
Abstract
Background
Different mosquito control strategies have been implemented to mitigate or prevent mosquito-related public health situations. Modern mosquito control largely relies on multiple approaches, including targeted, specific treatments. Given this, it is becoming increasingly important to supplement these activities with rapid and mobile diagnostic capacities for mosquito-borne diseases. We aimed to create and test the applicability of a rapid diagnostic system for West Nile virus that can be used under field conditions.
Methods
In this pilot study, various types of adult mosquito traps were applied within the regular mosquito monitoring activity framework for mosquito control. Then, the captured specimens were used for the detection of West Nile virus RNA under field conditions with a portable qRT-PCR approach within 3–4 h. Then, positive samples were subjected to confirmatory RT-PCR or NGS sequencing in the laboratory to obtain genome information of the virus. We implemented phylogenetic analysis to characterize circulating strains.
Results
A total of 356 mosquito individuals representing 7 species were processed in 54 pools, each containing up to 20 individuals. These pools were tested for the presence of West Nile virus, and two pools tested positive, containing specimens from the Culex pipiens and Anopheles atroparvus mosquito species. As a result of subsequent sequencing, we present the complete genome of West Nile virus and Bagaza virus.
Conclusions
The rapid identification of infected mosquitoes is the most important component of quick response adulticide or larvicide treatments to prevent human cases. The conceptual framework of real-time surveillance can be optimized for other pathogens and situations not only in relation to West Nile virus. We present an early warning system for mosquito-borne diseases and demonstrate its application to aid rapid-response mosquito control actions.
Graphical Abstract

Similar content being viewed by others

Background
There are multiple endemic mosquito-borne viruses in Europe with human or animal health relevance. In recent decades an increasing number of autochthonous cases were detected with these viruses [1, 2]. In addition, exotic viruses are also emerging in some parts of Europe, such as Dengue virus with local transmission in France in 2022 [3] and in Italy in 2023 [4].
One of the most significant endemic human pathogenic mosquito-borne viruses in Europe is the West Nile virus (WNV). WNV is a positive-sense RNA virus from the Flaviviridae family. The virus has a complex life cycle involving birds, mosquitoes, and humans. The virus is transmitted to humans through the bite of an infected mosquito. The mosquito gets the virus from biting an infected bird. The virus then replicates in the mosquito's body and is passed on to humans when the mosquito bites them. It can cause fever, headache, body aches, nausea, vomiting, and sometimes swollen lymph glands or a skin rash. In severe cases, West Nile virus can cause neurological illnesses such as encephalitis or meningitis [5,6,7].
It was first described in the continent in 1960; since then, the virus has appeared in many European countries such as France, Cyprus, Portugal, Hungary, etc. [8]. In these countries, genetic lineage 1 and lineage 2 of the virus have been responsible for human and animal infections so far [9].
In Spain, the most significant region of WNV circulation is the southern part of the country where multiple outbreaks were recorded based on the data from the last decade [2, 10,11,12]. The year 2020 was exceptional for WNV activity, when an outstanding WNV epidemic took place in southern Spain, affecting Andalusia, Seville, Catalonia, and Valencia, causing 77 human cases and 8 deaths [13,14,15].
It is most commonly found in Africa, the Middle East, and parts of Asia, but it has also been found in Europe, North America, and Australia.
West Nile virus has a complex life cycle involving birds, mosquitoes, and humans. The virus is transmitted to humans through the bite of an infected mosquito. The mosquito becomes infected when it feeds on an infected bird. The virus then replicates in the mosquito's body and is passed on to humans when the mosquito bites them.
Culex pipiens mosquito (Linnaeus, 1785) is considered as the primary vector of WNV but actual vector competence may vary between regions and other species can also contribute to its spread. Based on available literature data, some Aedes species, such as Aedes albopictus (Skuse, 1894), and members of the genus Anopheles (Meigen, 1818) can be considered competent vectors as well [16,17,18,19,20,21].
It is also a common attribute of Flaviviruses that multiple viruses are naturally co-circulating between avian and mosquito hosts in the same ecosystem. WNV often co-circulates with Bagaza or Usutu viruses [22]. Bagaza virus (BAGV) belongs to the Flaviviridae family, Ntaya serocomplex. It was first isolated from Culex mosquito species in the 1966 outbreak in Bagaza, Central Africa [23]. The virus is pathogenic in red-legged partridges and caused an outbreak in Cadiz, Spain, in 2019 [24].
There are multiple mosquito monitoring programmes in Europe, using different strategies, but most of the data still come from event-based (human cases) and indicator-based surveillance activities [25,26,27]. Whether it is mosquito surveillance or event-based data, the resulting data will not be immediately available; they will take time to be processed in the laboratory or made available in the ECDC (European Centre for Disease Prevention and Control) [25,26,27]. Thus, the necessary actions are not real time. In this study we demonstrate a WNV detection approach that uses on-site PCR technique to provide rapid surveillance results in 3–4 h, which is a rapid protocol that can be implemented in the field, followed immediately by the necessary targeted control after virus detection. We complemented the surveillance activity with additional complete genome sequencing in the laboratory and provided genome data of BAGV and WNV from southern Spain in 2021.
Methods
Trapping and virus detection
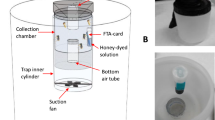
Adult mosquito collection was carried out in two Spanish regions: Valencia and Andalusia, during the WNV season, between 23 August and 8 September 2021 with overnight trapping. During overnight trapping (from 6 p.m. to 8 a.m.), trap nets were changed daily, except in a few cases because of the distance between sites and weather conditions. If a trap did not catch any or caught insufficient quantities on a given night, the net was left out for a longer period. Details of each sample location are summarized in Additional file 1: Table S1. Based on data from previous years, 25 sites were selected, mostly in suburban areas, with abundant local mosquito populations [28]. Trapping took place at 7 sites in the province of Valencia and 18 sites in Andalusia (Fig. 1). Various sampling methods were used mostly with BG-Sentinel traps (both with lure and CO2) and in some cases supplemented with CDC Light traps (with yeast as CO2 source) or BG-Mosquitaire (with lure). As the type of trap was not important in the testing of our protocol, any other type of adult mosquito trap can be used for the work. After collection from traps, a freezing box was used as an euthanization tool for the mosquitoes. The box operated with general ice accumulators, which were pre-frozen in −20 °C freezers. Following the freezing cycle, the mosquitoes underwent sex separation and morphological identification [29]. Sampling details are summarized in Table 1.

Mosquito sampling sites in the provinces of Valencia (7) and Andalusia (18), Spain
Female mosquitoes, due to their blood-feeding behaviour, play an active role in virus transmission. Analysing these mosquitoes for the presence of the virus provides direct evidence of virus circulation in the region. Therefore, we focused on processing female mosquitoes, systematically categorizing and grouping them by species, sampling site, and collection date. This approach was employed to facilitate targeted West Nile virus (WNV) testing in separate, well-defined pools. To align with our sampling numbers and ensure rapid and efficient sample processing, we decided to use 20 individuals per pool.
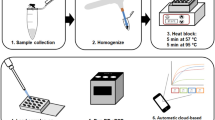
Our concept aimed to streamline the detection process using mobile devices and minimize the associated laboratory procedures, so we selected the following methods and types of equipment accordingly. Samples were homogenized manually (sterile quartz sand and 500 µl PBS buffer were added to each pool) using sterile single-use plastic sticks. Total RNA was extracted using the Beckman Coulter RNAdvance Viral XP 1.5-ml Tube Protocol (Beckman Coulter, Inc., CA, USA), following the manufacturer’s protocol. For the magnetic bead-based nucleic acid extraction we used the MagJET Separation Rack (Thermo Fisher Scientific™). Samples were tested for WNV RNA by qRT-PCR with previously published primers [30]. Primers WN10533-10552 (AAG TTG AGT AGA CGG TGC TG) and WN10625-10606 (AGA CGG TTC TGA GGG CTT AC) were used to amplify a conserved 92-bp region of the WNV 3—noncoding region. Besides probe WN10560-10579 (CTC AAC CCC AGG AGG ACT GG) was used for the qPCR. Briefly, the qRT-PCR was performed on MyGo® Mini S Real-Time PCR instrument (IT-IS Life Science Ltd.), a compact PCR machine that is suitable for use in the field, using the Brilliant III Ultra-Fast QPCR Master Mix (Agilent Technologies, CA, USA).
The total qRT-PCR master mix was 15 µl containing a total of 1 µl primers (50 µM), 0.25 µl probe (50 µM), 10 µl Mastermix, 0.2 µl dithiothreitol, 1 µl RT block enzyme, and 2.55 µl nuclease-free water. The PCR master mixes used in the field were prepared in advance and delivered frozen to the site; then, 5 µl extracted RNA of the samples (templates) was added to the master mix on site. The thermal cycling programme was set as follows: 10 min at 50 ℃ for reverse transcription, 3 min at 95 ℃ for denaturation, and 45 cycles of 10 s at 95 ℃ and 25 s at 60 ℃ for amplification [30]. All steps are field compatible, extraction does not require a homogenizer or centrifugal step because of the beads, and the PCR machine is compact and portable. Thanks to these features, the full protocol for in situ WNV detection from emptying the traps to PCR results takes about 3–4 h with the above-mentioned conditions, depending on the number of investigated mosquitoes.
To confirm the results of the above-mentioned in situ WNV detecting protocol, following the field-based surveillance activity, a previously published heminested RT-PCR was performed on the positive pools with 5 µl sample under laboratory conditions [31], and the amplified 250 bp of the NS5 gene was sent for Sanger sequencing (Eurofins Genomics Sequencing Laboratory, Germany). The results of the Sanger sequencing were inconclusive, showing mixed amplicons. Therefore, we decided to use Illumina sequencing on the Anopheles atroparvus pool to obtain the whole genomes and to distinguish possible mixed sequences.
Genome sequencing and analysis
Following the total nucleic acid isolation and reverse transcription, DNA was amplified by random PCR. RNA library was generated using the NEBNext Ultra II Directional RNA Library Prep for Illumina (NEB, Ipswitch, MA, USA). Briefly, 10 ng of total RNA was used as input for fragmentation step, and the cDNA generation was performed using random primers. Thereafter, the cDNA was end-prepped and adapter-ligated; then, the library was amplified according to the manufacturer’s instructions. The quality of the libraries was checked on Agilent 4200 TapeSation System using D1000 Screen Tape (Agilent Technologies, Palo Alto, CA, USA); the quantity was measured on Qubit 3.0 (Thermo Fisher Scientific, Waltham, MA, USA). Illumina sequencing was performed on the NovaSeq 6000 instrument (Illumina, San Diego, CA, USA) with 2 × 151 run configuration. Raw reads were quality checked with FastQC v0.12.1 and error corrected and quality trimmed with NanoFilt v2.8.0. Genomes and genome parts were de novo assembled with SPAdes v3.15.5 (raw reads as SPAdes has a built-in error correction and quality trimming function) and MEGAHIT v1.2.9 (corrected reads) and were mapped to the closest matches in Genbank in Geneious Prime v2023.1.1. Illumina reads were mapped to the consensus sequences from the former step and further corrected in Geneious Prime v2023.1.1. For multiple sequence alignments, sequence, and phylogenetic analyses, Geneious Prime 2023.1.1 and PhyML software version 3.0 were used. Phylogenetic analysis of WNV sequence was performed using PhyML software version 3.0. Model selection was accomplished using the model selection algorithm built into the software [32]. The model selection was run according to the Bayesian information criterion [33]. The best model was the GTR + G + I with 1000 bootstraps. For BAGV neighbour-joining phylogenetic tree was inferred with 1000 bootstrap replicates in Geneious Prime 2023.1.1.
Results and discussion
An increasing body of research is endorsing the transition from event-based surveillance to forecasting or early warning system approaches in surveillance practices [34, 35]. Most of the studies are based on virus surveillance by detecting WNV from human samples or by processing mosquitoes that have been collected over several years [36,37,38,39].
During the field sampling, 356 adult mosquitoes were trapped representing 7 species. Occasionally, where larvae were present on site, we collected them with larva dipping method, although only two pools were included in the study with this method (Additional file 1: Table S1). Among the 54 pools that were processed for WNV testing on site by qRT-PCR, two pools were positive for WNV RNA. The two positive pools contained two individuals of Culex pipiens (28 Ct) and three individuals of Anopheles atroparvus mosquitoes (17 Ct) from the village of Coria, Andalusia, respectively. This is in line with evidence showing that Andalusia became a WNV hotspot during the last few years [39]. During the identification process, we observed that the abdomens of the mosquitoes were empty. This observation led us to conclude that the positivity for the West Nile virus originated from the mosquitoes themselves rather than from a blood meal they may have ingested.
Only partial genomic data were recovered during the confirmatory PCR experiments from the pool containing Cx. pipiens, probably because of low virus titres. Therefore, this sample was not subjected to NGS sequencing. However, we were able to retrieve complete and nearly complete viral genomic sequences from the An. atroparvus sample. The mean coverage was 127.01X ± 594.42 and 111.49X ± 538.62 in case of WNV and Bagaza genomes, respectively. Illumina sequencing verified the mixed positive status of the sample containing West Nile and Bagaza viruses (Genbank accession numbers OR472391 and OR472392). Based on these results we documented the co-circulation of these viruses in the region within An. atroparvus species. Although members of the genus Anopheles are not considered as primary vectors for WNV, multiple literature data present positive specimens from Europe; therefore, their vector role cannot be ruled out [16, 18,19,20, 40]. In Portugal, the role of Anopheles mosquitoes in the spread of the virus was described in the 1970s and the ECDC also lists members of the genus as potential vectors of WNV [22, 41,42,43,44,45,46]. The fact that we detected the virus in this species does not prove beyond doubt that it is a vector of the virus, but it is important to draw attention to the possibility and the role of the species in the virus circulation.
Using the complete genome sequences of BAGV and WNV, we performed phylogenetic analyses (Figs. 2, 3).

PhyML tree of West Nile virus sequences with 1000 bootstrap replicates as the test of phylogeny. The novel sequence data of this study are presented in bold letters. Yellow and blue colours are used for visual clarity. They highlight the node tip labels on the phylogenetic tree that are closest to the sequences we have described

Neighbour-joining tree of Bagaza virus sequences. Neighbour-joining phylogenetic tree was inferred, using 1000 bootstrap replicates in Geneious Prime® 2023.1.1 Novel sequence data of this study are presented in bold letters. Yellow and blue colours are used for visual clarity. They highlight the node tip labels on the phylogenetic tree that are closest to the sequences we have described
Our results indicate that the WNV sequence belongs to Lineage 1 and clusters together with sequences from Culex perexiguus collected in 2020 and 2021 in Spain and human samples collected in 2020 in Spain. This novel sequence belongs to the same phylogenetic cluster, the WNV lineage 1, clade 1a, Mediterranean subtype. Among these, the WNV sequence of this study is more closely related to the Cx. perexiguus sequence from 2021 [39]. Similarly to our positive sample, sequences from both earlier Cx. perexiguus mosquitoes and the human samples originate from near Coria, one of the centres of the 2020 human outbreak. In addition, it is important to note that our sequences (both WNV and BAGV) were found in An. atroparvus, highlighting the potential of other mosquito species as vectors or players in sustaining the endemic transmission of the virus. Also, it highlights the importance of expanding the target species for future surveillance studies beyond Culex species.
The phylogenetic position of the newly described BAGV sequence aligns with that of a Portuguese red-legged partridge sequence from 2021, indicating its membership in the G4 group of the Ntaya serogroup, within the BAGV/ITV monophyletic cluster [2, 47, 48] (Fig. 3).
Co-circulation of different Flaviviruses in the same ecosystem was reported in multiple localities across Europe, usually involving Usutu, West Nile, and Bagaza viruses [11, 49, 50].
BAGV is a zoonosis which is increasingly gaining attention as a potential and more significant veterinary pathogen in Europe with increasing frequency of detections. The 2019 outbreak of BAGV in Spain occurred in the area where WNV, USUV, and BAGV were confirmed to co-circulate in 2011. Red-legged partridges found dead during the outbreak had enlarged livers and kidneys and other poor body condition. This is a cause for concern, among other things because of its role in the ecosystem. In addition to animal health relevance, during the Indian outbreak, anti-BAGV antibodies were detected in humans, raising awareness that humans may also be exposed to the virus at some level [11, 51].
Conclusions
In the present paper, we demonstrated the feasibility of our on-site surveillance line of action in mosquito-borne pathogen monitoring. It may be a valuable approach to aid rapid-response mosquito control actions and outbreak investigation activities or a good alternative for outbreak early warning systems, specifically in low-resource regions where mobile solutions can overcome logistic challenges. In addition to demonstrating this approach, we have released new genome sequences for both Bagaza and West Nile viruses from Europe. Moreover, we have identified another potential mosquito vector, An. atroparvus, in the region, thereby reinforcing the practicality and viability of these methods. To the best of our knowledge, this is the first in situ surveillance study for WNV.
Availability of data and materials
All data presented in this paper are available in the additional files. All of the DNA sequences are uploaded to the GenBank and accession numbers are publicly available (OR472391, OR472392).
References
Pierson TC, Diamond MS. The continued threat of emerging flaviviruses. Nat Microbiol. 2020;5:796–812.
Agüero M, Fernández-Pinero J, Buitrago D, Sánchez A, Elizalde M, San Miguel E, et al. Bagaza Virus in Partridges and Pheasants, Spain, 2010. Emerg Infect Dis. 2011;17(8):1498-1501.https://doi.org/10.3201/eid1708.110077
Cochet A, Calba C, Jourdain F, et al. Autochthonous dengue in mainland France, 2022: geographical extension and incidence increase. Euro Surveill. 2022;27(44):2200818. https://doi.org/10.2807/1560-7917.ES.2022.27.44.2200818.
Cassaniti I, Ferrari G, Senatore S, et al. Preliminary results on an autochthonous dengue outbreak in Lombardy Region, Italy, August 2023. Euro Surveill. 2023;28(37):2300471. https://doi.org/10.2807/1560-7917.ES.2023.28.37.2300471.
Campbell GL, Marfin AA, Lanciotti RS, Gubler DJ. West Nile virus. Lancet Infect Dis. 2002;2:519–29.
Nash D, Mostashari F, Fine A, Miller J, O’Leary D, Murray K, et al. The outbreak of West Nile virus infection in the New York city area in 1999. N Engl J Med. 2001;344:1807–14.
Sejvar JJ. West nile virus: an historical overview. Ochsner J. 2003;5:6–10.
Hubálek Z, Halouzka J. West Nile Fever–a Reemerging Mosquito-Borne Viral Disease in Europe. Emerg Infect Dis. 1999;5:643–50.
Kaaijk P, Luytjes W. Are we prepared for emerging flaviviruses in Europe? Challenges for vaccination. Hum Vaccin Immunother. 2018;14:337–44.
Cuervo PF, Artigas P, Mas-Coma S, Bargues MD. West Nile virus in Spain: forecasting the geographical distribution of risky areas with an ecological niche modelling approach. Transbound Emerg Dis. 2022;69:e1113–29.
Beck C, Jimenez-Clavero MA, Leblond A, Durand B, Nowotny N, Leparc-Goffart I, et al. Flaviviruses in Europe: complex circulation patterns and their consequences for the diagnosis and control of West Nile disease. Int J Environ Res Public Health. 2013;10:6049–83.
Vazquez A, Sanchez-Seco MP, Ruiz S, Molero F, Hernandez L, Moreno J, et al. Putative new lineage of West Nile virus, Spain. Emerg Infect Dis. 2010;16:549–52.
Figuerola J, Jiménez-Clavero MÁ, Ruíz-López MJ, Llorente F, Ruiz S, Hoefer A, et al. A one health view of the West Nile virus outbreak in Andalusia (Spain) in 2020. Emerg Microbes Infect. 2022;11:2570–8.
Aguilera-Sepúlveda P, Napp S, Llorente F, Solano-Manrique C, Molina-López R, Obón E, et al. West Nile Virus lineage 2 spreads westwards in Europe and overwinters in North-Eastern Spain (2017–2020). Viruses. 2022;14:569.
Macias A, Martín P, Pérez-Olmeda M, et al. West Nile virus emergence in humans in Extremadura, Spain 2020. Front Cell Infect Microbiol. 2023;13:1155867. https://doi.org/10.3389/fcimb.2023.1155867.
Kemenesi G, Krtinić B, Milankov V, Kutas A, Dallos B, Oldal M, et al. West Nile virus surveillance in mosquitoes, April to October 2013, Vojvodina province, Serbia: implications for the 2014 season. Euro Surveill. 2014;19:20779.
Fortuna C, Remoli ME, Severini F, di Luca M, Toma L, Fois F, et al. Evaluation of vector competence for West Nile virus in Italian Stegomyia albopicta (=Aedes albopictus) mosquitoes. Med Vet Entomol. 2015;29:430–3.
Mancini G, Montarsi F, Calzolari M, Capelli G, Dottori M, Ravagnan S, et al. Mosquito species involved in the circulation of West Nile and Usutu viruses in Italy. Vet Ital. 2017;53:97–110.
Maquart M, Boyer S, Rakotoharinome VM, Ravaomanana J, Tantely ML, Heraud JM, et al. High prevalence of West Nile Virus in domestic birds and detection in 2 new mosquito species in Madagascar. PLoS ONE. 2016;11:e0147589.
Nir Y, Goldwasser R, Lasowski Y, Margalit J. Isolation of West Nile virus strains from mosquitoes in Israel. Am J Epidemiol. 1968;87:496–501.
Shocket MS, Verwillow AB, Numazu MG, et al. Transmission of West Nile and five other temperate mosquito-borne viruses peaks at temperatures between 23°C and 26°C. Elife. 2020;9:e58511. https://doi.org/10.7554/eLife.58511.
Llorente F, García-Irazábal A, Pérez-Ramírez E, Cano-Gómez C, Sarasa M, Vázquez A, et al. Influence of flavivirus co-circulation in serological diagnostics and surveillance: a model of study using West Nile, Usutu and Bagaza viruses. Transbound Emerg Dis. 2019;66:2100–6.
Bondre VP, Sapkal GN, Yergolkar PN, Fulmali P, v, Sankararaman V, Ayachit VM, et al. Genetic characterization of Bagaza virus (BAGV) isolated in India and evidence of anti-BAGV antibodies in sera collected from encephalitis patients. J Gen Virol. 2009;90:2644–9.
Höfle U, Cardona Cabrera T, Sánchez-Cano A, et al. Bagaza virus and Plasmodium spp. coinfection in red-legged partridges (Alectoris rufa), in Southern Spain 2019. Transbound Emerg Dis. 2022;69(5):e3393-e3399. https://doi.org/10.1111/tbed.14658.
Bakonyi T, Haussig JM. West Nile virus keeps on moving up in Europe. Euro Surveill. 2020;25:2–5.
Young JJ, Haussig JM, Aberle SW, et al. Epidemiology of human West Nile virus infections in the European Union and European Union enlargement countries, 2010 to 2018 [published correction appears in Euro Surveill. 2021 May;26(20):]. Euro Surveill. 2021;26(19):2001095. https://doi.org/10.2807/1560-7917.ES.2021.26.19.2001095.
Gossner CM, Marrama L, Carson M, Allerberger F, Calistri P, Dilaveris D, et al. West Nile virus surveillance in Europe: moving towards an integrated animal-human-vector approach. Euro Surveill. 2017;22:10–9.
ECDC- European Centre for Disease Prevention and Control. https://www.ecdc.europa.eu/en. Accessed 11 Aug 2023.
Schaffner F, Angel G, Geoffroy B, Hervy JP, Rhaiem A, Brunhes J. The Mosquitoes of Europe. An identification and training programme. Montpellier: IRD Editions & EID Méditerranée; 2001.
Tang Y, Anne Hapip C, Liu B, Fang CT. Highly sensitive TaqMan RT-PCR assay for detection and quantification of both lineages of West Nile virus RNA. J Clin Virol. 2006;36:177–82.
Scaramozzino N, Crance JM, Jouan A, DeBriel DA, Stoll F, Garin D. Comparison of flavivirus universal primer pairs and development of a rapid, highly sensitive heminested reverse transcription-PCR assay for detection of flaviviruses targeted to a conserved region of the NS5 gene sequences. J Clin Microbiol. 2001;39:1922–7.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Lefort V, Longueville JE, Gascuel O. SMS: Smart Model Selection in PhyML. Mol Biol Evol. 2017;34:2422–4.
World Health Organization. Early Warning, Alert and Response System (EWARS). Accessed 10 Oct 2023.
Ricks PM, Njie GJ, Dawood FS, Blain AE, Winstead A, Popoola A, et al. Lessons learned from CDC’s Global COVID-19 early warning and response surveillance system. Emerg Infect Dis. 2022;28:8–16.
Szentpáli-Gavallér K, Antal L, Tóth M, Kemenesi G, Soltész Z, Dán A, et al. Monitoring of West Nile virus in mosquitoes between 2011–2012 in Hungary. Vector Borne Zoonotic Dis. 2014;14:648–55.
Lanciotti RS, Kerst AJ, Nasci RS, Godsey MS, Mitchell CJ, Savage HM, et al. Rapid detection of west nile virus from human clinical specimens, field-collected mosquitoes, and avian samples by a TaqMan reverse transcriptase-PCR assay. J Clin Microbiol. 2000;38:4066–71.
González-Reiche AS, de Monzón-Pineda ML, Johnson BW, Morales-Betoulle ME. Detection of West Nile viral RNA from field-collected mosquitoes in tropical regions by conventional and real-time RT-PCR. Methods Mol Biol. 2010;630:109–24.
Ruiz-López MJ, Muñoz-Chimeno M, Figuerola J, Gavilán AM, Varona S, Cuesta I, et al. Genomic analysis of West Nile Virus lineage 1 detected in mosquitoes during the 2020–2021 outbreaks in Andalusia, Spain. Viruses. 2023;15:266.
Zana B, Erdélyi K, Nagy A, Mezei E, Nagy O, Takács M, et al. Multi-approach investigation regarding the West Nile virus situation in Hungary, 2018. Viruses [Internet]. 2020;12(1):123. https://doi.org/10.3390/v12010123.
Magallanes S, Llorente F, Ruiz-López MJ, Martínez-de la Puente J, Soriguer R, Calderon J, et al. Long-term serological surveillance for West Nile and Usutu virus in horses in south-West Spain. One Health. 2023;17:100578.
Beck C, Desprès P, Paulous S, Vanhomwegen J, Lowenski S, Nowotny N, et al. A high-performance multiplex immunoassay for serodiagnosis of flavivirus-associated neurological diseases in horses. Biomed Res Int. 2015;2015:678084.
Llorente F, Pérez-Ramírez E, Fernández-Pinero J, Soriguer R, Figuerola J, Jiménez-Clavero MÁ. Flaviviruses in game birds, Southern Spain, 2011–2012. Emerg Infect Dis. 2013;19:1023–5.
Khan SA, Chowdhury P, Choudhury P, Dutta P. Detection of West Nile virus in six mosquito species in synchrony with seroconversion among sentinel chickens in India. Parasit Vectors. 2017;10:13.
Engler O, Savini G, Papa A, Figuerola J, Groschup M, Kampen H, et al. European surveillance for West Nile virus in mosquito populations. Int J Environ Res Public Health. 2013;10:4869–95.
ECDC-European Centre for Disease Prevention and Control-Anopheles atroparvus - Factsheet for experts. https://www.ecdc.europa.eu/en/disease-vectors/facts/mosquito-factsheets/anopheles-atroparvus. Accessed 12 Sept 2023.
NCBI. National Center for Biotechnology Information (NCBI). Bethesda (MD): National Library of Medicine (US). 1988. https://www.ncbi.nlm.nih.gov. Accessed 06 Apr 2023.
Falcão M, Barros M, Duarte MD, dos Santos FA, Fagulha T, Henriques M, et al. Genome characterization and spaciotemporal dispersal analysis of Bagaza virus detected in Portugal, 2021. Pathogens. 2023;12:150.
Guerrero-Carvajal F, Bravo-Barriga D, Martín-Cuervo M, Aguilera-Sepúlveda P, Ferraguti M, Jiménez-Clavero MÁ, et al. Serological evidence of co-circulation of West Nile and Usutu viruses in equids from western Spain. Transbound Emerg Dis. 2021;68:1432–44.
Scaramozzino P, Carvelli A, Bruni G, Cappiello G, Censi F, Magliano A, et al. West Nile and Usutu viruses co-circulation in central Italy: outcomes of the 2018 integrated surveillance. Parasit Vectors. 2021;14:243.
Bondre VP, Sapkal GN, Yergolkar PN, Fulmali PV, Sankararaman V, Ayachit VM, et al. Genetic characterization of Bagaza virus (BAGV) isolated in India and evidence of anti-BAGV antibodies in sera collected from encephalitis patients. J Gen Virol. 2009;90:2644–9.
Acknowledgements
We express our gratitude to the mosquito control experts who provided invaluable assistance during our fieldwork and to Juan Pablo Serna, field mosquito control technician in Lokímica, for preparing the maps of the trapping sites. The research was performed in collaboration with the Hungarian Centre for Genomics and Bioinformatics at the Szentágothai Research Centre of the University of Pécs. Zsaklin Varga was supported by the Biological and Sportbiological Doctoral School of the University of Pécs, Hungary. The field part of the study, including trapping and displacements, was financed by Lokímica in the framework of the mosquito surveillance programmes conducted in the investigated areas. Zsaklin Varga was supported by the ÚNKP-23-3-II-PTE-1758 New National Excellence Program of the Ministry for Culture and Innovation from the source of the National Research, Development and Innovation Fund.
Funding
Open access funding provided by University of Pécs. The work was supported by the National Research, Development and Innovation Office, grant number: FK-138563 and by RRF-2.3.1-21-2022-00010 “National Laboratory of Virology”.
Author information
Authors and Affiliations
Contributions
ZSV: investigation, field testing of the protocol, laboratory analyses, writing the original draft, visualization. RBM: field resources, supervision of fieldwork. JRI: fieldwork, mosquito collection and identification. KK: supervision, funding acquisition, review and editing the manuscript. GET: sequencing and bioinformatic analysis. BZ: phylogenetic analysis, laboratory work. SZ: phylogenetic analysis. TG: bioinformatic analysis. FJ: funding acquisition. GK: conceptualization, supervision, writing and editing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors consent the publication of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Mosquito trapping data. Table S2. NGS sequencing genome coverage plot and raw data for West Nile virus. Table S3. NGS sequencing genome coverage plot and raw data for Bagaza virus.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Varga, Z., Bueno-Marí, R., Risueño Iranzo, J. et al. Accelerating targeted mosquito control efforts through mobile West Nile virus detection. Parasites Vectors 17, 140 (2024). https://doi.org/10.1186/s13071-024-06231-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-024-06231-7