
Abstract
Histone methyltransferase SETDB1 (SET domain bifurcated histone lysine methyltransferase 1, also known as ESET or KMT1E) is known to be involved in the deposition of the di- and tri-methyl marks on H3K9 (H3K9me2 and H3K9me3), which are associated with transcription repression. SETDB1 exerts an essential role in the silencing of endogenous retroviruses (ERVs) in embryonic stem cells (mESCs) by tri-methylating H3K9 (H3K9me3) and interacting with DNA methyltransferases (DNMTs). Additionally, SETDB1 is engaged in regulating multiple biological processes and diseases, such as ageing, tumors, and inflammatory bowel disease (IBD), by methylating both histones and non-histone proteins. In this review, we provide an overview of the complex biology of SETDB1, review the upstream regulatory mechanisms of SETDB1 and its partners, discuss the functions and molecular mechanisms of SETDB1 in cell fate determination and stem cell, as well as in tumors and other diseases. Finally, we discuss the current challenges and prospects of targeting SETDB1 for the treatment of different diseases, and we also suggest some future research directions in the field of SETDB1 research.
Similar content being viewed by others

Introduction
Protein methylation, a covalent modification on proteins, is dynamically regulated by protein methyltransferases and demethylases, and S-adenosyl-l-methionine (AdoMet) is the main methyl group donor [1,2,3]. Protein methylation occurs mainly at lysine or arginine residues of histones and non-histones and has been demonstrated to be implicated in the regulation of many biological processes by affecting the activity, subcellular localization, or stability of proteins [4,5,6,7]. In recent years, major advancements in our understanding of protein methylation have been made, including not only its regulatory mechanisms, but also its pathophysiological functions [8,9,10,11].
The SET domain bifurcated histone lysine methyltransferase 1 (SETDB1), also known as lysine N-methyltransferase 1E (KMT1E) or Erg-associated SET domain (ESET), is a family member of the SET domain-containing histone methyltransferases. SETDB1 deposits di- and tri-methyl marks on H3K9 (H3K9me2 and H3K9me3) which are transcriptional repression marks [12,13,14,15,16]. Additionally, SETDB1 has been found to methylate non-histones, such as tri-methylating AKT at K64 and K140 (AKTK64me3 and AKTK140me3), and di-methylating P53 at K370 (P53K370me2) [17,18,19]. SETDB1 has been shown to be involved in maintaining endogenous retroviruses (ERVs) silencing in embryonic stem cells (mESCs) [20,21,22,23], as well as in cell fate determination and tumorigenesis [24,25,26,27,28,29]. In this review, we summarize the structure features of SETDB1, the upstream regulatory mechanisms controlling SETDB1 expression and activity, and the partners of SETDB1, as well as the pivotal roles of SETDB1 in cancer progression, inflammatory bowel disease, ageing, and embryonic stem cells by regulating methylation of H3K9 and non-histone proteins.
Structure of SETDB1
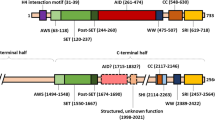
SETDB1 was first identified by Harte et al. in 1999, and they revealed that Setdb1 gene localized to human chromosome band 1q21 [30]. By using the N-terminal region of ERG as a bait to screen a yeast two-hybrid mouse cDNA library, Yang et al. isolated a 4.6 kb full-length mouse cDNA encoding a protein of 1307 amino acids, referred to as ESET [31]. Mouse ESET has 92% similarity to human protein SETDB1, and SETDB1 is a histone H3K9-specific methyltransferase contributing to heterochromatin protein 1a (HP1a)-mediated silencing of euchromatic genes [16, 32]. The evolutionarily conserved SET, pre-SET, and post-SET domains comprise the C-terminal region of SETDB1, which is necessary for its methyltransferase activity. The N-terminal region of SETDB1 contains two consecutive tudor domains (TUDs) and a methyl-CpG binding domain (MBD) that interacts with chromatin modifying enzymes, such as DNA methyltransferases (DNMTs), to participate in DNA silencing [12, 33, 34] (Fig. 1). Three isoforms of Setdb1 gene have been identified, of which isoform 1 is encoded by the longest transcript containing all intact domains, and is widely expressed [32, 35]. Although isoform 2 is a shorter splice variant, it still has all the important domains similar to those of isoform 1. However, in contrast to isoform 1, isoform 3 contains only 400 amino acids at the N-terminus (Fig. 1) [12, 33].

Schematic representation of human SETDB1 and its isoforms. The domains are indicated as different colors, and the SET domain is the major catalytic domain. NES nuclear export sequence, NLS nuclear localization sequence, MBD methyl-CpG binding domain, SET Su(var)3–9, Enhancer-of-zeste and Trithorax
The upstream regulatory mechanisms of SETDB1
The expression and activity of SETDB1 is regulated at multiple levels (Fig. 2). Compared with control, the protein levels of SETDB1 and its substrate H3K9me3 are obviously increased in the striatal neurons of Huntington’s disease patients and transgenic R6/2 (a Huntington’s disease mouse model) mice, and both specificity protein 1 (Sp1) and Sp3 can bind to the Setdb1 promoter to activate the transcription of Setdb1 gene [36]. Mithramycin is an antibiotic agent that has been revealed to suppress the growth of cancers by preventing the binding of Sp-family transcription factors to the DNA of gene promoters [37]. Unsurprisingly, mithramycin inhibits the basal promoter activity of Setdb1 gene in a dose-dependent manner, and in addition, combined treatment with mithramycin and cystamine extends the lifespan of R6/2 mice by 40% and obviously improves the behavioral and neuropathological phenotype [36]. In addition, both mithramycin A and its analog (mithralog) EC-8042 effectively suppress SETDB1 expression in melanoma cells and enhance the efficacy of mitogen-activated protein kinase-inhibitor-based therapies for melanoma [38]. In gastric cancer, transcription factor 4 (TCF4) directly bind to the promoter of Setdb1 gene (binding motif, CAAAG) to enhance the expression of SETDB1, and approximately 90% of patients with gastric cancer (GC) are caused by Helicobacter pylori infection, which promotes SETDB1 expression in a TCF4-dependent manner [39]. Furthermore, elevated SETDB1 interacts with ERG to promote gastric carcinogenesis and metastasis by binding to the promoter regions of matrix metalloproteinase 9 (MMP9) and cyclin D1 (CCND1) to accelerate their transcription [39]. In breast cancer cells, SETDB1 increases the expression of c-MYC to promote cell cycle progression, enhanced growth, and colony formation, and increased c-MYC positive feedback regulates the expression of SETDB1 by directly binding to Setdb1 promoter and enhances its transcription [40].

The upstream regulatory mechanisms of SETDB1. The expression of SETDB1 is regulated by transcription factors TCF4, C-MYC, and SP1/SP3. miR-409-3p, miR-621, and miR-29 directly target the 3′-UTR of Setdb1 and inhibits its expression. ATF7IP and UBE2Es directly interact with and monoubiquitinate SETDB1 to enhance the methyltransferase activity of SETDB1. Mithramycin is an inhibitor of SP1/SP3
MicroRNAs are a class of small non-coding RNAs that negatively regulate gene expression by targeting the 3′-UTRs of mRNAs and causing their mRNAs degradation [41]. Under certain conditions, microRNAs can affect the expression of SETDB1 by targeting its mRNA. For example, miR-621 and miR-29 bind directly to the 3′-UTR of Setdb1 and inhibit its expression. Moreover, the miR-621-SETDB1 strengthens radiosensitivity in hepatocellular carcinoma (HCC) through activation of the p53 signaling pathway [42]. miR-409-3p has been reported to negatively regulate the expression of SETDB1 in non-small cell lung cancer (NSCLC) [43].
The methyltransferase activity, stability, and subcellular localization of SETDB1 protein are essential for its function. For example, SETDB1 in the nucleus is necessary for the expansion of adult muscle stem cells and the suppression of skeletal myoblast terminal differentiation, and Wnt3a-dependent cytoplasmic SETDB1 relocalization and genomic release from certain target genes promote myogenesis [44]. Activating transcription factor 7-interacting protein (ATF7IP) mediates nuclear retention of SETDB1 by binding to the N-terminal region of SETDB1 to inhibit its nuclear export, and increase the K885 ubiquitination of SETDB1 to enhance its methyltransferase activity in HEK293T cells [45]. The deficiency of ATF7IP facilitates proteasomal degradation of nuclear SETDB1 protein, implying that stability of SETDB1 regulated by ATF7IP is essential for heterochromatin formation [46]. Furthermore, the disruption of either SETDB1 or ATF7IP in tumor cells restores tumor antigen expression and augments tumor immunogenicity [47]. Similar to K885 ubiquitination of SETDB1 in humans, the K867 in the SET-insertion domain of SETDB1 can also be monoubiquitinated by UBE2E family of E2 enzymes in an E3-independent manner in mESCs, which is indispensable for methyltransferase activity of SETDB1 and its role in endogenous retrovirus silencing [48]. The monoubiquitinated Egg/Eggless (the ortholog of SETDB1 in Drosophila) is required for piRNA-mediated transposon repression [49]. Windei/Wed (ATF7IP ortholog in Drosophila) controls nuclear retention of Egg/Eggless and recruits mUb-Egg to transposon loci for silencing [49]. These studies demonstrate that the monoubiquitination of SETDB1 is critical for its methyltransferase activity, nuclear localization, and function. Therefore, further identification of the enzymes that regulate the monoubiquitination of SETDB1 and the partners that control its nuclear localization is vital for exploring the biological function of SETDB1.
The partners of SETDB1
As is well known, H3K9 can be methylated by a variety of histone methyltransferases [50, 51], and how these methyltransferases work together to dynamically regulate the methylation of H3K9 is a topic worthy of further investigation. Recent studies have shown that certain H3K9 methyltransferases, such as SETDB1/KMT1E, G9a/KMT1C, Suv39h1/KMT1A, and GLP/KMT1D, cooperate to form a megacomplex and function in gene silencing [52]. Methylation of H3K9 is usually linked to heterochromatin formation and gene silencing, and H3K9me3 is highly enriched in heterochromatic regions, while H3K9me1/2 is enriched in silent euchromatin regions [52]. HP1a is a common partner of SETDB1 and is essential for the formation and maintenance of heterochromatin in both Drosophila and mammals [53,54,55,56]. In Drosophila, mutants with hypomorphic or null expression of dSETDB1 result in the loss of most H3K9 methylation marks and HP1-binding on chromosome 4, a condensed heterochromatin region [57]. SETDB1, HP1a, and Su(var)3–9 inhibit the same genes on chromosome 4, and genes that non-ubiquitously expressed are preferentially targeted, and then stimulate genes in pericentromeric regions [53]. Furthermore, Maksimov et al. revealed that in Drosophila, Su(var)3–9 binds to the majority of single-copy genes in euchromatin only in the presence of dSETDB1, but is largely dSETDB1-independent at repeated sequences in heterochromatin [58, 59]. Similar to Drosophila, SETDB1 physically associates with HP1 and KAP1 around the euchromatic promoter, establishing a silenced state that is epigenetically heritable in mammalian cells [54]. Moreover, HP1 interaction-defective Setdb1 protein is subject to protein degradation by the proteasome pathway in mESCs, and HP1 mutants unable to recognize H3K9me2/3 or dimerize fail to stabilize Setdb1 [56]. HP1 deficiency results in the loss of pluripotency of mESCs and a reciprocal gain of lineage-specific features, which can be restored by overexpression of Setdb1, Nanog and Oct4 [55]. In addition, the heterochromatic chromatin assembly factor 1 (CAF1)-HP1a-SETDB1 complex monomethylates K9 on non-nucleosomal histone H3, which may subsequently trimethylated by SUV39h1/h2 in pericentric regions in HeLa cell line [60], and KRAB-ZFP-associated protein 1 (KAP-1) is a molecular scaffold that coordinates histone methylation and deposition of HP1 protein to repress gene expression [16]. Heterochromatin-inducible activity is inhibited by mTOR-mediated phosphorylation on KAP1, and KAP1 knockdown or drug-induced phosphorylation of KAP1 can force human cytomegalovirus out of latency in human hematopoietic stem cell [61]. In addition, Müller et al. reported that SETDB1 recruits the chromodomain protein M-phase phosphoprotein 8 (MPP8) to its genomic target loci and maintains transcriptional repression of LINE1 elements without preserving H3K9me3 levels, which is critical for maintaining self-renewal of ground-state pluripotent stem cells [62].
H3K9 methylation is associated with DNA methylation, which is inherited after mitosis in a manner coupled to DNA methylation, suggesting that SETDB1 may function in conjunction with DNMTs [52]. Deletion of Setdb1 reduces the levels of H3K9me3 and loci-specific DNA methylation, while increasing 5-hydroxymethylation (5hmC) and binding of ten-eleven translocation 1 in mESCs [63]. The silencing of hypermethylated germline “genome-defence” genes is dependent upon SETDB1, PRC1.6/RING1B and DNA methylation in epiblast-like cells [64]. In contrast, in pre-implantation embryos and naïve ESCs, H3K9me3 and RING1B-dependent H2AK119ub1 are enriched at the hypomethylated promoters of germline genes that bind by the PRC1.6 DNA-binding subunits MGA/MAX/E2F6 [64]. These studies demonstrated that SETDB1 usually functions in the euchromatic region with multiple partners, and represses gene expression. Interestingly, recent studies suggest that DNA methylation does not appear to contribute to the maintenance of H3K9me3, as Dnmt1 KO cells that have greatly reduced levels of DNA methylation on endogenous retroviruses can still maintain H3K9me3 [65]. Long terminal repeat (LTR) expression correlated with loss of H3K9me3, but loss of H3K9me3 did not always lead to transcriptional activation, probably due to DNA methylation [66, 67]. Therefore, the relationship between SETDB1 (H3K9me3) and DNA methylation is still controversial and more studies are needed for in-depth investigation.
The functions of SETDB1 in biological processes and diseases
Current studies have shown that H3K9 is the main substrate of SETDB1, and SETDB1 regulates multiple biological processes (e.g., embryonic stem cell and aging) and various diseases (e.g., tumors and inflammatory bowel disease) by tri-methylating H3K9.
SETDB1 in cell fate determination and stem cell
Cell fate determination
Meiosis is a biological process in which diploid germ cells undergo one round of DNA replication followed by two rounds of division to produce haploid gametes [68]. Chen et al. demonstrated that SETDB1-catalyzed H3K9me3 is indispensable for the formation of bivalent in early meiosis [68]. Spermatocytes with Setdb1 deficiency displayed aberrant centromere clustering and bouquet formation, failure of homologous chromosome pairing and synapsis, and impaired meiotic silencing of unsynapsed chromatin, leading to meiotic arrest before pachytene and spermatocytes apoptosis [68, 69]. In addition, SETDB1 was also identified as a maternal transcriptional co-regulator of genes contributed to mitosis during early embryos in mice [70].
In Drosophila, H3K9me3 chromatin has been found to be critical for the maintenance of female germ cell fate [71]. SETDB1, its binding partner Windei/Wed and HP1a were found to be necessary for silencing testis gene transcription (e.g., phf7) by enhancing H3K9me3 at these genes in female germ cells [71]. In mice, SETDB1 has been reported to act as a bridge between the meiotic DNA damage response and sex chromosome silencing, and meiotic Setdb1 deletion induces midpachytene apoptosis by perturbing meiotic sex chromosome remodeling and silencing during male meiosis [69]. Additionally, SETDB1 has been found to associate with the topological regulator Cohesin to regulate embryonic stem cell pluripotency and lineage development by affecting the topological structures of related genes [25, 72].
Embryonic stem cell
Retrotransposons constitute nearly forty percent of the mouse genome [73], and are mostly transcriptionally silenced during early embryogenesis [74]. In mESCs and in early embryos, deletion of KAP1 (also known as TRIM28), a partner of SETDB1, led to a marked upregulation of a range of ERVs, particularly intracisternal A-type particles (IAP) elements, by downregulating H3K9me3 levels in these regions [23, 74]. Germline-specific conditional knockout of Setdb1 produced a reduction in methylated long terminal repeats (LTRs) and LINE1 elements, as well as DNA methylation at H3K9me3-enriched retrotransposon families, which results in concomitant derepression of marked IAP, ETn, and ERVK10C elements, as well as ERV-proximal genes in Setdb1 deficiency E13.5 primordial germ cells (PGCs) of mice [73]. Interestingly, Setdb1 knockout mice showed a reduced number of male E13.5 PGCs, while postnatal hypogonadism in both sexes [73]. Furthermore, SETDB1 has been found to inhibit the expression of Dppa2 (developmental pluripotency associated 2), Otx2 (orthodenticle homeobox 2) and Utf1 (undifferentiated embryonic cell transcription factor 1), while activating the BMP/SMAD pathway genes Acvrl1 and Smad in developing PGCs, which indicates that SETDB1 is crucial for PGC fate determination of epiblast cells (Fig. 3) [75].

The mechanisms underlying the role of SETDB1 in cell fate determination and stem cell. SETDB1 trimethylates H3K9 (H3K9me3) to suppress the expression of DPPA2, OTX2, and UTF1 to regulate primordial germ cells (PGC) formation, and to inhibit CEBPβ and CDKN1α, FBP1 and FBP2 expression to affect the hematopoietic stem and progenitor cells (HSPCs), and to repress LINE1 transcript expression to facilitate stem cell self-renewal
Although histone methylation and DNA methylation are essential for the repression of ERVs transcription, the genes upregulated after Setdb1 deletion differ from those derepressed genes in mESCs with Dnmt1, Dnmt3a, and Dnmt3b deficient, with the exception of a small number of primarily germline-specific genes [22]. This paradoxical phenotype may be due to an ectopic interaction between SETDB1 and NP95/UHRF1. Under normal conditions, SETDB1 maintains silencing of ERVs, while in the absence of DNMT1, prolonged binding of NP95/UHRF1 to hemimethylated DNA transiently disrupts SETDB1-dependent deposition of H3K9me3 in these regions [76]. In naïve ESCs with SETDB1 deficiency, Tet methylcytosine dioxygenase 2 (TET2) activates IAP elements in a catalytic-dependent manner. Surprisingly, TET2 has no effect on DNA methylation levels at IAPs, but regulates these retrotransposons in a TET2-dependent loss of H4R3me2s manner [77]. In addition, the physically interaction between SETDB1 and methyltransferase-like 3 (METTL3), an RNA N6-methyladenosine (m6A) methyltransferase, is important for the integrity of IAP heterochromatin in mESCs [78,79,80,81].
Hematopoietic stem and progenitor cell
Additionally, Setdb1 has been found to be critical for the maintenance of hematopoietic stem and progenitor cells (HSPCs) in mice, as demonstrated by the rapid depletion of hematopoietic stem, HSPCs, and leukemic stem cells after Setdb1 deletion, which is caused by ectopic activation of nonhematopoietic genes (e.g., gluconeogenic enzyme genes fructose-1,6-bisphosphatase 1 (Fbp1) and Fbp2) (Fig. 3) [82]. Conditional ablation of Setdb1 in pro-B cells (Mb1-Cre) has a significant impact on the pro-B cell compartment, and the B cell populations in the spleen and bone marrow that correspond to later developmental stages are virtually eliminated in mice [83]. Further evidence suggests that these effects of Setdb1 deficiency on B cells may be associated with derepression of endogenous murine leukemia virus (MLV) copies and subsequent activation of the unfolded protein response pathway and apoptosis [84]. In addition, SETDB1 limits the priming of T helper 1 (Th1) cells and maintains the integrity of Th2 cells by repressing a repertoire of ERVs in a H3K9me3-dependent manner [85].
Similarly, in zebrafishes with Setdb1 or Atf7ip deficiency, excessive myeloid differentiation with impaired HSPC expansion is observed, leading to a decrease in T cell and erythroid lineage [86]. Mechanistically, Setdb1 and Atf7ip interaction facilitates H3K9me3 deposition in cebpβ and cdkn1a to inhibit their expression (Fig. 3) [86]. Concomitantly, deletion of Atf7ip or Setdb1 derepresses retrotransposons, thereby inhibiting human leukemia cell growth and inducing myeloid differentiation and inflammation by activating the viral sensor Mda5/Rig-I like receptor signaling [86].
Other stem cell
Fei et al. demonstrated that SETDB1 works in coordination with Polycomb repressive complex 2 (PRC2) to suppress neural differentiation independently of H3K9me3 [87]. Moreover, ERVs are heavily DNA methylated in both ESCs and differentiated somatic cells, but distinctive sets of ERVs are reactivated in different types of Setdb1-deficient somatic cells in an H3K9me3-dependent or -independent manner [21].
In conclusion, SETDB1 plays an important role in cell fate determination, stem cell differentiation and function, but the regulatory mechanisms are heterogeneous and cell-specific. For example, transcriptomic results showed that Setdb1 deletion significantly induced the expression of ERV families such as the murine leukemia virus (MLV), mouse mammary tumor virus (MMTV) and VL30 in pro-B cells, whereas these ERVs remained silent or expressed at low levels in SETDB1-deficient ESCs and PGCs, which expressed ERV families such as IAPE-z, GLn and ETn/MusD [83]. Thus, further studies are needed to further elucidate the specific mechanisms by which SETDB1 regulates cell fate determination and stem cell differentiation, and how this cell-specific mechanism is achieved, e.g., is it dependent on H3K9me3? and which regulators are involved in determining this cell-specific mechanism.
SETDB1 in tumors
According to Global Cancer Statistics, there were an estimated 19.3 million new cancer cases and 10 million cancer-related deaths worldwide in 2020, with lung cancer being the most commonly diagnosed cancer in both sexes [88]. SETDB1 has been found to be amplified in lung cancer cell lines and primary tumors, resulting in increased mRNA and protein levels, which contributes to tumor growth and invasion [89]. SETDB1 promotes the expression of IGFBP4 (insulin like growth factor binding protein 4), LRP8 (LDL receptor related protein 8), and FZD1 (frizzled class receptor 1), but inhibits APOE (Apolipoprotein E) expression, thereby activating WNT-β-catenin pathway and suppressing P53 expression to enhance NSCLC growth in vitro and in vivo [90]. Recently, Zakharova et al. demonstrated that SETDB1 plays an essential role in epigenome, 3D genome organization, and chromatin architecture in the determination of lung adenocarcinoma programs [91]. Due to its high expression in lung cancer, SETDB1 can be used as a diagnostic biomarker for NSCLC with an area under the curve of 0.7741 [92]. SETDB1 also reinforces invadopodia formation and extracellular matrix degradation by suppressing forkhead box A2 (FOXA2) expression, which facilitates migration and invasion capabilities of NSCLC cells [93]. However, Wu et al. demonstrated that in highly metastatic lung cancer cells, SETDB1 is downregulated and interacts with SMAD2/3 to repress metastasis of lung cancer through ANXA2 (Annexin A2) [94]. Similarly, during breast cancer, TGF-β activates SMAD3, which recruits SETDB1 to methylate H3K9, while repressing H3K9 acetylation to inhibit the transcription of Snai1 (snail family transcriptional repressor 1) gene [95]. SNAIL1 is the “master” transcription factor that regulates epithelial–mesenchymal transition (EMT), cancer stem cell properties, cancer dissemination, and patient survival [96]. Thus, SETDB1 impairs TGF-β-induced EMT by interacting with SMAD3 and then downregulating SNAIL1 (Fig. 4). In squamous cell carcinoma, enhancer of zeste homolog 2 (EZH2) inhibits RUNX3 (RUNX Family Transcription Factor 3) expression to activate both SETDB1 and ΔNp63α, which drives an aggressive cancer stem cell phenotype [97]. In contrast, Xiao et al. demonstrated that SETDB1 facilitates the translation of c-MYC and Cyclin D1 mRNAs to promote cell cycle progression of breast cancer cells. Moreover, c-MYC directly binds to the promoter regions of Bmi1 and Setdb1 to enhance their expression, suggesting a positive feedback loop between SETDB1 and c-MYC [40]. Overall, these studies indicate that SETDB1 may act as an oncogenic driver, but is a tumor metastasis suppressor in lung and breast cancers (Fig. 4).

The mechanisms mediating the function of SETDB1 in tumors. SETDB1 directly inhibits or cooperates with other modulators (e.g., Smad3 and ERG) to regulate the genes-related to tumorigenesis and tumor progression. CRC colorectal cancer, EMT epithelial–mesenchymal transition, NSCLC non-small cell lung cancer, PDAC pancreatic ductal adenocarcinomas
In hepatitis B-associated human HCC, SETDB1 is upregulated and has been linked to HCC disease progression and poorer prognosis [98]. Sp1, a positive regulator of SETDB1, is hyperactivated, while miR-29, a negative regulator of SETDB1 was downregulated in HCC, and they jointly contribute to the high expression of STEDB1 [98]. In addition to miR-29, miR-621 has been shown to reduce the expression of SETDB1 by directly targeting its 3′ UTR and enhancing the radiosensitivity of HCC cells, which is mediated by the P53 signaling pathway [42]. Furthermore, Setdb1 deficiency has been found to increase apoptosis to prevent formation of pancreatic ductal adenocarcinomas (PDACs) by binding to p53 promoter regions and directly regulating its expression in the context of heterozygous p53 deletion (Fig. 4) [99].
SETDB1 has been found to be highly amplified in tumors of melanoma patients and melanoma cell lines, with overexpression contributing to a more aggressive phenotype in vivo and in vitro studies by upregulating thrombospondin 1 (THBS1) expression in a H3K4me1-dependent manner [100]. Ceol et al. demonstrated that SETDB1 accelerates formation of melanoma in a zebrafish model by trimethylating H3K9, which binds to the HOXA genes to enhance its expression [101]. Aberrant overexpression of SETDB1 is also detected in colorectal cancer (CRC), promoting CRC cell proliferation, migration, and invasion [102]. However, SETDB1 deficiency arrests CRC cells in G1 phase to inhibit cell proliferation and CRC tumorigenesis by decreasing H3K9me3 enrichment at the promoter of p21 and then suppressing p21 expression (Fig. 4) [102]. Similarly, Helicobacter pylori (H. pylori) infection induces SETDB1 expression in a TCF4-dependent manner, which contributes to gastric cancer formation [39]. Increased SETDB1 directly interacts with ERG (ETS transcription factor) to facilitate the expression of CCND1 and MMP9 to promote cell proliferation and metastasis (Fig. 4) [39]. Co-administration of CDK4/6 inhibitor palbociclib obviously enhanced the therapeutic efficacy of SETDB1 depletion on tumor growth by protecting TRIM28-mediated p-RB from proteasomal degradation both in vitro and in vivo [103]. On the contrary, SETDB1 inhibits the expression of genes associated with acute myeloid leukemia (AML), such as Dock1, Hoxa9, and Six1, to delay MLL-AF9-mediated disease progression by promoting the differentiation of leukemia cells [104].
It is reported that the protein levels of H3K9me3 and SETDB1 were increased in patients with pediatric high-grade gliomas [105]. SETDB1 silent attenuated cell viability, proliferation and migration, while increased apoptosis in two patient-derived high-grade gliomas cell lines [105]. The same conclusion was reached in another study using glioma cell lines (GOS-3, 1321N1, T98G, and U87MG) [106]. In this study, the authors also showed that another H3K9me3 methyltransferase, SUV39H1, also has the ability to regulate glial cell proliferation, migration, and colony-forming capacity [106]. Similarly, inhibition of SUV39H1 by Chaetocin significantly inhibited proliferation, clonogenic potential, and migration ability of T98G cells [107]. Furthermore, increased nuclear SUV39H1 expression was correlated with adverse prognosis of glioblastomas patients [107]. Thus, these studies indicated that H3K9me3, as well as its methyltransferases SETDB1 and SUV39H1, play important regulatory roles in glioma development and progression and may represent novel targets for targeted therapy of glioma.
Immunotherapies have shown considerable efficacy in treating several types of cancer. Amplification of SETDB1 in human tumors has been reported to contribute to immune exclusion and resistance to immune checkpoint blockade [108]. Screening chromatin regulators by CRISPR-Cas9, Griffin et al. identified that SETDB1 and other members of the HUSH and KAP1 complexes act as mediators of immune escape in a mouse tumor model treated with immune checkpoint blockade [109]. They further found that SETDB1 represses broad domains that are enriched in immune clusters and transposable elements associated with segmental duplication events, thereby suppressing transposable element-encoded retroviral antigens, latent transposable element-derived regulatory elements, immunostimulatory genes, and triggering transposable element-specific cytotoxic T cell responses in vivo [109]. Monoclonal antibodies (mAbs) targeting the programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) axis have shown striking clinical benefit in several types of cancer [110]. SETDB1-TRIM28 inhibition combined with elevated PD-L1 facilitated the formation of micronuclei in the cytoplasm, thereby activating the cGAS (cyclic GMP-AMP synthase)-STING (stimulator of interferon genes) innate immune response pathway and increasing infiltration of CD8+ T cells [108]. Furthermore, SETDB1 deficiency has been found to improve the antitumor effects of anti-PD-L1 [108]. Lysine Demethylase 5B (KDM5B) recruits SETDB1 to inhibit endogenous retroelements in a demethylase-independent manner, thereby inhibiting the cGAS-STING pathway and type-I interferon response, which contributes to tumor growth and immune memory inhibition [97]. Similarly, in acute myeloid leukemia, SETDB1 has been found to function as a novel negative innate immune regulator by suppressing type I interferon response, thereby contributing to immune evasion and oncogenic cellular state [111]. In NSCLC and melanoma patients, SETDB1 expression level is shown to be negatively correlated with radiotherapy efficacy. Moreover, SETDB1 inhibition markedly improves radiotherapy efficacy by facilitating the expression of basal and radiation-induced ERVs, enhancing MDA5/MAVS signaling, and upregulating type I interferons expression [112]. These studies suggest that SETDB1 is a negative regulator of tumor-intrinsic immunogenicity and thus a potential target for immunotherapy.
SETDB1 in other biological processes and diseases
It has been reported that approximately 10–15% of tumor cells lengthen telomeres by the alternative lengthening of telomeres (ALT) mechanism, in which TRIM28 protects the telomeric histone methyltransferase SETDB1 from degradation, thereby maintaining the H3K9me3 heterochromatin state of the telomeric DNA, which prevents telomere shortening and reduces telomeric sister chromatid exchange in cells [113]. Consistently, SETDB1 deficiency disrupts telomeric heterochromatin and abrogates ALT [114]. Telomere length is strongly associated with aging, which is the greatest risk factor for cancer. Hallmarks of aging include genomic instability, telomere attrition, mitochondrial dysfunction, cellular senescence, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, stem cell exhaustion, and altered intercellular communication [115]. The expression of SETDB1 is decreased significantly with age, and SETDB1 affects aging by regulating mitochondrial function [116]. Aging also causes a reduction in the function of spermatogonial stem cell and an increase in the risk of paternal age-related genetic diseases. SETDB1 deficiency impairs cell proliferation and cell–cell adhesion in spermatogonial stem cells by depositing H3K9me3 to inhibit MMP3/10 expression [117].
Chronic inflammation in the gastrointestinal tract is a key feature of inflammatory bowel disease (IBD), and patients with IBD are at increased risk of developing CRC [118, 119]. The expression level of SETDB1 is decreased and rare missense variants of SETDB1 are over-represented in patients with IBD [120, 121]. Moreover, Setdb1 deficiency in mouse intestinal epithelial cells is associated with barrier disruption, defective intestinal epithelial differentiation, inflammation and mortality, indicating that SETDB1 is essential for intestinal epithelial homeostasis [120]. Furthermore, mice with downregulated SETDB1 expression in intestinal stem cells develop spontaneous terminal ileitis and colitis by triggering Z-DNA-binding protein 1 (ZBP1)-dependent necroptosis via de-silencing endogenous retroviruses [121]. Thus, targeting SETDB1 or necroptosis of intestinal stem cells may be potential novel strategies for the treatment of severe IBD in humans. Interestingly, conditional knockout of Setdb1 (Setdb1-NS-cKO) in mouse neural progenitor cells showed that SETDB1 represses 5-hydroxytryptamine receptor 3A (Htr3a) transcription through endogenous retroviral sequence RMER21B-mediated distal chromatin interactions in the embryonic ganglionic eminence, thereby modulating mood behaviors and cortical Htr3a-positive interneurons development [122]. Moreover, SETDB1 was also reported to be involved in neural progenitor cells switch differentiation, and forebrain-specific Setdb1 knockout mice showed early neurogenesis impairment, severe brain defects and early lethality [123]. Additionally, SETDB1 and ATF7IP were found to cooperatively promote osteoblast proliferation by catalyzing H3K9me3 in Macrod2 (mono-ADP ribosylhydrolase 2) promoter region to suppress its expression under mechanical unloading [124]. Therefore, these studies indicated that SETDB1 is a versatile epigenetic regulator.
The functions of SETDB1 by methylating non-histone proteins
In addition to histone methylation, accumulating evidence has demonstrated that non-histone proteins can also be methylated by histone methyltransferases, for example, histone methyltransferase SMYD2 mono-methylates K370 site of P53 in tumor cells [3, 125, 126]. SETDB1 is known to be not only a methyltransferase that tri-methylates H3K9 but also methylates non-histone proteins (e.g., P53 and AKT) to participate in tumorigenesis (Fig. 5) [17,18,19, 127]. Moderate copy number gain of SETDB1 leads to its overexpression in HCC, which catalyzes the K370 site of P53 di-methylation by interacting with P53 to increase recognition and degradation of P53 by MDM2 [19]. In addition, SETDB1 binds to Trp53 promoter to inhibit its expression, decreasing apoptosis and increasing growth of human PDACs [99]. These studies indicate that SETDB1 kills two birds with one stone, that is, SETDB1 not only suppresses P53 expression at the transcriptional level, but also methylates P53 to promote its degradation, and then facilitating tumor development.

The role of SETDB1 in tumors by methylating nonhistones. SETDB1 trimethylates AKT at K64, K140 or K142 to increase the phosphorylation and activity of AKT, which accelerates non-small cell lung cancer and colorectal cancer or skin tumorigenesis, respectively. In liver cancer, SETDB1 dimethylates gain-of-function (GOF) mutant P53 at K370 to prevent its degradation by ubiquitination, and then promotes liver cancer cell growth
In addition to P53, AKT is also a substrate that is methylated by SETDB1 and involved in tumorigenesis (Fig. 5) [17, 18, 127]. SETDB1 tri-methylates the K140 and K142 sites of AKT to promote its phosphorylation on T308 and S473 sites and activation, which is antagonized by lysine demethylase 4B (KDM4B). Moreover, non-methylated mutant Akt1 knock-in mice not only have reduced body size and weight, but also less susceptible to carcinogen-induced skin tumorigenesis [18]. In NSCLC, SETDB1 triggers di- and tri-methylation of the AKT K64 site followed by initiation of K63-linked AKT ubiquitination by recruiting Jumonji Domain-Containing Protein 2A (JMJD2A) and E3 ligases (e.g., TRAF6 and Skp2-SCF) to the AKT complex, which in turn leads to cell membrane recruitment, T308 phosphorylation and subsequently activation of AKT. AKT hyperactivation promotes NSCLC progression and predicts poor outcome in NSCLC patients [17]. Similarly, in colorectal cancer, SETDB1 overexpression facilitates cell proliferation by activating AKT, and inhibition of SETDB1 augments the sensitivity of cetuximab in colorectal cancer [127]. Therefore, targeting SETDB1-mediated AKT methylation is a promising strategy for the treatment of cancers (such as skin tumor, NSCLC, and colorectal cancer).
Conclusions and perspectives
The recent surge of studies on SETDB1 has highlighted its vital role in regulating multiple diseases, embryonic development, and stem cells by methylating H3K9 or non-histone proteins. SETDB1 plays an indispensable role in cell fate determination by regulating ERVs silencing in H3K9 methylation-dependent or -independent manner. However, further research is needed to understand how SETDB1 works with different partners to regulate cell fate under different pathophysiological conditions. Interestingly, SETDB1 has been found to be upregulated in most cancers, and its overexpression has been significantly associated with cancer aggressiveness and poorer prognosis. However, in some cancers, SETDB1 functions as an oncogenic driver, while in others it is a tumor metastasis suppressor. The relevant molecular mechanisms deserve further exploration. Although some studies have revealed the role of SETDB1 in diseases such as IBD, the effects and regulatory mechanisms of SETDB1 on diseases need to be further elucidated. In addition to methylating H3K9, some recent studies have found that SETDB1 can methylate non-histone proteins (e.g., P53 and AKT), which further expands the downstream mechanisms of SETDB1 and deepens our understanding of SETDB1. There is an urgent need to clarify the function of SETDB1 under distinct physiological or pathological conditions and to ascertain how the specificity of SETDB1 binding to a specific substrate (histones or non-histone proteins) is determined. Most importantly, SETDB1 inhibition has been found to significantly enhance the efficacy of radiotherapy and immunotherapy in cancers, suggesting that the development of SETDB1 inhibitors/drugs with high specificity, low toxicity, and high efficiency could provide new options for tumor treatment by targeting SETDB1. Additionally, since SETDB1 is an important regulator that controls the development of many diseases, therapeutic RNAs targeting SETDB1, including small interfering RNAs (siRNAs), antisense oligonucleotides (ASOs), or large RNAs such as mRNAs, long non-coding RNAs (lncRNAs), and cyclic RNAs, represent an alternative strategy for developing treatments for diseases.
Availability of data and materials
All the materials will be provided from the corresponding author on reasonable request.
Abbreviations
- SETDB1:
-
SET domain bifurcated histone lysine methyltransferase 1
- ERVs:
-
Endogenous retroviruses
- mESCs:
-
Embryonic stem cells
- DNMTs:
-
DNA methyltransferases
- IBD:
-
Inflammatory bowel disease
- AdoMet:
-
S-Adenosyl-l-methionine
- ESET:
-
Erg-associated SET domain
- KMT1E:
-
Lysine N-methyltransferase 1E
- HP1a:
-
Heterochromatin protein 1a
- TUDs:
-
Tudor domains
- MBD:
-
Methyl-CpG binding domain
- Sp1:
-
Specificity protein 1
- MAPK:
-
Mitogen-activated protein kinase
- TCF4:
-
Transcription factor 4
- GC:
-
Gastric cancer
- MMP9:
-
Matrix metalloproteinase 9
- CCND1:
-
Cyclin D1
- HCC:
-
Hepatocellular carcinoma
- NSCLC:
-
Non-small cell lung cancer
- ATF7IP:
-
Activating transcription factor 7-interacting protein
- CAF1:
-
Chromatin assembly factor 1
- KAP-1:
-
KRAB-ZFP-associated protein 1
- IAP:
-
Intracisternal A-type particles
- LTRs:
-
Long terminal repeats
- PGCs:
-
Primordial germ cells
- Dppa2:
-
Developmental pluripotency associated 2
- Otx2:
-
Orthodenticle homeobox 2
- Utf1:
-
Undifferentiated embryonic cell transcription factor 1
- MPP8:
-
M-phase phosphoprotein 8
- TET2:
-
Tet methylcytosine dioxygenase 2
- HSPCs:
-
Hematopoietic stem and progenitor cells
- METTL3:
-
Methyltransferase-like 3
- PRC2:
-
Polycomb repressive complex 2
- IGFBP4:
-
Insulin like growth factor binding protein 4
- LRP8:
-
LDL receptor related protein 8
- FZD1:
-
Frizzled class receptor 1
- APOE:
-
Apolipoprotein E
- FOXA2:
-
Forkhead box A2
- ANXA2:
-
Annexin A2
- EMT:
-
Epithelial–mesenchymal transition
- Snai1:
-
Snail family transcriptional repressor 1
- EZH2:
-
Enhancer of zeste homolog 2
- RUNX3:
-
RUNX Family Transcription Factor 3
- PDACs:
-
Pancreatic ductal adenocarcinomas
- THBS1:
-
Thrombospondin 1
- CRC:
-
Colorectal cancer
- AML:
-
Acute myeloid leukemia
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed cell death ligand 1
- cGAS:
-
Cyclic GMP-AMP synthase
- STING:
-
Stimulator of interferon genes
- KDM5B:
-
Lysine demethylase 5B
- ALT:
-
Alternative lengthening of telomeres
- ZBP1:
-
Z-DNA-binding protein 1
- Htr3a:
-
5-Hydroxytryptamine receptor 3A
- Macrod2:
-
Mono-ADP Ribosylhydrolase 2
- KDM4B:
-
Lysine demethylase 4B
- JMJD2A:
-
Jumonji Domain-Containing Protein 2A
References
Biggar KK, Li SS. Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol. 2015;16(1):5–17.
Li R, Wei X, Jiang DS. Protein methylation functions as the posttranslational modification switch to regulate autophagy. Cell Mol Life Sci. 2019;76(19):3711–22.
Yi X, Jiang XJ, Fang ZM. Histone methyltransferase SMYD2: ubiquitous regulator of disease. Clin Epigenet. 2019;11(1):112.
Wu Q, Schapira M, Arrowsmith CH, Barsyte-Lovejoy D. Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat Rev Drug Discov. 2021;20(7):509–30.
Feldman D, Ziv C, Gorovits R, Efrat M, Yarden O. Neurospora crassa protein arginine methyl transferases are involved in growth and development and interact with the NDR kinase COT1. PLoS ONE. 2013;8(11): e80756.
Dilworth D, Barsyte-Lovejoy D. Targeting protein methylation: from chemical tools to precision medicines. Cell Mol Life Sci. 2019;76(15):2967–85.
Li R, Yi X, Wei X, Huo B, Guo X, Cheng C, Fang ZM, Wang J, Feng X, Zheng P, et al. EZH2 inhibits autophagic cell death of aortic vascular smooth muscle cells to affect aortic dissection. Cell Death Dis. 2018;9(2):180.
Yi X, Jiang XJ, Li XY, Jiang DS. Histone methyltransferases: novel targets for tumor and developmental defects. Am J Transl Res. 2015;7(11):2159–75.
Yi X, Jiang X, Li X, Jiang DS. Histone lysine methylation and congenital heart disease: from bench to bedside (Review). Int J Mol Med. 2017;40(4):953–64.
Jiang DS, Yi X, Li R, Su YS, Wang J, Chen ML, Liu LG, Hu M, Cheng C, Zheng P, et al. The histone methyltransferase mixed lineage leukemia (MLL) 3 may play a potential role on clinical dilated cardiomyopathy. Mol Med. 2017;23:196–203.
Wei X, Yi X, Zhu XH, Jiang DS. Histone methylation and vascular biology. Clin Epigenet. 2020;12(1):30.
Markouli M, Strepkos D, Chlamydas S, Piperi C. Histone lysine methyltransferase SETDB1 as a novel target for central nervous system diseases. Prog Neurobiol. 2021;200: 101968.
Fukuda K, Shinkai Y. SETDB1-mediated silencing of retroelements. Viruses. 2020;12(6):596.
Fukuda K, Shimura C, Miura H, Tanigawa A, Suzuki T, Dohmae N, Hiratani I, Shinkai Y. Regulation of mammalian 3D genome organization and histone H3K9 dimethylation by H3K9 methyltransferases. Commun Biol. 2021;4(1):571.
Zeller P, Padeken J, van Schendel R, Kalck V, Tijsterman M, Gasser SM. Histone H3K9 methylation is dispensable for caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat Genet. 2016;48(11):1385–95.
Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ 3rd. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16(8):919–32.
Wang G, Long J, Gao Y, Zhang W, Han F, Xu C, Sun L, Yang SC, Lan J, Hou Z, et al. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat Cell Biol. 2019;21(2):214–25.
Guo J, Dai X, Laurent B, Zheng N, Gan W, Zhang J, Guo A, Yuan M, Liu P, Asara JM, et al. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat Cell Biol. 2019;21(2):226–37.
Fei Q, Shang K, Zhang J, Chuai S, Kong D, Zhou T, Fu S, Liang Y, Li C, Chen Z, et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat Commun. 2015;6:8651.
Barral A, Pozo G, Ducrot L, Papadopoulos GL, Sauzet S, Oldfield AJ, Cavalli G, Dejardin J. SETDB1/NSD-dependent H3K9me3/H3K36me3 dual heterochromatin maintains gene expression profiles by bookmarking poised enhancers. Mol Cell. 2022;82(4):816-832.e812.
Kato M, Takemoto K, Shinkai Y. A somatic role for the histone methyltransferase Setdb1 in endogenous retrovirus silencing. Nat Commun. 2018;9(1):1683.
Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8(6):676–87.
Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464(7290):927–31.
Wu K, Liu H, Wang Y, He J, Xu S, Chen Y, Kuang J, Liu J, Guo L, Li D, et al. SETDB1-mediated cell fate transition between 2C-like and pluripotent states. Cell Rep. 2020;30(1):25-36.e26.
Warrier T, El Farran C, Zeng Y, Ho BSQ, Bao Q, Zheng ZH, Bi X, Ng HH, Ong DST, Chu JJH, et al. SETDB1 acts as a topological accessory to Cohesin via an H3K9me3-independent, genomic shunt for regulating cell fates. Nucleic Acids Res. 2022;50(13):7326–49.
Strepkos D, Markouli M, Klonou A, Papavassiliou AG, Piperi C. Histone methyltransferase SETDB1: a common denominator of tumorigenesis with therapeutic potential. Cancer Res. 2021;81(3):525–34.
Zhao Z, Feng L, Peng X, Ma T, Tong R, Zhong L. Role of histone methyltransferase SETDB1 in regulation of tumourigenesis and immune response. Front Pharmacol. 2022;13:1073713.
Torrano J, Al Emran A, Hammerlindl H, Schaider H. Emerging roles of H3K9me3, SETDB1 and SETDB2 in therapy-induced cellular reprogramming. Clin Epigenet. 2019;11(1):43.
Vural S, Palmisano A, Reinhold WC, Pommier Y, Teicher BA, Krushkal J. Association of expression of epigenetic molecular factors with DNA methylation and sensitivity to chemotherapeutic agents in cancer cell lines. Clin Epigenet. 2021;13(1):49.
Harte PJ, Wu W, Carrasquillo MM, Matera AG. Assignment of a novel bifurcated SET domain gene, SETDB1, to human chromosome band 1q21 by in situ hybridization and radiation hybrids. Cytogenet Cell Genet. 1999;84(1–2):83–6.
Yang L, Xia L, Wu DY, Wang H, Chansky HA, Schubach WH, Hickstein DD, Zhang Y. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene. 2002;21(1):148–52.
Blackburn ML, Chansky HA, Zielinska-Kwiatkowska A, Matsui Y, Yang L. Genomic structure and expression of the mouse ESET gene encoding an ERG-associated histone methyltransferase with a SET domain. Biochim Biophys Acta. 2003;1629(1–3):8–14.
Karanth AV, Maniswami RR, Prashanth S, Govindaraj H, Padmavathy R, Jegatheesan SK, Mullangi R, Rajagopal S. Emerging role of SETDB1 as a therapeutic target. Expert Opin Ther Targets. 2017;21(3):319–31.
Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. 2006;281(28):19489–500.
Markouli M, Strepkos D, Piperi C. Structure, activity and function of the SETDB1 protein methyltransferase. Life (Basel). 2021;11(8):817.
Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc Natl Acad Sci USA. 2006;103(50):19176–81.
Osada N, Kosuge Y, Ishige K, Ito Y. Mithramycin, an agent for developing new therapeutic drugs for neurodegenerative diseases. J Pharmacol Sci. 2013;122(4):251–6.
Federico A, Steinfass T, Larribere L, Novak D, Moris F, Nunez LE, Umansky V, Utikal J. Mithramycin A and mithralog EC-8042 inhibit SETDB1 expression and its oncogenic activity in malignant melanoma. Mol Ther Oncolytics. 2020;18:83–99.
Shang W, Wang Y, Liang X, Li T, Shao W, Liu F, Cui X, Wang Y, Lv L, Chai L, et al. SETDB1 promotes gastric carcinogenesis and metastasis via upregulation of CCND1 and MMP9 expression. J Pathol. 2021;253(2):148–59.
Xiao JF, Sun QY, Ding LW, Chien W, Liu XY, Mayakonda A, Jiang YY, Loh XY, Ran XB, Doan NB, et al. The c-MYC-BMI1 axis is essential for SETDB1-mediated breast tumourigenesis. J Pathol. 2018;246(1):89–102.
Kim WR, Park EG, Lee HE, Park SJ, Huh JW, Kim JN, Kim HS. Hsa-miR-422a originated from short interspersed nuclear element increases ARID5B expression by collaborating with NF-E2. Mol Cells. 2022;45(7):465–78.
Shao Y, Song X, Jiang W, Chen Y, Ning Z, Gu W, Jiang J. MicroRNA-621 acts as a tumor radiosensitizer by directly targeting SETDB1 in hepatocellular carcinoma. Mol Ther. 2019;27(2):355–64.
Liu S, Li B, Xu J, Hu S, Zhan N, Wang H, Gao C, Li J, Xu X. SOD1 promotes cell proliferation and metastasis in non-small cell lung cancer via an miR-409-3p/SOD1/SETDB1 epigenetic regulatory feedforward loop. Front Cell Dev Biol. 2020;8:213.
Beyer S, Pontis J, Schirwis E, Battisti V, Rudolf A, Le Grand F, Ait-Si-Ali S. Canonical Wnt signalling regulates nuclear export of Setdb1 during skeletal muscle terminal differentiation. Cell Discov. 2016;2:16037.
Tsusaka T, Shimura C, Shinkai Y. ATF7IP regulates SETDB1 nuclear localization and increases its ubiquitination. EMBO Rep. 2019;20(12): e48297.
Timms RT, Tchasovnikarova IA, Antrobus R, Dougan G, Lehner PJ. ATF7IP-mediated stabilization of the histone methyltransferase SETDB1 is essential for heterochromatin formation by the HUSH complex. Cell Rep. 2016;17(3):653–9.
Hu H, Khodadadi-Jamayran A, Dolgalev I, Cho H, Badri S, Chiriboga LA, Zeck B, Lopez De Rodas Gregorio M, Dowling CM, Labbe K, et al. Targeting the Atf7ip-Setdb1 complex augments antitumor immunity by boosting tumor immunogenicity. Cancer Immunol Res. 2021;9(11):1298–315.
Sun L, Fang J. E3-independent constitutive monoubiquitination complements histone methyltransferase activity of SETDB1. Mol Cell. 2016;62(6):958–66.
Osumi K, Sato K, Murano K, Siomi H, Siomi MC. Essential roles of Windei and nuclear monoubiquitination of Eggless/SETDB1 in transposon silencing. EMBO Rep. 2019;20(12): e48296.
Montavon T, Shukeir N, Erikson G, Engist B, Onishi-Seebacher M, Ryan D, Musa Y, Mittler G, Meyer AG, Genoud C, et al. Complete loss of H3K9 methylation dissolves mouse heterochromatin organization. Nat Commun. 2021;12(1):4359.
Padeken J, Methot S, Zeller P, Delaney CE, Kalck V, Gasser SM. Argonaute NRDE-3 and MBT domain protein LIN-61 redundantly recruit an H3K9me3 HMT to prevent embryonic lethality and transposon expression. Genes Dev. 2021;35(1–2):82–101.
Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C, Harel-Bellan A, Ameyar-Zazoua M, Ait-Si-Ali S. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell. 2010;37(1):46–56.
Lundberg LE, Stenberg P, Larsson J. HP1a, Su(var)3–9, SETDB1 and POF stimulate or repress gene expression depending on genomic position, gene length and expression pattern in Drosophila melanogaster. Nucleic Acids Res. 2013;41(8):4481–94.
Ayyanathan K, Lechner MS, Bell P, Maul GG, Schultz DC, Yamada Y, Tanaka K, Torigoe K, Rauscher FJ 3rd. Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev. 2003;17(15):1855–69.
Dong L, Liao H, Zhao L, Wang J, Wang C, Wang B, Sun Y, Xu L, Xia Y, Ling S, et al. A functional crosstalk between the H3K9 methylation writers and their reader HP1 in safeguarding embryonic stem cell identity. Stem Cell Rep. 2023;18(9):1775–92.
Maeda R, Tachibana M. HP1 maintains protein stability of H3K9 methyltransferases and demethylases. EMBO Rep. 2022;23(4): e53581.
Tzeng TY, Lee CH, Chan LW, Shen CK. Epigenetic regulation of the Drosophila chromosome 4 by the histone H3K9 methyltransferase dSETDB1. Proc Natl Acad Sci USA. 2007;104(31):12691–6.
Maksimov DA, Koryakov DE. Binding of SU(VAR)3–9 partially depends on SETDB1 in the chromosomes of Drosophila melanogaster. Cells. 2019;8(9):1030.
Seller CA, Cho CY, O’Farrell PH. Rapid embryonic cell cycles defer the establishment of heterochromatin by eggless/SetDB1 in Drosophila. Genes Dev. 2019;33(7–8):403–17.
Loyola A, Tagami H, Bonaldi T, Roche D, Quivy JP, Imhof A, Nakatani Y, Dent SY, Almouzni G. The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 2009;10(7):769–75.
Rauwel B, Jang SM, Cassano M, Kapopoulou A, Barde I, Trono D. Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. Elife. 2015;4: e06068.
Muller I, Moroni AS, Shlyueva D, Sahadevan S, Schoof EM, Radzisheuskaya A, Hojfeldt JW, Tatar T, Koche RP, Huang C, et al. MPP8 is essential for sustaining self-renewal of ground-state pluripotent stem cells. Nat Commun. 2021;12(1):3034.
Leung D, Du T, Wagner U, Xie W, Lee AY, Goyal P, Li Y, Szulwach KE, Jin P, Lorincz MC, et al. Regulation of DNA methylation turnover at LTR retrotransposons and imprinted loci by the histone methyltransferase Setdb1. Proc Natl Acad Sci USA. 2014;111(18):6690–5.
Mochizuki K, Sharif J, Shirane K, Uranishi K, Bogutz AB, Janssen SM, Suzuki A, Okuda A, Koseki H, Lorincz MC. Repression of germline genes by PRC1.6 and SETDB1 in the early embryo precedes DNA methylation-mediated silencing. Nat Commun. 2021;12(1):7020.
Wang Z, Fan R, Russo A, Cernilogar FM, Nuber A, Schirge S, Shcherbakova I, Dzhilyanova I, Ugur E, Anton T, et al. Dominant role of DNA methylation over H3K9me3 for IAP silencing in endoderm. Nat Commun. 2022;13(1):5447.
Wenger A, Biran A, Alcaraz N, Redo-Riveiro A, Sell AC, Krautz R, Flury V, Reveron-Gomez N, Solis-Mezarino V, Volker-Albert M, et al. Symmetric inheritance of parental histones governs epigenome maintenance and embryonic stem cell identity. Nat Genet. 2023;55(9):1567–78.
Wen Q, Zhou J, Tian C, Li X, Song G, Gao Y, Sun Y, Ma C, Yao S, Liang X, et al. Symmetric inheritance of parental histones contributes to safeguarding the fate of mouse embryonic stem cells during differentiation. Nat Genet. 2023;55(9):1555–66.
Cheng EC, Hsieh CL, Liu N, Wang J, Zhong M, Chen T, Li E, Lin H. The essential function of SETDB1 in homologous chromosome pairing and synapsis during meiosis. Cell Rep. 2021;34(1): 108575.
Hirota T, Blakeley P, Sangrithi MN, Mahadevaiah SK, Encheva V, Snijders AP, ElInati E, Ojarikre OA, de Rooij DG, Niakan KK, et al. SETDB1 links the meiotic DNA damage response to sex chromosome silencing in mice. Dev Cell. 2018;47(5):645-659.e646.
Eymery A, Liu Z, Ozonov EA, Stadler MB, Peters AH. The methyltransferase Setdb1 is essential for meiosis and mitosis in mouse oocytes and early embryos. Development. 2016;143(15):2767–79.
Smolko AE, Shapiro-Kulnane L, Salz HK. The H3K9 methyltransferase SETDB1 maintains female identity in Drosophila germ cells. Nat Commun. 2018;9(1):4155.
Gualdrini F, Polletti S, Simonatto M, Prosperini E, Pileri F, Natoli G. H3K9 trimethylation in active chromatin restricts the usage of functional CTCF sites in SINE B2 repeats. Genes Dev. 2022;36(7–8):414–32.
Liu S, Brind’Amour J, Karimi MM, Shirane K, Bogutz A, Lefebvre L, Sasaki H, Shinkai Y, Lorincz MC. Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev. 2014;28(18):2041–55.
Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010;463(7278):237–40.
Mochizuki K, Tando Y, Sekinaka T, Otsuka K, Hayashi Y, Kobayashi H, Kamio A, Ito-Matsuoka Y, Takehara A, Kono T, et al. SETDB1 is essential for mouse primordial germ cell fate determination by ensuring BMP signaling. Development. 2018;145(23):dev164160.
Sharif J, Endo TA, Nakayama M, Karimi MM, Shimada M, Katsuyama K, Goyal P, Brind’Amour J, Sun MA, Sun Z, et al. Activation of endogenous retroviruses in Dnmt1(−/−) ESCs involves disruption of SETDB1-mediated repression by NP95 binding to hemimethylated DNA. Cell Stem Cell. 2016;19(1):81–94.
Deniz O, de la Rica L, Cheng KCL, Spensberger D, Branco MR. SETDB1 prevents TET2-dependent activation of IAP retroelements in naive embryonic stem cells. Genome Biol. 2018;19(1):6.
Xu W, Li J, He C, Wen J, Ma H, Rong B, Diao J, Wang L, Wang J, Wu F, et al. METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature. 2021;591(7849):317–21.
Fang ZM, Zhang SM, Luo H, Jiang DS, Huo B, Zhong X, Feng X, Cheng W, Chen Y, Feng G, et al. Methyltransferase-like 3 suppresses phenotypic switching of vascular smooth muscle cells by activating autophagosome formation. Cell Prolif. 2022;56: e13386.
Li N, Yi X, He Y, Huo B, Chen Y, Zhang Z, Wang Q, Li Y, Zhong X, Li R, et al. Targeting ferroptosis as a novel approach to alleviate aortic dissection. Int J Biol Sci. 2022;18(10):4118–34.
Chen J, Wei X, Yi X, Jiang DS. RNA modification by m(6)A methylation in cardiovascular disease. Oxid Med Cell Longev. 2021;2021:8813909.
Koide S, Oshima M, Takubo K, Yamazaki S, Nitta E, Saraya A, Aoyama K, Kato Y, Miyagi S, Nakajima-Takagi Y, et al. Setdb1 maintains hematopoietic stem and progenitor cells by restricting the ectopic activation of nonhematopoietic genes. Blood. 2016;128(5):638–49.
Collins PL, Kyle KE, Egawa T, Shinkai Y, Oltz EM. The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc Natl Acad Sci USA. 2015;112(27):8367–72.
Pasquarella A, Ebert A, Pereira de Almeida G, Hinterberger M, Kazerani M, Nuber A, Ellwart J, Klein L, Busslinger M, Schotta G. Retrotransposon derepression leads to activation of the unfolded protein response and apoptosis in pro-B cells. Development. 2016;143(10):1788–99.
Adoue V, Binet B, Malbec A, Fourquet J, Romagnoli P, van Meerwijk JPM, Amigorena S, Joffre OP. The histone methyltransferase SETDB1 controls T helper cell lineage integrity by repressing endogenous retroviruses. Immunity. 2019;50(3):629-644.e628.
Wu J, Li J, Chen K, Liu G, Zhou Y, Chen W, Zhu X, Ni TT, Zhang B, Jin D, et al. Atf7ip and Setdb1 interaction orchestrates the hematopoietic stem and progenitor cell state with diverse lineage differentiation. Proc Natl Acad Sci USA. 2023;120(1): e2209062120.
Fei Q, Yang X, Jiang H, Wang Q, Yu Y, Yu Y, Yi W, Zhou S, Chen T, Lu C, et al. SETDB1 modulates PRC2 activity at developmental genes independently of H3K9 trimethylation in mouse ES cells. Genome Res. 2015;25(9):1325–35.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Rodriguez-Paredes M, Martinez de Paz A, Simo-Riudalbas L, Sayols S, Moutinho C, Moran S, Villanueva A, Vazquez-Cedeira M, Lazo PA, Carneiro F, et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene. 2014;33(21):2807–13.
Sun QY, Ding LW, Xiao JF, Chien W, Lim SL, Hattori N, Goodglick L, Chia D, Mah V, Alavi M, et al. SETDB1 accelerates tumourigenesis by regulating the WNT signalling pathway. J Pathol. 2015;235(4):559–70.
Zakharova VV, Magnitov MD, Del Maestro L, Ulianov SV, Glentis A, Uyanik B, Williart A, Karpukhina A, Demidov O, Joliot V, et al. SETDB1 fuels the lung cancer phenotype by modulating epigenome, 3D genome organization and chromatin mechanical properties. Nucleic Acids Res. 2022;50(8):4389–413.
Cruz-Tapias P, Zakharova V, Perez-Fernandez OM, Mantilla W, Ram I-CS, Ait-Si-Ali S. Expression of the major and pro-oncogenic H3K9 lysine methyltransferase SETDB1 in non-small cell lung cancer. Cancers (Basel). 2019;11(8):1134.
Ueshima S, Fang J. Histone H3K9 methyltransferase SETDB1 augments invadopodia formation to promote tumor metastasis. Oncogene. 2022;41(24):3370–80.
Wu PC, Lu JW, Yang JY, Lin IH, Ou DL, Lin YH, Chou KH, Huang WF, Wang WP, Huang YL, et al. H3K9 histone methyltransferase, KMT1E/SETDB1, cooperates with the SMAD2/3 pathway to suppress lung cancer metastasis. Cancer Res. 2014;74(24):7333–43.
Du D, Katsuno Y, Meyer D, Budi EH, Chen SH, Koeppen H, Wang H, Akhurst RJ, Derynck R. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial–mesenchymal transition. EMBO Rep. 2018;19(1):135–55.
Tang X, Sui X, Weng L, Liu Y. SNAIL1: linking tumor metastasis to immune evasion. Front Immunol. 2021;12: 724200.
Balinth S, Fisher ML, Hwangbo Y, Wu C, Ballon C, Sun X, Mills AA. EZH2 regulates a SETDB1/DeltaNp63alpha axis via RUNX3 to drive a cancer stem cell phenotype in squamous cell carcinoma. Oncogene. 2022;41(35):4130–44.
Wong CM, Wei L, Law CT, Ho DW, Tsang FH, Au SL, Sze KM, Lee JM, Wong CC, Ng IO. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology. 2016;63(2):474–87.
Ogawa S, Fukuda A, Matsumoto Y, Hanyu Y, Sono M, Fukunaga Y, Masuda T, Araki O, Nagao M, Yoshikawa T, et al. SETDB1 inhibits p53-mediated apoptosis and is required for formation of pancreatic ductal adenocarcinomas in mice. Gastroenterology. 2020;159(2):682-696.e613.
Orouji E, Federico A, Larribere L, Novak D, Lipka DB, Assenov Y, Sachindra S, Huser L, Granados K, Gebhardt C, et al. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int J Cancer. 2019;145(12):3462–77.
Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferre F, et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471(7339):513–7.
Cao N, Yu Y, Zhu H, Chen M, Chen P, Zhuo M, Mao Y, Li L, Zhao Q, Wu M, et al. SETDB1 promotes the progression of colorectal cancer via epigenetically silencing p21 expression. Cell Death Dis. 2020;11(5):351.
Huang Z, Li X, Tang B, Li H, Zhang J, Sun R, Ma J, Pan Y, Yan B, Zhou Y, et al. SETDB1 modulates degradation of phosphorylated RB and anti-cancer efficacy of CDK4/6 inhibitors. Cancer Res. 2023;83(6):875–89.
Ropa J, Saha N, Hu H, Peterson LF, Talpaz M, Muntean AG. SETDB1 mediated histone H3 lysine 9 methylation suppresses MLL-fusion target expression and leukemic transformation. Haematologica. 2020;105(9):2273–85.
Klonou A, Korkolopoulou P, Giannopoulou AI, Kanakoglou DS, Pampalou A, Gargalionis AN, Sarantis P, Mitsios A, Sgouros S, Papavassiliou AG, et al. Histone H3K9 methyltransferase SETDB1 overexpression correlates with pediatric high-grade gliomas progression and prognosis. J Mol Med (Berl). 2023;101(4):387–401.
Spyropoulou A, Gargalionis A, Dalagiorgou G, Adamopoulos C, Papavassiliou KA, Lea RW, Piperi C, Papavassiliou AG. Role of histone lysine methyltransferases SUV39H1 and SETDB1 in gliomagenesis: modulation of cell proliferation, migration, and colony formation. Neuromol Med. 2014;16(1):70–82.
Sepsa A, Levidou G, Gargalionis A, Adamopoulos C, Spyropoulou A, Dalagiorgou G, Thymara I, Boviatsis E, Themistocleous MS, Petraki K, et al. Emerging role of linker histone variant H1x as a biomarker with prognostic value in astrocytic gliomas. A multivariate analysis including trimethylation of H3K9 and H4K20. PLoS ONE. 2015;10(1): e0115101.
Lin J, Guo D, Liu H, Zhou W, Wang C, Muller I, Kossenkov AV, Drapkin R, Bitler BG, Helin K, et al. The SETDB1-TRIM28 complex suppresses antitumor immunity. Cancer Immunol Res. 2021;9(12):1413–24.
Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, Dele-Oni D, Du PP, Halawi AG, Ishizuka JJ, et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature. 2021;595(7866):309–14.
Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y, Xia Y. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J Hematol Oncol. 2022;15(1):24.
Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, Janakiraman V, Durinck S, Stinson J, Arnott D, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol. 2017;216(11):3535–49.
Pan D, Bao X, Hu M, Jiao M, Li F, Li CY. SETDB1 restrains endogenous retrovirus expression and antitumor immunity during radiotherapy. Cancer Res. 2022;82(15):2748–60.
Wang C, Songyang Z, Huang Y. TRIM28 inhibits alternative lengthening of telomere phenotypes by protecting SETDB1 from degradation. Cell Biosci. 2021;11(1):149.
Gauchier M, Kan S, Barral A, Sauzet S, Agirre E, Bonnell E, Saksouk N, Barth TK, Ide S, Urbach S, et al. SETDB1-dependent heterochromatin stimulates alternative lengthening of telomeres. Sci Adv. 2019;5(5): eaav3673.
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217.
Lee JH, Demarest TG, Babbar M, Kim EW, Okur MN, De S, Croteau DL, Bohr VA. Cockayne syndrome group B deficiency reduces H3K9me3 chromatin remodeler SETDB1 and exacerbates cellular aging. Nucleic Acids Res. 2019;47(16):8548–62.
Liu R, Liu Z, Guo M, Zeng W, Zheng Y. SETDB1 regulates porcine spermatogonial adhesion and proliferation through modulating MMP3/10 transcription. Cells. 2022;11(3):370.
Liu D, Saikam V, Skrada KA, Merlin D, Iyer SS. Inflammatory bowel disease biomarkers. Med Res Rev. 2022;42(5):1856–87.
Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology. 2022;162(3):715-730.e713.
Juznic L, Peuker K, Strigli A, Brosch M, Herrmann A, Hasler R, Koch M, Matthiesen L, Zeissig Y, Loscher BS, et al. SETDB1 is required for intestinal epithelial differentiation and the prevention of intestinal inflammation. Gut. 2021;70(3):485–98.
Wang R, Li H, Wu J, Cai ZY, Li B, Ni H, Qiu X, Chen H, Liu W, Yang ZH, et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature. 2020;580(7803):386–90.
Li J, Zheng S, Dong Y, Xu H, Zhu Y, Weng J, Sun D, Wang S, Xiao L, Jiang Y. Histone methyltransferase SETDB1 regulates the development of cortical Htr3a-positive interneurons and mood behaviors. Biol Psychiatry. 2023;93(3):279–90.
Tan SL, Nishi M, Ohtsuka T, Matsui T, Takemoto K, Kamio-Miura A, Aburatani H, Shinkai Y, Kageyama R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development. 2012;139(20):3806–16.
Zhang L, Xu L, Zhang X, Wang K, Tan Y, Li G, Wang Y, Xue T, Sun Q, Cao X, et al. Methyltransferase Setdb1 promotes osteoblast proliferation by epigenetically silencing Macrod2 with the assistance of Atf7ip. Cells. 2022;11(16):2580.
He Y, Yi X, Zhang Z, Luo H, Li R, Feng X, Fang ZM, Zhu XH, Cheng W, Jiang DS, et al. JIB-04, a histone demethylase Jumonji C domain inhibitor, regulates phenotypic switching of vascular smooth muscle cells. Clin Epigenet. 2022;14(1):101.
Chen Y, Yi X, Huo B, He Y, Guo X, Zhang Z, Zhong X, Feng X, Fang ZM, Zhu XH, et al. BRD4770 functions as a novel ferroptosis inhibitor to protect against aortic dissection. Pharmacol Res. 2022;177: 106122.
Hou Z, Sun L, Xu F, Hu F, Lan J, Song D, Feng Y, Wang J, Luo X, Hu J, et al. Blocking histone methyltransferase SETDB1 inhibits tumorigenesis and enhances cetuximab sensitivity in colorectal cancer. Cancer Lett. 2020;487:63–73.
Acknowledgements
We apologize to authors whose papers could not be cited due to space restrictions.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 82070306, No. 81873456, and No. 82170502).
Author information
Authors and Affiliations
Contributions
DSJ and DD supervised the general concept of this review. HL and XW wrote the manuscript. XHZ and XY participated in the revision and literature search. All authors contributed to the editing process and read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors provided consent for publication of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Luo, H., Wu, X., Zhu, XH. et al. The functions of SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) in biological process and disease. Epigenetics & Chromatin 16, 47 (2023). https://doi.org/10.1186/s13072-023-00519-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13072-023-00519-1