
Yamamoto T, Miyoshi H, Yamamoto N, Yamamoto N, Inoue J, Tsunetsugu-Yokota Y. Lentivirus vectors expressing short hairpin RNAs against the U3-overlapping region of HIV nef inhibit HIV replication and infectivity in primary macrophages. Blood. 2006;108:3305–12.
ter Brake O, Legrand N, von Eije KJ, Centlivre M, Spits H, Weijer K, et al. Evaluation of safety and efficacy of RNAi against HIV-1 in the human immune system (Rag-2(−/−)gammac(−/−)) mouse model. Gene Ther. 2009;16:148–53.
Nishitsuji H, Kohara M, Kannagi M, Masuda T. Effective suppression of human immunodeficiency virus type 1 through a combination of short- or long-hairpin RNAs targeting essential sequences for retroviral integration. J Virol. 2006;80:7658–66.
Jacque JM, Triques K, Stevenson M. Modulation of HIV-1 replication by RNA interference. Nature. 2002;418:435–8.
Martinez MA, Gutierrez A, Armand-Ugon M, Blanco J, Parera M, Gomez J, et al. Suppression of chemokine receptor expression by RNA interference allows for inhibition of HIV-1 replication. AIDS. 2002;16:2385–90.
Xu WW, Han MJ, Chen D, Chen L, Guo Y, Willden A, et al. Genome-wide search for the genes accountable for the induced resistance to HIV-1 infection in activated CD4+ T cells: apparent transcriptional signatures, co-expression networks and possible cellular processes. BMC Med Genomics. 2013;6:15.
Christ F, Debyser Z. The LEDGF/p75 integrase interaction, a novel target for anti-HIV therapy. Virology. 2013;435:102–9.
Zaitseva L, Cherepanov P, Leyens L, Wilson SJ, Rasaiyaah J, Fassati A. HIV-1 exploits importin 7 to maximize nuclear import of its DNA genome. Retrovirology. 2009;6:11.
Al-Mawsawi LQ, Neamati N. Blocking interactions between HIV-1 integrase and cellular cofactors: an emerging anti-retroviral strategy. Trends Pharmacol Sci. 2007;28:526–35.
Chiu YL, Cao H, Jacque JM, Stevenson M, Rana TM. Inhibition of Human Immunodeficiency Virus Type 1 replication by RNA interference directed against Human Transcription Elongation Factor P-TEFb (CDK9/CyclinT1). J Virol. 2004;78:2517–29.
Ping YH, Chu CY, Cao H, Jacque JM, Stevenson M, Rana TM. Modulating HIV-1 replication by RNA interference directed against human transcription elongation factor SPT5. Retrovirology. 2004;1:46.
Acknowledgements
This work was supported by the defeatHIV-1 Delaney Cell and Genome Engineering Initiative (NIH U19 AI096111) to JR and by NIH grant AI029329 to JB and JR.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Bobbin, M.L., Burnett, J.C. & Rossi, J.J. RNA interference approaches for treatment of HIV-1 infection. Genome Med 7, 50 (2015). https://doi.org/10.1186/s13073-015-0174-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-015-0174-y