
Abstract
Background
The study of parasites provides insight into intricate ecological relationships in ecosystem dynamics, food web structures, and evolution on multiple scales. Hepatozoon (Eucoccidiorida: Hepatozoidae) is a genus of protozoan hemoparasites with heteroxenous life cycles that switch infections between vertebrates and blood-feeding invertebrates. The most comprehensive review of the genus was published 26 years ago, and currently there are no harmonized data on the epizootiology, diagnostics, genotyping methods, evolutionary relationships, and genetic diversity of Hepatozoon in the Americas.
Methods
Here, we provide a comprehensive review based on the PRISMA method regarding Hepatozoon in wild mammals within the American continent, in order to generate a framework for future research.
Results
11 out of the 35 countries of the Americas (31.4%) had data on Hepatozoon, with Carnivora and Rodentia orders having the most characterizations. Bats, ungulates, and shrews were the least affected groups. While Hepatozoon americanum, H. americanum-like, H. canis, H. didelphydis, H. felis, H. milleri, H. griseisciuri, and H. procyonis correspond to the identified species, a plethora of genospecies is pending for a formal description combining morphology and genetics. Most of the vectors of Hepatozoon in the Americas are unknown, but some flea, mite, and tick species have been confirmed. The detection of Hepatozoon has relied mostly on conventional polymerase chain reaction (PCR), and the implementation of specific real time PCR for the genus needs to be employed to improve its diagnosis in wild animals in the future. From a genetic perspective, the V4 region of the 18S rRNA gene has been widely sequenced for the identification of Hepatozoon in wild animals. However, mitochondrial and apicoplast markers should also be targeted to truly determine different species in the genus. A phylogenetic analysis of herein retrieved 18S ribosomal DNA (rDNA) sequences showed two main clades of Hepatozoon: Clade I associated with small mammals, birds, and herpetozoa, and Clade II associated with Carnivora. The topology of the tree is also reflected in the haplotype network.
Conclusions
Finally, our review emphasizes Hepatozoon as a potential disease agent in threatened wild mammals and the role of wild canids as spreaders of Hepatozoon infections in the Americas.
Graphical Abstract

Similar content being viewed by others


Background
The ongoing sixth mass extinction has driven efforts in documenting and understanding animal biodiversity for conservation and management [1]. Wild vertebrates and their parasites maintain infectious agents such as viruses, bacteria, and protozoa that may eventually spill over to humans or domestic animals, leading to outbreaks or epidemics [2,3,4]. As most species on earth are parasites, their role as indicators of the ecosystem’s health has gained attention in recent years [5, 6]. To complete biological cycles, parasites rely on the trophic interactions of their hosts [7]. Indeed, while some parasites co-evolve with their hosts, causing minimal damage, others are etiological agents [8,9,10]. This duality in the biology of parasites is important to understanding intricate ecological relationships, ecosystem dynamics, food web structure, and evolution across multiple scales [9, 10]. Additionally, infectious diseases caused by parasites pose significant conservation challenges due to their impact on wild animals [11, 12]. Therefore, identifying the parasites that act as etiological agents is crucial in defining the threats to wildlife conservation [13, 14].
Hepatozoon (Eucoccidiorida: Hepatozoidae) is a genus of protozoa that invades red and white blood cells of vertebrates [15, 16]. Hepatozoon spp. are commonly found in wildlife perpetuating in enzootic cycles [15, 17]. These hemoparasites have heteroxenous life cycles involving a vast array of intermediate hosts (vertebrates) and definitive hosts (blood-feeding invertebrates) [15, 16], a fact that has shaped the remarkable biological diversity of the genus.
After its original description by Miller in 1908, the most comprehensive review on the genus Hepatozoon was published 90 years later [15]. Further reviews added data mainly on canine hepatozoonosis [16, 18, 19], and there are currently no harmonized data on the epizootiology of the genus Hepatozoon in the Americas. Here we provide a comprehensive review, based on the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) method [20], of Hepatozoon in wild mammals of the American continent, with the following objectives: (i) to catalogue the publications since the original description of the taxon, (ii) to compare the publications by country, (iii) to compile the nature of biological samples and the techniques employed to detect and genetically characterize the parasite, (iv) to assess the conservation status of wild mammals with confirmed infection, and (v) to analyze available genetic data. Collectively, the objectives of this review were to generate a framework of reference for future investigation on Hepatozoon in the Americas.
Methods
Literature search and database
We recovered literature on Hepatozoon published between 1916 and December 31, 2022, through an exhaustive review using reputable sources including PubMed (https://pubmed.ncbi.nlm.nih.gov/), SciELO [Scientific Electronic Library Online] (https://scielo.org/es/), Scopus (https://www.scopus.com/home.uri), and Web of Science (https://webofknowledge.com/UA). Only peer-reviewed publications were included. Data from books, gray literature such as scientific conferences, and theses were excluded. Data on Hepatozoon retrieved from domestic animals or invasive rodents were not quantified, but molecular sequences were included in genetic analyses.
To mitigate bias and maximize the number of available studies, we used targeted search strings and title-abstract-keywords unit searches, incorporating the terms “Hepatozoon,” “hepatozoonosis,” “wild mammals,” “wildlife,” “wild animals,” and “mammals,” and names of the countries in the American continent, along with names of orders and groups of mammals native to the Americas, as detailed in the Handbook of the Mammals of the World [21]. To hone our search, we applied Boolean operators “AND” and “OR,” and followed the advanced search protocols suggested in the selected databases.
Compiled papers were subjected to the PRISMA protocol [20], with the following inclusion criteria: (i) reports of Hepatozoon in mammals of the Americas and (ii) studies on ectoparasites extracted from mammals of the Americas that assessed the presence of Hepatozoon. The documents lacking these criteria were excluded. The metadata matrix included the publication year, country, vertebrate host order, family, species, type of sample, diagnostic technique, conservation and population statuses of the hosts according to the International Union for Conservation of Nature (IUCN) Red List (https://www.iucnredlist.org/), primers used in molecular diagnostics, and study references. Scientific names and taxonomy of the hosts species referenced in this review followed the Integrative Taxonomic Information System (ITIS) (https://www.itis.gov/).
Analysis of sequences
Sequence selection and alignment
To define the final dataset included in this study, sequences of Hepatozoon and associated metadata were sourced from the GenBank database (https://ncbi.nlm.nih.gov/nucleotide) up to December 31, 2022, using “Hepatozoon” as the search criterion. Subsequently, the sequence filtering was carried out in four steps. First, only 18S ribosomal DNA (rDNA) sequences were included. Second, sequences annotated with a nomenclature other than Hepatozoon were excluded. Third, each sequence was individually aligned against an annotated 18S rDNA reference sequence of Hepatozoon (MH615006) to identify those sequences that showed more than 50% coverage for a given region of the 18S rRNA gene. Fourth, each sequence that satisfied the aforementioned criteria was recorded in a table, specifying the GenBank accession number, submission date, sequence length, and the flanked region of the 18S rRNA gene. Finally, all sequence datasets were aligned with MAFFT using the G-INS-i algorithm [22].
Phylogenetic analyses
Phylogeny was inferred using maximum likelihood (ML [23]) and Bayesian inference (BI [24, 25]) methods in IQ-TREE version (v.) 1.6.12 and [26] MrBayes v. 3.2.6 [27], respectively. The best ML and BI evolutionary models and phylogenetic reconstructions were calculated using the “-m TESTNEWONLY -mrate G” and “lset nst=mixed rates=gamma” commands, implemented in IQ-TREE [28] and MrBayes [27, 29], respectively. All best-fit models were selected under the Bayesian information criterion (BIC) [30].
Rapid hill-climbing and stochastic disturbance methods were employed for ML phylogenies, with 1,000 ultra-fast bootstrapping pseudo-replicates to evaluate the inferred tree robustness. Ultra-fast bootstrap values < 70%, 70–94%, and ≥ 95% were considered non-significant, moderate, and high statistical support, respectively [31]. Regarding the BI phylogenies, two independent tests of 20 × 106 generations and four Markov chain Monte Carlo (MCMC) chains were implemented, sampling trees every 1,000 generations and removing the first 25% as burn-in. Tracer v. 1.7.1 [32] was employed to confirm the correlation and effective sample size (ESS) of the MCMCs. Bayesian posterior probabilities (BPP) > 0.70 at nodes were considered high statistical support [33]. Trees were visualized and edited with FigTree v. 1.4.1 (http://tree.bio.ed.ac.uk/software/figtree/) and Inkscape v 1.1 (https://inkscape.org/es/). A consensus phylogram for both ML and BI was generated following the approach outlined by Santodomingo et al. [34].
Haplotype analyses
A haplotype analysis of Hepatozoon genotypes was intended to evaluate genetic variants circulating among wild mammals in the Americas. According to the National Human Genome Research Institute (https://www.genome.gov/), a genotype is defined as a DNA sequence at a specific locus, whereas a haplotype is a set of genetic variants, or polymorphisms, that tend to be inherited together. To calculate the number of polymorphic sites (S), haplotypes (H), haplotype diversity (Hd), nucleotide diversity (π), and haplotype frequencies, the gametic phase was inferred using the PHASE module in DNAsp v. 6.12.03 software [35]. Nucleotide polymorphism analyses excluded gaps and considered invariable sites. A median-joining (MJ) haplotype network [36] was constructed using PopART v. 1.7 (http://popart.otago.ac.nz) to display haplotype frequencies.
General findings
A total of 1,406 studies were identified as potentially useful for the review. After thoroughly reviewing the papers meeting the selection criteria, 84 studies (5.97%) were selected, as they contained information on Hepatozoon in wild mammals in the Americas and their ectoparasites (Fig. 1). Eleven out of the 35 countries of the Americas (31.4%), including Argentina, Brazil, Canada, Chile, Colombia, Costa Rica, French Guiana, Panama, Uruguay, the USA, and Venezuela, had data on Hepatozoon (Additional file 1: Table S1). No data were found for the other 24 American countries.

Flowchart of the current systematic review on Hepatozoon infections in wild mammals and their ectoparasites across the Americas (constructed with draw.io v. 22.0.3; https://www.drawio.com/)
The majority of studies on Hepatozoon in wild mammals were concentrated in Brazil and the USA, with 36 (42.9%) and 30 (35.7%) papers, respectively (Additional file 1: Table S1). In contrast, other countries in the Americas yielded less than five studies (~ 6%), underscoring a significant gap in the literature. In fact, research in the Americas has largely focused on domestic animals [16, 19, 37, 38]. This disparity not only underscores the need for a broader geographical scope in future studies, but also calls for assessing the occurrence of Hepatozoon in wild mammals in regions that currently lack data. In this regard, Chile displays a recent uptick in research on Hepatozoon infections in wildlife (Additional file 1: Table S1).
Regarding the number of studies on Hepatozoon among mammalian orders, Carnivora and Rodentia had the most, with 43 (51.2%) and 35 (41.7%) reports, respectively. Didelphimorphia followed, with 10 studies (11.9%), Chiroptera with two studies (2.4%), and Artiodactyla with two studies (2.4%). Lagomorpha, Microbiotheria, Perissodactyla, and Soricomorpha each had one study (1.2%) (Fig. 2). The disproportional distribution of studies by country—predominantly in Brazil and the USA—indicates a geographical bias. This fact not only skews the distribution of Hepatozoon spp., but also highlights the lack of a comprehensive understanding of infection by this protozoan in other mammalian orders. Furthermore, it denotes the limited research efforts on these hemoparasites in other American countries.

Temporal patterns of published articles regarding Hepatozoon organized by mammal orders in the American Continent
Overall, the number of studies on Hepatozoon in mammals of the Americas has displayed a notable upswing in recent decades (Fig. 2). Undoubtedly, the availability of molecular diagnostic tools contributed to this increase [39]. Indeed, molecular tools enhance sensitivity in detecting Hepatozoon from different types of biological samples, thus facilitating the identification of new species and previously unrecognized infections in wild mammals. The potential impact of these infections on wildlife health, coupled with the growing interest in arthropod-borne diseases, particularly those transmitted by ticks, has also contributed to the increase in research regarding Hepatozoon in wild mammals.
Epizootiology of Hepatozoon in mammals of the Americas
Hepatozoon spp. thrive in enzootic cycles [15, 17], which are ecological systems involving vertebrate hosts, vectors, environmental components, and the critical community size of hosts required to maintain the infectious agent indefinitely [2, 40]. Hepatozoon spp. are sustained in both vertebrate and invertebrate hosts [15], with transmission pathways that shift depending on the specific parasite-host interactions.
In vertebrates, the transmission routes of Hepatozoon primarily include the ingestion of infected ectoparasites containing mature oocysts, often during grooming [15, 16, 41, 42], and, to a lesser extent, the consumption of infected prey tissue carrying meronts or cystozoites (predation) or ectoparasites attached to their prey [43,44,45,46]. In mammals, transmission of macromeronts also occurs through the placenta (transplacental) [47,48,49]. However, this route was only confirmed in Hepatozoon canis, but it probably also occurs in Hepatozoon americanum [50]. Conversely, in ectoparasites, transstadial perpetuation has been documented only for H. canis [51] and H. americanum in ticks [52]. Although transovarial transmission has not been demonstrated for the Hepatozoon genus [51, 53, 54], the documented transmission strategies highlight the evolutionary success of Hepatozoon to complete life cycles.
Hepatozoon spp. infect a wide range of vertebrate hosts, including herpetozoa [15, 17, 55], birds [56, 57], and mammals [58, 59]. Additionally, blood-sucking arthropods such as flies, triatomines [15, 17, 60], mosquitoes [55, 61], ticks [52, 62, 63], fleas, lice, and mites [15, 64, 65] serve as vectors [15]. A total of 6,631 mammals have been analyzed in 81 studies across the American continent, with 1,789 animals testing positive for Hepatozoon, yielding a cumulative infection frequency (IF) of 26.93%. Notably, Hepatozoon IFs varied across mammalian orders (see Additional file 1: Table S1). Microbiotheria and Carnivora exhibited the highest IFs, with 86.67% (65/75) and 42.12% (730/1,733), respectively. Didelphimorphia had an IF of 25.95% (144/555), while Rodentia had an IF of 22.87% (798/3,489). Chiroptera displayed an IF of 11.21% (12/107), whereas Artiodactyla had an IF of 5.42% (33/609). In contrast, Soricomorpha had a remarkably low IF of 1.69% (1/59). Unfortunately, the sample sizes for the orders Lagomorpha and Perissodactyla were too small to provide a significant IF (Additional file 1: Table S1).
To date, 107 species of mammals from nine orders have been screened for Hepatozoon. Of these, two species of carnivores were found infected with H. americanum (1.89%) and five species with H. americanum-like (4.72%) (Additional file 1: Table S1). The former species is known for its virulence in canids [66]. The latter was considered an emerging South American variant of H. americanum [67]; however, H. americanum-like exploits a niche with different vectors and hosts, so it should be considered a different species. Moreover, pathogenic effects of H. americanum-like still need to be assessed in detail. In contrast, H. canis, which appears well adapted to its canine hosts [66, 67], has also been found infecting nine out of ten canid species in the continent (80%). Notably, some species of Artiodactyla, Chiroptera, Didelphimorphia, and Rodentia orders may also be susceptible to H. canis (Additional file 1: Table S1), a fact that underlines the generalist and opportunistic nature of this species.
Hepatozoon felis, the causative agent of feline hepatozoonosis [68], has been identified in six felids (5.66%), four canids (3.77%), two procyonids (1.96%), and one mustelid (0.94%) (Additional file 1: Table S1). Hepatozoon felis is the predominant species of Hepatozoon infecting wild felids worldwide [68]. Therefore, it is not unexpected to observe a higher IF in felids (49/158; 31.01%) compared to other animals (83/317; 26.18%) (Additional file 1: Table S1). However, the IF of H. felis in non-felid species may suggest that the hemoparasite could vary in host specificity, possibly due to transmission by ubiquitous vectors (such as flea, mite, or tick) or carnivorism among mammal groups [68]. These findings also raise questions about the potential shift that transmission dynamics of feline hepatozoonosis could undergo given the current population decline of South American felids (see Additional file 1: Table S1) [69].
A total of 60 mammal species (56.60%) were found to be infected with Hepatozoon spp. such as Hepatozoon didelphydis, H. griseisciuri, H. milleri, and H. procyonis, each one exhibiting a different IF and infecting specific mammal groups (as shown in Table S1). Furthermore, lineages of Hepatozoon have been documented, covering species associated with carnivores, herpetozoa and small mammals, opossums, reptiles, and rodents (Additional file 1: Table S1). According to Dupré [70], a “lineage” is defined as an independent evolutionary line that extends back in time from a current species to its ancestors. In this sense, Hepatozoon lineages reflect unique evolutionary histories and specific adaptations of Hepatozoon spp. to different hosts, suggesting putatively novel species associated with a diverse array of wild mammals in the Americas.
According to the data gathered (Additional file 1: Table S1), the IF of Hepatozoon spp. varies among mammals. Mustelids (Carnivora: Mustelidae) had the highest IF of 57.9% (11/19), followed by raccoons with 46.48% (317/682) and canids with 41.54% (339/816). Opossums showed an IF of 33.17% (209/630), while felids had lower IF of 29.17% (63/216). Regarding rodents, Sciuromorpha (Sciuridae) had the highest IF of 50.10% (247/493), while Hystricomorpha (Caviidae, Cuniculidae, and Echimyidae) and Myomorpha (Cricetidae) had IF of 31% (137/442) and 16.14% (412/2,552), respectively. Bats, ungulates, and shrews were the least affected groups, with IFs of 11.21% (12/107), 5.56% (34/611), and 1.7% (1/59), respectively.
Regarding specific Hepatozoon spp., H. milleri had the highest IF of 90.9% (10/11), followed by H. griseisciuri with 49.80% (244/490), H. americanum with 44.1% (41/93), and H. procyonis with 47.97% (296/617). In contrast, H. didelphydis, H. canis, and H. americanum-like had low IFs of 24.7% (23/93), 20.24% (133/657), and 11.43% (44/385), respectively. It is important to note that the small sample size may have biased the IF in H. milleri, and a similar reason could explain the high IF of Hepatozoon in mustelids.
Blood-sucking arthropods associated with the epizootiology of Hepatozoon
Ticks, fleas, lice, and mites have been suggested as potential vectors of Hepatozoon spp. in mammals [15]. For instance, Hepatozoon DNA has typically been detected in mammal-associated ticks of the genera Amblyomma [71, 72], Dermacentor [73, 74], Haemaphysalis [62, 72, 73], Ixodes [75,76,77,78,79], and Rhipicephalus [42, 80]. Although the detection of Hepatozoon DNA in blood-sucking arthropods does not definitively prove any role in transmission [54], ticks harboring Hepatozoon DNA should not be ruled out as potential vectors [79].
Currently, several tick species, including Amblyomma ovale [81, 82], Haemaphysalis longicornis, Haemaphysalis flava [62], Ixodes ricinus [54], Rhipicephalus microplus [80], and Rhipicephalus sanguineus group [42, 53, 83], have been identified as vectors for H. canis. Additionally, Amblyomma maculatum is a recognized vector for H. americanum [52, 63, 84,85,86]. These ticks are frequently found on carnivores and ruminants, and their role in the spread of Hepatozoon spp. towards other mammal groups remains unclear.
Concerning mammal species in the Americas, blood-sucking ectoparasites that could be related to the epizootiology of the Hepatozoon species include ticks such as Amblyomma dubitatum, A. maculatum, A. tigrinum, and A. sculptum in carnivores [13, 71, 87,88,89]; Ixodes neuquenensis in Microbiotheria [75]; and Amblyomma fuscum and species of the Ixodes sigelos group in rodents [79, 90]. To better understand the implications of these ticks in the Hepatozoon epizootiology among the Americas’ ecosystems, experimental transmission studies are necessary to clarify these associations. Transstadial detection of oocysts in tick hemolymph would also contribute to elucidate vector roles.
While fleas, lice, and mites are also considered vectors or definitive hosts of Hepatozoon spp. associated with rodents [64, 65], only one flea species (Megabothris abantis) and two mite species (Euhaemogamasus ambulans and Echinolaelaps echidninus) have been reported as vectors for Hepatozoon spp. in rodents of the Americas [64]. Moreover, the fleas Amalaraeus dissimilis and Peromyscopsylla ostsibirica are possible vectors of Hepatozoon in rodents of the genus Microtus in Alaska [91]. The limited knowledge on Hepatozoon vectors stems from the challenges that the detection of Hepatozoon oocysts in blood-sucking arthropods pose [64, 92]. These may include finding the ectoparasites on their hosts, maintaining the vectors alive in laboratory conditions, and submitting their hemolymph to microscopical analyses for the detection of oocysts and sporocysts, and eventually genetic identification.
Hepatozoon and concurrent infections
Hepatozoon infections can occur simultaneously with other infectious diseases and are common in regions where vector-borne diseases prevail. Blood-sucking arthropods are known to transmit a plethora of pathogenic bacteria and protozoa [93]. In the Americas, Hepatozoon infections in wild mammals have been observed alongside other infectious agents, including some with zoonotic potential. For example, concurrent infections of Hepatozoon with Piroplasmida spp. [94], Rangelia vitalii, Leishmania sp., [95, 96], Anaplasma sp., Babesia sp., and Ehrlichia spp. [97, 98] have been found in canids. Rodents have shown co-infections with Babesia spp., Trypanosoma sp. [99,100,101,102], Anaplasma sp., Bartonella spp., Ehrlichia spp., and Theileria sp. [98], while detections in felids point to co-infections with Cytauxzoon felis, Cytauxzoon sp., Piroplasmida sp., and Theileria sp. [94, 98, 100].
Additionally, procyonids have shown co-infections with Babesia microti [103], Trypanosoma cruzi [104,105,106], Anaplasma sp., Babesia sp., Ehrlichia spp., and Theileria sp. [98], while opossums have concurrent infections with Babesia spp., Ehrlichia spp., Piroplasmida sp., and Theileria sp. [94, 98, 107, 108]. On the other hand, tapirids have been reportedly co-infected with Theileria sp. exclusively [94]. Furthermore, when Hepatozoon infections co-occur with other infections, they may lead to exacerbation of pre-existing conditions, resulting in severe morbidity, prolonged duration of clinical manifestations, and interference between diagnosis and treatment [92, 94, 108, 109]. Overall, the IF of Hepatozoon in mammals in the Americas varies between studies, as shown in Additional file 1: Table S1. The IFs of Hepatozoon in each mammalian order (Fig. 3A) and the IF of Hepatozoon spp. are illustrated by country (Fig. 3B).

Map of the American Continent showing the infection rates (IRs) of Hepatozoon by mammalian order (A), and occurrence of Hepatozoon spp. in American country (B). Maps were constructed with Quantum Geographic Information System (QGIS) v. 3.18.1- Zürich (https://www.gnu.org/licenses)
Detection and characterization of Hepatozoon in mammals of the Americas
Biological samples used to detect Hepatozoon
Blood was the main sample employed to detect and characterize Hepatozoon, with 56 studies (66.7%). The liver was the second most frequently used sample, accounting for 13 studies (15.5%), followed by the spleen with 12 studies (14.3%). Other samples included lung (10 studies; 11.9%), heart (eight studies; 9.5%), and muscle (seven studies; 8.3%). Less commonly employed samples were bone marrow, tail, skeletal muscle, kidney, and synovial fluid (ranging from 1.2% to 3.6%) (Additional file 1: Table S1). Moreover, as ticks feed on vertebrate blood, they were used as sentinels to assess the presence of Hepatozoon in wild mammals in three studies (3.6%) [78, 79, 90]. Therefore, the variety of tissues that Hepatozoon spp. may attain in the vertebrate host, including their ectoparasites, maximizes the chance of detection.
Detecting gamonts in blood is frequently used as a quick diagnosis of Hepatozoon infection in vertebrates [39]. However, the spleen is more sensitive, because it harbors meronts (groups of meront cells in multiple division) and subsequently higher loads of parasites; therefore, it is considered the best target for detection [42, 109]. Meronts have also been observed in bone marrow of red foxes (Vulpes vulpes) [48, 110]. Depending on the Hepatozoon species, alternative samples for detection and histopathology include biopsies of bone marrow or skeletal muscle for H. canis [66, 83, 111], skeletal muscle for H. americanum and H. felis [66, 68], and cardiac muscle for H. felis [68].
Diagnostic techniques to detect Hepatozoon
The most frequently used methods were conventional polymerase chain reaction (cPCR), employed in 47 studies (56%), blood smear in 28 studies (33.3%), and histology in 26 studies (30.9%). In contrast, nested PCR (nPCR) was less frequently employed, accounting for 4.8% (four studies), while bone marrow smear and leukocyte-platelet layer smear were used in two studies (2.4%) and one study (1.2%), respectively (Additional file 1: Table S1). The effectiveness of each technique varies depending on the intensity of infection. For instance, while observing blood smears is sensitive in animals with high parasitemia, histology is valuable in detecting subclinical infections, in which Hepatozoon encysts in different organs and fewer gamonts circulate in blood [39]. This is particularly relevant in H. americanum and H. americanum-like infection, two species that produce very low parasitemia during the clinical disease [16, 39, 84]. PCR stands out as the gold standard for diagnosing and characterizing Hepatozoon spp., particularly in cases of subclinical infection with low parasitemia [39, 112, 113], where nPCR can significantly improve the specificity [39, 89]. Although real-time PCR is highly sensitive, none of the reviewed studies implemented this technique. Applying real-time PCR to surveil Hepatozoon infections in wildlife could significantly expand diagnostic capabilities. This approach not only will enable the rapid processing of large sample volumes, reducing the time and costs of diagnosis, but also will improve the sensitivity of cPCR in detecting Hepatozoon infections [114, 115].
Hepatozoon genotyping
Molecular analyses of the phylum Apicomplexa have provided valuable insight into the genomic composition and genetic structure of the group, revealing the existence of three kinds of genomes: nuclear, mitochondrial, and the apicoplast [116,117,118]. The first genetic characterizations of the genus Hepatozoon relied on partial sequences of the apicoplast-encoded 16S rRNA gene [119], the nuclear ribosomal DNA internal transcribed spacer 1 (ITS-1) [120, 121], and the 18S rRNA gene [58, 59, 122, 123]. Since then, 18S rDNA sequences have been widely used for genotyping and molecular systematics of the genus [124,125,126].
Extrachromosomal genomes are often detected in significant quantities, with two or even 15 copies per cell [127, 128]. For the genus Hepatozoon, the first assembled mitochondrial genome (mitogenome) belonged to Hepatozoon catesbianae, a species that infects the frog Lithobates catesbeianus [129]. Moreover, the apicoplast genome of H. canis [128] and the mitogenome of Hepatozoon spp. associated with rodents were recently sequenced [125, 126]. It was not unexpected that the mitochondrial cytochrome c oxidase I (cox1) locus had a higher nucleotide divergence (≥ 0.08) if compared with the 18S rRNA gene (≥ 0.012), becoming the marker of choice to distinguish Hepatozoon species [125, 126, 128]. Nevertheless, the paucity of mitochondrial and apicoplast sequences in online databases represents a significant gap that needs to be addressed in order to unravel the intricate diversity and systematics of the genus. In this sense, the works of Léveillé et al. [125, 128, 129] and Hrazdilová et al. [126] are groundbreaking on a global scale.
Only one out of 84 reviewed articles used primers targeting the mitogenome of Hepatozoon in wild rodents [125]. In contrast, the remaining studies employed primers for the 18S rRNA gene to detect and characterize Hepatozoon. This approach has been the primary method applied in mammals of the Americas since 2006 (Additional file 1: Tables S1, S3). Overall, the 18S rRNA gene is conserved and frequently used in Hepatozoon phylogenies because it yields good resolution to the genus or even species level in the Apicomplexa order [123, 124, 126, 130]. Furthermore, the most abundant collection of Hepatozoon sequences in public database derives from this marker (Additional file 1: Tables S2, S3).
It is important to keep in mind that an accurate molecular detection of Hepatozoon spp. relies mostly in the selection of primers and an optimal thermal cycling condition [39]. Reported sequences in the reviewed studies were obtained through different PCR protocols, using 23 distinct primer sets (refer to Fig. 4 and Additional file 1: Table S2 for details). These protocols yielded sequences ranging from 300 to 1,816 base pairs (bp), with most primers targeting the V4 region of the 18S rRNA gene (Fig. 4).

Primers and flanked regions (V1 to V9) used for the molecular diagnosis and genotyping of Hepatozoon in wild mammals in the Americas. The position of each primer was aligned with the 18S rDNA complete reference sequence of Hepatozoon canis detected in a domestic dog from Israel (MH615006, 1,816 bp; shown in black). For details on the primers, refer to Table S3
A correct choice of primers for amplifying specific DNA fragments is crucial to achieving the objectives in a given study [39, 131]. A key aspect in primer selection is using those primers with the lowest potential to form secondary structures, such as hairpins or dimers (Additional file 1: Tables S2). For molecular diagnostics, it is preferable to opt for primers that amplify shorter fragments to maximize the sensitivity of the results [39]. Conversely, when conducting phylogenetic inferences, it is recommended to choose primers that yield longer fragments or even a complete gene sequence (Fig. 4, Additional file 1: Table S2) [39]. Nevertheless, some studies constructed phylogenies of the genus Hepatozoon incorporating short and long sequences, and the trees yielded congruent topologies consistent with the evolutionary history of the genus [124, 130, 132, 133]. It is important to note that these studies implemented short sequences above 387 bp flanking the IV region of the 18S rRNA gene.
Evolutionary relationships and haplotype diversity of Hepatozoon spp. in mammals of the Americas
The small subunit of the ribosomal RNA gene (SSU rRNA or 18S rRNA) encompasses nine regions (V1–V9), with the regions V2, V4, and V9 the most hypervariable [131]. As of December 31, 2022, the GenBank database lists over 2,916 18S rDNA sequences (≥ 387 bp) associated with Hepatozoon, flanking one or more 18S rRNA gene variable regions (Additional file 1: Table S3). For the V4 region, 2,596 sequences (390–1816 bp) showed over 50% coverage, and 247 have been recovered from 48 wild mammal species across the Americas (Additional file 1: Table S3). Conversely, Hepatozoon sequences corresponding to other regions of the gene were less represented (Additional file 1: Table S3). Therefore, analyses of genotypes incorporated a final curated dataset of 115 sequences obtained from mammals in the Americas, along with 126 Hepatozoon sequences recovered from birds, small mammals, herpetozoa, and carnivores worldwide. In contrast, haplotype analyses were confined to the aforementioned 115 sequences. All sequences overlapped in the V4 region of the 18S rRNA gene, each with ≥ 500-bp length, similar to the approach applied by Vásquez-Aguilar et al. [38].
Before detailing the results of genotype and haplotype analyses, a brief recall on the definitions of these terms is given for clarity of interpretation. A genotype refers to the specific DNA sequence of a given locus, while a haplotype encapsulates a collection of genetic variants, typically co-inherited. These definitions are instrumental to analyze and understand the phylogenetic relationships and genetic variability within the Hepatozoon genus, enabling a deeper comprehension of the evolutionary dynamics shaping the species among diverse ecological niches.
Phylogenetic inferences and haplotype diversity
Both ML and BI trees revealed two main clades with strong node support: (i) Hepatozoon spp. associated with small mammals, birds, and herpetozoa (Fig. 5, Clade I), and (ii) Hepatozoon spp. related to carnivores (Fig. 6, Clade II). However, Hemolivia and Karyolysus render the genus Hepatozoon non-monophyletic (Figs. 5, 6). Previous studies also support these evolutionary relationships [120, 130, 134]. In the meantime, the paraphyly of the genus Hepatozoon remains unsolved, and a denser taxon sampling and additional molecular markers are required [124, 126, 134].

Phylogeny of Hepatozoon spp. associated with small mammals, birds, and herpetozoa (Clade I). Maximum likelihood (ML) and Bayesian inference (BI) 18S rRNA gene consensus tree constructed for a subset of Hepatozoon spp. using 241 sequences and an alignment of 2,001 bp. Best-fit evolutionary models calculated for ML and BI methods were TVM+F+G4 and M134, M85, M15, respectively. Ultrafast-bootstrap values and Bayesian posterior probabilities are indicated above or below each branch. Asterisks (*) indicate node support of 100/1 for ML and BI, respectively. GenBank accession numbers are located at the end of tip labels. The scale bar indicates the number of nucleotide substitutions per site. The phylogeny on the left represents a section of the complete Hepatozoon phylogeny. A dashed branch symbolizes the connection between this section and the remaining of the phylogeny

Phylogeny of Hepatozoon spp. associated with carnivores (Clade II). Maximum likelihood (ML) and Bayesian inference (BI) 18S rRNA gene consensus tree constructed for a subset of Hepatozoon spp. using 241 sequences and an alignment of 2,001 bp. Best-fit evolutionary models calculated for ML and BI methods were TVM+F+G4 and M134, M85, M15, respectively. Ultrafast-bootstrap values and Bayesian posterior probabilities are indicated above or below each branch. Asterisks (*) indicate node support of 100/1 for ML and BI, respectively. GenBank accession numbers are located at the end of tip labels. The scale bar indicates number of nucleotide substitutions per site. The phylogeny on the left represents a section of the complete Hepatozoon phylogeny. A dashed branch symbolizes the connection between this section and the remaining of the phylogeny
The nucleotide alignment polymorphism analysis of Hepatozoon 18S rDNA sequences yielded 87 haplotypes (Fig. 7; Additional file 1: Table S4), Hd = 0.986 ± 0.002, with π = 0.04217 ± 0.00102, and S = 178. Moreover, the haplotype network showed a correspondence between haplogroups (Fig. 7) and the two major clades depicted in the phylogenies (Figs. 5, 6). Our haplotype network was similar to that reported by Perles et al. [109], which showed a bipartite split between haplotypes of (i) Hepatozoon spp. obtained from rodents and herpetozoa, and (ii) Hepatozoon spp. found in carnivores.

Haplotype network inferred for a subset of Hepatozoon 18S rDNA sequences obtained from wild mammals in the Americas using 115 sequences and an alignment of 621 bp. For details of haplotypes of Hepatozoon circulating among wild hosts refer to Table S4
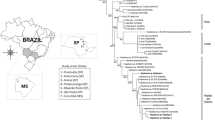
Hepatozoon spp. Clade I
Clade I includes genotypes (Fig. 5) and haplotypes (Fig. 7) of Hepatozoon spp. recovered from mammals in the Americas belonging to the orders Carnivora (Canidae), Didelphimorphia (Didelphidae), Rodentia (Cricetidae, Echimyidae, and Sciuridae), and Microbiotheria (Microbiotheriidae). Although Clade I includes Hepatozoon spp. associated with small mammals, birds, and herpetozoa, two genotypes of Hepatozoon (MW633709, MW633710), found in South American gray foxes (Lycalopex grisea) in Chile [88], clustered with Hepatozoon genotypes associated with Patagonia green racer snakes (Philodryas patagoniensis) [135] (Fig. 5, subclade D). In addition, our BLASTn comparisons for these genotypes revealed a similarity of 99.84–99.68% with Hepatozoon genotypes recovered from Patagonia green racer snakes in Uruguay (99–100% query cover, 0 gaps, 0 E-value) [135]. Additionally, these associations were supported by the haplotype network (Fig. 7, Clade I, haplotypes H22 and H21).
Likewise, one Hepatozoon genotype (KC127680) characterized in a crab-eating fox (Cerdocyon thous) [97] and another genotype (OM033664) from a gray short-tailed opossum (Monodelphis domestica) [136] in Brazil, cluster with Hepatozoon genotypes related to snakes (Fig. 5, subclade D). Additionally, the haplotype network places H24 and H23 haplotypes on the same branch as H19 and H20 (Fig. 7, Clade I), which suggests a common parasitic pathway.
Our results collectively reveal that the Hepatozoon genotypes found in South American gray foxes in Chile, crab-eating fox, and gray short-tailed opossum in Brazil could have been acquired by preying on infected snakes [137,138,139,140]. However, rodents that frequently constitute the primary diet of both South American gray foxes [137, 141] and crab-eating foxes [140] may act as paratenic hosts for Hepatozoon species infectin snakes and lizards [142]. Moreover, crab-eating foxes also prey on frogs [143], which are also considered paratenic hosts for Hepatozoon spp. of reptiles [15]. However, infective cystic stages have been found in rodents from Brazil, suggesting that they are paratenic hosts in the transmission of Hepatozoon towards predators [144, 145].
A Hepatozoon genotype (KX776354) recovered from a Paraguayan fat-tailed mouse opossum (Thylamys macrurus) in Brazil [145] clustered into the clade of Hepatozoon spp. detected in rodents (Fig. 5, subclade of Brazilian rodents). This opossum species has been reported to be in syntopy with several rodent species, such as Oecomys mamorae [146], in which Hepatozoon has been also documented [136, 145], a fact that would explain a common haplotype (H17) among rodents and opossums (Fig. 7, Clade I). Therefore, it is likely that these small mammal species may share ectoparasites that could facilitate accidental cross-species infections [145].
Hepatozoon lineages found in South American cricetids (Rodentia: Cricetidae) and echimyids (Rodentia: Echimyidae) formed two clearly separated clades (Fig. 5, subclade sigmodontine rodents and subclade H). In this context, the Hepatozoon sp. infecting the Paraguayan punaré (Thrichomys pachyurus) appears to be a lineage specific to echimyids, as suggested for South American cricetid rodents [79, 102]. Likewise, the haplotype network supports the separation of Hepatozoon spp. from echimyids (Fig. 7, haplotypes H27 and H26). However, the inclusion of more Hepatozoon genotypes associated with South American echimyid rodents within this phylogenetic framework is needed to substantiate this phylogenetic hypothesis.
Regarding opossums, although the phylogeny does not relate Hepatozoon genotypes associated with the monito del monte (Dromiciops gliroides) and the big-eared opossum (Didelphis aurita) (Fig. 5, subclades G and I), the haplotype network places the haplotypes characterized from these opossum species at the same origin node (Fig. 7, haplotypes H29, H30, H31, H28, and H87). These findings suggest Hepatozoon lineages with a genetic structure associated with South American opossums. However, to further support this hypothesis, additional data are needed.
Hepatozoon spp. Clade II
Clade II of Hepatozoon is composed of genotypes and haplotypes related to mammals of the orders Carnivora (Canidae and Felidae), Didelphimorphia (Didelphidae), Lagomorpha (Leporidae), and Rodentia (Cricetidae and Caviidae) (Fig. 6 and Fig. 7). Although this clade primarily comprises Hepatozoon spp. that infect carnivores, the inclusion of Hepatozoon genotypes related to Didelphimorphia, Lagomorpha, and Rodentia suggest that they correspond to Hepatozoon spp. that infected carnivores via predation [44, 45, 58, 59, 147]. This hypothesis would explain the shared haplotypes among canids and opossums (H51), and the clustering of rodent haplotypes with canids (Fig. 7, Clade II, haplotypes H55, H53, and H62).
On the other hand, experimental studies have shown that rabbits (Lagomorpha) serve as paratenic hosts for Hepatozoon spp. that infect carnivores [44]. But Allen et al. [59] proposed that some undescribed species of Hepatozoon may cycle in lagomorphs. However, the initial claim is supported by the close evolutionary relationships observed in Hepatozoon genotypes found in rabbits and felids (Fig. 6, subclade P), as well as the shared node of origin among rabbit and felid haplotypes (Fig. 7, Clade II, haplotypes H50, H48, H49, H33, H32, and H47). These associations could suggest that rabbits might serve as paratenic hosts for Hepatozoon spp. related to felids.
Regarding Hepatozoon genotypes and haplotypes of Didelphimorphia and Rodentia, their clustering into Clade II suggests that the sampled mammals came from regions endemic for canine hepatozoonosis; moreover, it demonstrates the potential of these mammalian groups to act as paratenic hosts for Hepatozoon species that infect canids [148,149,150]. Particularly, rodents have been implicated as vertebrate reservoirs of Hepatozoon in wildlife [15, 147]. Indeed, infective cystic stages in rodents facilitate the persistence of Hepatozoon spp. in the ecosystem [144, 147], including H. americanum [45]. This fact might account for the shared genotypes or haplotypes among canid and rodents, for both H. americanum (Fig. 6, subclade K; and Fig. 7, haplotype H62) and H. canis (Fig. 6, subclade L; and Fig. 7, haplotypes H53 and H55). Furthermore, it could suggest that rodents might also act as paratenic hosts for H. canis in Brazil [148]. However, this hypothesis remains unclear [109, 144].
Overall, our results reveal previously unreported associations between Hepatozoon genotypes in both distantly related taxa (Fig. 5, subclade D; and Fig. 6, subclades K, L, and P) and closely related mammalian groups (Fig. 5, subclades Sigmodontine and North American rodents), as well as in predator–prey relationships (Fig. 5, subclade D; and Fig. 6, subclades K, L, and P). This wide host spectrum suggests that the diversity and biogeographical patterns of Hepatozoon spp. in wild mammals across the Americas are more complex than currently understood. In this sense, the studies of Di Cataldo et al. [88] and Weck et al. [136] may not accurately reflect the relationships among Hepatozoon genotypes found in foxes in Chile and fat-tailed mouse opossum in Brazil due to lack of data in their phylogenetic framework. Therefore, incorporating more Hepatozoon sequences into an alignment could support with more confidence the monophyly of new genotypes and clarify their evolutionary relationships and ecological associations, as our analyses demonstrate.
Factors driving the diversity and transmission of Hepatozoon spp. in mammals of the Americas
The genetic diversity of a given species is modulated by multiple processes that include mutation, recombination, and biodemography [151, 152]. For Hepatozoon spp., the life cycle, transmission dynamics, and dispersion capacity are factors that shape their diversity as well [38]. Moreover, the transmission of Hepatozoon species may be facilitated by syntopy of hosts sharing ectoparasites [145], the distribution of suitable vectors [52, 62, 63, 153], and through ingestion of infective cystozoites by carnivorism [44, 45, 142] or ectoparasites attached to prey [18, 45]. However, it seems that host specificity and food webs play a crucial role in the transmission of Hepatozoon species, as corroborated by previous studies [147, 154].
Small mammals and herpetozoa are typical prey of carnivores, and those harboring cysts are likely to transmit Hepatozoon spp. to their predators. This ecological pattern is shown in Fig. 5, where occasional infections of canids with snake-related genotypes of Hepatozoon can be observed (subclade D), suggesting a low host specificity in certain Hepatozoon spp. However, given that vector capacity—defined as the daily rate of effective infections spread by a specific arthropod population and influenced by vector density, behavior, longevity, vector-host encounter rates, and vector competence—varies among invertebrate hosts [155,156,157,158], predators can be considered dead-end hosts for these Hepatozoon spp., thus affecting the transmission dynamics. In this context, predators may contribute to reducing the transmission of the parasite (dilution effect) [41, 147]. Nevertheless, these infections may affect the immune response of predators, potentially increasing their susceptibility to other infectious agents [93, 159, 160].
In addition, Hepatozoon spp. exhibit greater host specificity in invertebrates (definitive host) than in vertebrates (intermediate host) [15, 17, 123]. Therefore, the degree of specificity that an invertebrate parasite shows to its vertebrate host will define the chance for a given Hepatozoon sp. to find and colonize a suitable vertebrate host [157, 161]. Colonization may be affected by the immune response of the vertebrate hosts [8]. Thus, considering that the composition of parasite communities is primarily structured by host species (both intermediated and definitive), phylogenetically related hosts are more likely to share parasite species since they exhibit similar immunological pathways and ecological and evolutionary processes [8, 162]. These interactions with invertebrate and vertebrate hosts play a significant role in the diversification and dissemination of Hepatozoon spp.
Some studies have proposed that Hepatozoon genotypes exhibit close phylogenetic relationships and a genetic structure according to the vertebrate groups that they parasitize [109, 144, 163]. Notably, rodent-associated Hepatozoon spp. seem to be specific, in contrast to those species that infect reptiles [164]. Likewise, studies have confirmed a degree of genetic diversity in Hepatozoon spp. infecting rodents [144, 145]. Although some genotypes of Hepatozoon found in rodents are shared with reptiles, they are considered to be Hepatozoon spp. of reptiles using rodents as paratenic hosts within their life cycle [142].
In particular, the findings in Chile suggest discrete lineages of Hepatozoon spp. associated with the native rodent genera Abrothrix, Oligoryzomys, and Phyllotis [75, 79, 102, 132], and with the ancient marsupial monito del monte (D. gliroides) [75], suggesting that Hepatozoon co-evolved with these mammals [102, 165, 166]. Thus, the evolutionary history and diversification dynamics of these hosts could be shaping the phylogenetic relationships and genetic structure of the Hepatozoon lineages characterized in Chile [166]. However, a denser sampling across hosts and the inclusion of both 18Sr DNA and cox1 sequences in the genetic analyses are needed to support this hypothesis.
The knowledge of the Hepatozoon genotypes and haplotypes circulating among wildlife mammals in the Americas provides valuable insight into the epidemiology of this hemoparasite, shedding light on exposed or susceptible hosts. Also, it facilitates tracking the spread and occurrence of unique haplotypes of Hepatozoon among mammalian groups (Fig. 7), especially in threatened host species. Although a greater diversity of Hepatozoon spp. is found in canids and rodents, this may reflect a sampling bias. Overall, these results reveal a significant lack of data and highlight the need for a comprehensive sampling of less prospected mammals in the Americas.
Would Hepatozoon spp. pose a risk to mammals of the Americas?
Infectious diseases pose a significant threat to wildlife, leading to population decline, biodiversity loss, ecological disruptions, and an increased risk of disease transmission [11, 12, 167]. Therefore, studying infectious diseases in wildlife is crucial for the conservation of animal biodiversity and human health, particularly in the context of global climate change [3, 168]. In the Americas between 1916 and 2022, Hepatozoon spp. have been reported in 107 species of mammals, belonging to 62 genera, 18 families, and nine orders (Table S1). This fact highlights the broad range of susceptible mammals and the potential impact on health that Hepatozoon spp. in the wildlife of the Americas could pose.
Based on our findings, H. canis and H. felis emerge as the most widespread species among wild mammals in the Americas. Hepatozoon canis was identified in 13 wild mammal species, infecting 133/657 individuals (20.24%), while H. felis was found in 12 species, infecting 132/475 individuals (27.97%) (Additional file 1: Table S1). It is recognized that H. canis infection in canids might persist sub-clinically, and its severity can range from mild to life-threatening [16, 53, 66, 83], while H. felis seems well suited to felid hosts [68]. However, IFs of H. canis and H. felis among non-canid and non-felid hosts (Additional file 1: Table S1) raise concerns and warrant further investigation because of the potential risks they might pose to other mammalian groups, particularly those with conservation threats [169].
Remarkably, H. americanum exhibited IFs of 44.09% (41/93 individuals) across three wild mammal species, while H. americanum-like showed IFs of 11.43% (44/385 individuals) in five canid species (Additional file 1: Table S1). The first species is considered the primary cause of American canine hepatozoonosis (ACH) in North America [16, 38, 66, 170]. Meanwhile, the latter is a species closely related to H. americanum, and is emerging in South American canids [13, 67, 171]. Although H. americanum was found infecting only two mammal species in North America, it showed higher IFs.
While H. americanum and H. americanum-like were found in one North American and four South American canid species, respectively, H. canis was found in nine species of canids, including three in North America and six in South America (Additional file 1: Table S1). Additionally, H. felis was found in three canid species and six felid species across the Americas (Additional file 1: Table S1). Further assessment is necessary to understand the spread and potential impact of these infections among canid and felid populations, given that carnivores play an essential role in maintaining balance of the ecosystems as predators within the food webs, and are threatened species, [172,173,174,175,176,177].
The high occurrence of Hepatozoon infection in both North American and South American canids can be linked to their presence in endemic areas for canine hepatozoonosis [18, 58, 59, 144, 153]. It is worth mentioning that some canids, such as the coyote (Canis latrans) and crab-eating fox, are expanding beyond their natural ranges [178]. This fact, coupled with cross-breeding events with domestic dogs [178, 179], poses a significant risk for spillover of parasites between domestic animals and wildlife, as well as the emergence of new endemic canine hepatozoonosis foci [180].
Indeed, coyotes are commonly reported to be infected with H. americanum in North America [87, 153, 181,182,183,184], while crab-eating foxes are associated with H. canis and H. americanum-like infections in South American ecosystems [67, 171, 185, 186]. Both H. americanum and H. canis are recognized for their virulence in canids [16, 66, 84], as well as for their diverse modes of transmission between hosts and ticks, that are common ectoparasites of canids in the Americas [52, 63, 187].
Notably, Hepatozoon was primarily observed in Carnivora (42.12%; 730/1,733); however, Rodentia exhibited the highest number of species (62.26%; 66/106) infected with Hepatozoon (Additional file 1: Table S1). This can be attributed to the diverse ecological traits of rodents, such as their fast life pace, terrestriality, high population densities, various activity cycles, and diet breadth [188]. Likewise, being one of the most geographically widespread, diverse, and abundant mammalian orders, Rodentia influences parasite richness and transmission [3, 21, 189]. Our findings highlight the importance of both carnivores and rodents to understanding the epizootiology and transmission of Hepatozoon spp. Moreover, they suggest that rodents are key in maintaining Hepatozoon spp. in the ecosystems of the American continent.
Despite the fact that certain etiological agents in wildlife may not cause diseases in their hosts due to a harmonious parasite–host relationship (co-evolution) [190, 191], it is worth mentioning that while Hepatozoon infections in wildlife are often subclinical, they may vary from mild to severe [67, 182, 183, 192]. Indeed, these hemoparasites become pathogenic and opportunistic in immunocompromised individuals, exhibiting high virulence in concurrent infections, thereby increasing susceptibility to other vector-borne agents [66, 181, 193,194,195]. Furthermore, spillover of Hepatozoon spp. in atypical hosts may lead to infections more virulent than those observed in natural hosts [196,197,198].
Hepatozoon infections have been associated with mortality and clinical diseases in hyenas (Crocuta crocuta) and coyotes [183, 199, 200], with recent evidence linking the infection to myocarditis and myositis in coyotes [153]. In crab-eating foxes, Hepatozoon infections result in mild anemia, abnormal blood values, liver degeneration, and splenic growth [185]. For instance, in impalas (Aepyceros melampus), symptoms of mild hepatitis and lymphadenitis have been associated to Hepatozoon-like infection [201, 202]. Moreover, in rodents, high parasitemia in specific tissues (bone marrow, lung, and liver) post-second merogony [42] leads to anemia, fatigue, inflammation, and sometimes death [159, 203,204,205,206].
Hepatozoon transmission in vertebrates primarily occurs through the ingestion of an infected blood-sucking invertebrate. However, the second merogony—a phase of asexual reproduction producing cystozoites—seems pivotal for some Hepatozoon spp., as cystozoites are the infectious forms leveraging transmission through predation, thereby facilitating its spread across food webs [15, 42]. Rodents often act as paratenic hosts in this mode of infection [15, 147, 207, 208]. In fact, studies indicate that carnivores may acquire Hepatozoon infection by preying on rodents [147, 207]. Nevertheless, the role of rodents in the epidemiology of Hepatozoon species detected in carnivores is still unclear [145, 209].
Over 42,100 species are at risk of extinction, of which 11,367 (27%) are mammals [210]. Among 107 mammal species documented with Hepatozoon infections, 10 (9.35%) face potential extinction, two (20%) are vulnerable (VU), and eight (80%) are near threatened (NT); 86 (80.37%) are categorized as least concern (LC); three (2.80%) have data-deficient status (DD), and nine (8.41%) lack available data. Of the species positive for Hepatozoon, 49 (45.79%) have stable populations, 22 (20.56%) are decreasing, seven (6.54%) are increasing, 22 (20.56%) have unknown population status, and nine (8.41%) lack accessible data (see Additional file 1: Table S1).
A pre-existing infection can aid the establishment of a new infectious agent which otherwise might have been cleared by the host’s immune system; consequently, the acquisition of a novel infectious agent may promote the dissemination of an existing latent or dormant infection in the host, acting synergistically to enhance pathogenic processes, parasite transmission, and disease severity [93, 159, 160].
Hepatozoon infection in mammals of the Americas requires comprehensive attention, considering factors such as the availability of suitable arthropod vectors, the prevalence of infection in intermediate hosts, and the susceptibility of different mammal species. In addition, it is important to recognize other threats to wildlife in the Americas, including habitat loss (by urbanization, livestock, and farming), pollution, climate change, and tick-borne diseases [156, 211]. The conjunction of these factors may influence the spillover of Hepatozoon infections among wild and domestic animals. To date, only one study has monitored the fluctuation and assessed the impact of Hepatozoon infection on health in wildlife of the Americas [89], for which additional studies of this kind are necessary.
Given that parasites pose a risk to species susceptible to extinction [212], detecting and monitoring Hepatozoon spp. in wildlife becomes necessary. Genetic screening can reveal the patterns of dispersion of Hepatozoon lineages among hosts, enriching our knowledge of their roles in the Hepatozoon epizootiology [169, 212, 213]. It is crucial for further research and surveillance to comprehensively understand the dynamics of Hepatozoon infections in mammal populations of the Americas and assess their potential as a significant threat [88].
Conclusions and future perspectives
This review provides valuable insight into the distribution of Hepatozoon among mammalian hosts and potential vectors in the Americas, establishing a foundation for subsequent research. Notably, this review represents the first comprehensive summary of Hepatozoon infection in wild mammals in the region. However, numerous questions remain unanswered, particularly regarding the impact of hemoparasites on the health of wild mammals in the Americas.
Given the expanding distribution of certain canids, combined with hybridization events between some species and the diverse transmission modes of Hepatozoon, there is a risk for spillover between wildlife and domestic animals. This could also lead to the emergence of new areas with endemic foci for canine and felid hepatozoonosis. Canid species such as coyotes, crab-eating foxes, South American gray foxes, and Pampas foxes may serve as effective sentinels to track the expansion of H. americanum, H. canis, H. americanum-like, and H. felis in ecosystems across the American continent. Finally, genomic data and new molecular markers are urgently needed for the implementation of effective strategies to detect, control, and manage Hepatozoon infections.
Data availability
The datasets generated and analyzed in this review are available in the Additional file 1 (Tables S1, S2, S3, and S4).
Change history
16 May 2024
A Correction to this paper has been published: https://doi.org/10.1186/s13071-024-06309-2
References
Cowie RH, Bouchet P, Fontaine B. The sixth mass extinction: fact, fiction or speculation? Biol Rev. 2022;97:640–63. https://doi.org/10.1111/brv.12816.
Haydon DT, Cleaveland S, Taylor LH, Laurenson MK. Identifying reservoirs of infection: a conceptual and practical challenge. Emerg Infect Dis. 2002;8:1468–73. https://doi.org/10.3201/eid0812.010317.
Karesh WB, Dobson A, Lloyd-Smith JO, Lubroth J, Dixon MA, Bennett M, et al. Ecology of zoonoses: natural and unnatural histories. The Lancet. 2012;380:1936–45. https://doi.org/10.1016/S0140-6736(12)61678-X.
White RJ, Razgour O. Emerging zoonotic diseases originating in mammals: a systematic review of effects of anthropogenic land-use change. Mamm Rev. 2020;50:336–52. https://doi.org/10.1111/mam.12201.
Windsor DA. Controversies in parasitology, most of the species on earth are parasites. Int J Parasitol. 1998;28:1939–41. https://doi.org/10.1016/s0020-7519(98)00153-2.
Carlson CJ, Hopkins S, Bell KC, Doña J, Godfrey SS, Kwak ML, et al. A global parasite conservation plan. Biol Conserv. 2020;250:108596. https://doi.org/10.1016/j.biocon.2020.108596.
Poulin R. The Many Roads to Parasitism: A Tale of Convergence. In: Rollinson D, Hay SI, editors. Advances in Parasitology. Cambridge: Academic Press; 2010. p. 1–40. https://doi.org/10.1016/B978-0-12-385897-9.00001-X
Poulin R, Keeney DB. Host specificity under molecular and experimental scrutiny. Trends Parasitol. 2008;24:24–8. https://doi.org/10.1016/j.pt.2007.10.002.
Penczykowski RM, Laine AL, Koskella B. Understanding the ecology and evolution of host–parasite interactions across scales. Evol Appl. 2016;9:37. https://doi.org/10.1111/eva.12294.
Brian JI. Parasites in biodiversity conservation: friend or foe? Trends Parasitol. 2023;39:618–21. https://doi.org/10.1016/j.pt.2023.05.005.
Daszak P, Cunningham AA, Hyatt AD. Emerging infectious diseases of wildlife—threats to biodiversity and human health. Science. 1979;2000:443–9. https://doi.org/10.1126/science.287.5452.443.
Scott ME. The impact of infection and disease on animal populations: Implications for conservation biology. Conserv Biol. 1988;2:40–56. https://doi.org/10.1111/j.1523-1739.1988.tb00334.x.
Millán J, Travaini A, Cevidanes A, Sacristán I, Rodríguez A. Assessing the natural circulation of canine vector-borne pathogens in foxes, ticks and fleas in protected areas of Argentine Patagonia with negligible dog participation. Int J Parasitol Parasites Wildl. 2019;8:63–70. https://doi.org/10.1016/j.ijppaw.2018.11.007.
Valenzuela-Sánchez A, Medina-Voge G. Importance of infectious disease for the conservation of Chilean threatened wildlife. Gayana (Concepción). 2014;78:57–69. https://doi.org/10.4067/S0717-65382014000100008.
Smith TG. The genus Hepatozoon (Apicomplexa: Adeleina). J Parasitol. 1996;82:565–85. https://doi.org/10.2307/3283781.
Baneth G. Perspectives on canine and feline hepatozoonosis. Vet Parasitol. 2011;181:3–11. https://doi.org/10.1016/j.vetpar.2011.04.015.
Sam Telford. The Hemogregarines Hemoparasites of the Reptilia: color atlas and text. Boca Raton: CRC Press; 2009.
Ewing SA, Panciera RJ. American canine hepatozoonosis. Clin Microbiol Rev. 2003;16:688–97. https://doi.org/10.1128/cmr.16.4.688-697.2003.
Potter TM, Macintire DK. Hepatozoon americanum: an emerging disease in the south-central/southeastern United States. J Vet Emerg Crit Care. 2010;20:70–6. https://doi.org/10.1111/j.1476-4431.2009.00508.x.
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700. https://doi.org/10.1136/bmj.b2700.
Wilson DE, Lacher TE, Mittermeier RA. Handbook of the Mammals of the World. Barcelona: Lynx Edici; 2017.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. https://doi.org/10.1093/molbev/mst010.
Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–76. https://doi.org/10.1007/bf01734359.
Rannala B, Yang Z. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J Mol Evol. 1996;43:304–11. https://doi.org/10.1007/bf02338839.
Yang Z, Rannala B. Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo method. Mol Biol Evol. 1997;14:717–24. https://doi.org/10.1093/oxfordjournals.molbev.a025811.
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. https://doi.org/10.1093/molbev/msu300.
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42. https://doi.org/10.1093/sysbio/sys029.
Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9. https://doi.org/10.1038/nmeth.4285.
Huelsenbeck JP, Larget B, Alfaro ME. Bayesian phylogenetic model selection using reversible jump Markov chain Monte Carlo. Mol Biol Evol. 2004;21:1123–33. https://doi.org/10.1093/molbev/msh123.
Schwarz G. Estimating the dimension of a model. Ann Statist. 1978;6:461–4.
Minh BQ, Nguyen MAT, Von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30:1188–95. https://doi.org/10.1093/molbev/mst024.
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 17. Syst Biol. 2018;67:901–4. https://doi.org/10.1093/sysbio/syy032.
Huelsenbeck JP, Rannala B. Frequentist properties of Bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models. Syst Biol. 2004;53:904–13. https://doi.org/10.1080/10635150490522629.
Santodomingo A, Robbiano S, Thomas R, Parragué-Migone C, Cabello-Stom J, Vera-Otarola F, et al. A search for piroplasmids and spirochetes in threatened pudu (Pudu puda) and associated ticks from Southern Chile unveils a novel Babesia sp. and a variant of Borrelia chilensis. Transbound Emerg Dis. 2022;69:3737–48. https://doi.org/10.1111/tbed.14743.
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–302. https://doi.org/10.1093/molbev/msx248.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036.
O’Dwyer LH. Brazilian canine hepatozoonosis. Rev Bras Parasitol Vet. 2011;20:181–93. https://doi.org/10.1590/S1984-29612011000300002.
Vásquez-Aguilar AA, Barbachano-Guerrero A, Angulo DF, Jarquín-Díaz VH. Phylogeography and population differentiation in Hepatozoon canis (Apicomplexa: Hepatozoidae) reveal expansion and gene flow in world populations. Parasit Vectors. 2021;14:1–14. https://doi.org/10.1186/s13071-021-04924-x.
Modrý D, Beck R, Hrazdilová K, Baneth G. A review of methods for detection of Hepatozoon infection in carnivores and arthropod vectors. Vector Borne Zoonotic Dis. 2017;17:66–72. https://doi.org/10.1089/vbz.2016.1963.
Ashford RW. When is a reservoir not a reservoir? Emerg Infect Dis. 2003;9:1945.
Johnson PTJ, Dobson A, Lafferty KD, Marcogliese DJ, Memmott J, Orlofske SA, et al. When parasites become prey: ecological and epidemiological significance of eating parasites. Trends Ecol Evol. 2010;25:362–71. https://doi.org/10.1016/j.tree.2010.01.005.
Baneth G, Samish M, Shkap V. Life cycle of Hepatozoon canis (Apicomplexa: Adeleorina: Hepatozoidae) in the tick Rhipicephalus sanguineus and domestic dog (Canis familiaris). J Parasitol. 2007;93:283–99. https://doi.org/10.1645/GE-494R.1.
Johnson EM, Allen KE, Panciera RJ, Ewing SA, Little SE, Reichard MV. Field survey of rodents for Hepatozoon infections in an endemic focus of American canine hepatozoonosis. Vet Parasitol. 2007;150:27–32. https://doi.org/10.1016/j.vetpar.2007.08.050.
Johnson EM, Allen KE, Panciera RJ, Ewing SA, Little SE. Experimental transmission of Hepatozoon americanum to New Zealand White rabbits (Oryctolagus cuniculus) and infectivity of cystozoites for a dog. Vet Parasitol. 2009;164:162–6. https://doi.org/10.1016/j.vetpar.2009.05.028.
Johnson EM, Panciera RJ, Allen KE, Sheets ME, Beal JD, Ewing SA, et al. Alternate pathway of infection with Hepatozoon americanum and the epidemiologic importance of predation. J Vet Intern Med. 2009;23:1315–8. https://doi.org/10.1111/j.1939-1676.2009.0375.x.
Duscher GG, Leschnik M, Fuehrer HP, Joachim A. Wildlife reservoirs for vector-borne canine, feline and zoonotic infections in Austria. Int J Parasitol Parasites Wildl. 2015;4:88–96. https://doi.org/10.1016/j.ijppaw.2014.12.001.
Murata T, Taura Y, Nakama S, Inoue M, Tateyama S. Vertical transmission of Hepatozoon canis in dogs. J Vet Med Sci. 1993;55:867–8. https://doi.org/10.1292/jvms.55.867.
Hodžic A, Mrowietz N, Cézanne R, Bruckschwaiger P, Punz S, Habler VE, et al. Occurrence and diversity of arthropod-transmitted pathogens in red foxes (Vulpes vulpes) in western Austria, and possible vertical (transplacental) transmission of Hepatozoon canis. Parasitology. 2017;145:335–44. https://doi.org/10.1017/S0031182017001536.
Schäfer I, Müller E, Nijhof AM, Aupperle-Lellbach H, Loesenbeck G, Cramer S, et al. First evidence of vertical Hepatozoon canis transmission in dogs in Europe. Parasit Vectors. 2022;15:1–9. https://doi.org/10.1186/s13071-022-05392-7.
Vincent-Johnson NA. American canine hepatozoonosis. Vet Clim North Am Small Anim Pract. 2003;33:905–20. https://doi.org/10.1016/s0195-5616(03)00028-7.
Giannelli A, Ramos RAN, Di Paola G, Mencke N, Dantas-Torres F, Baneth G, et al. Transstadial transmission of Hepatozoon canis from larvae to nymphs of Rhipicephalus sanguineus. Vet Parasitol. 2013;196:1–5. https://doi.org/10.1016/j.vetpar.2013.02.017.
Ewing SA, DuBois JG, Mathew JS, Panciera RJ. Larval Gulf Coast ticks (Amblyomma maculatum) [Acari: Ixodidae] as host for Hepatozoon americanum [Apicomplexa: Adeleorina]. Vet Parasitol. 2002;103:43–51. https://doi.org/10.1016/s0304-4017(01)00572-6.
Baneth G, Samish M, Alekseev E, Aroch I, Shkap V. Transmission of Hepatozoon canis to dogs by naturally-fed or percutaneously-injected Rhipicephalus sanguineus ticks. J Parasitol. 2001;87:606–11. https://doi.org/10.1645/0022-3395(2001)087[0606:TOHCTD]2.0.co;2.
Giannelli A, Ramos RAN, Dantas-Torres F, Mencke N, Baneth G, Otranto D. Experimental evidence against transmission of Hepatozoon canis by Ixodes ricinus. Ticks Tick Borne Dis. 2013;4:391–4. https://doi.org/10.1016/j.ttbdis.2013.03.001.
Viana LA, Soares P, Silva JE, Paiva F, Coutinho ME. Anurans as paratenic hosts in the transmission of Hepatozoon caimani to caimans Caiman yacare and Caiman latirostris. Parasitol Res. 2012;110:883–6. https://doi.org/10.1007/s00436-011-2570-6.
Valkiūnas G, Mobley K, Iezhova TA. Hepatozoon ellisgreineri n sp (Hepatozoidae): description of the first avian apicomplexan blood parasite inhabiting granulocytes. Parasitol Res. 2016;115:609–13. https://doi.org/10.1007/s00436-015-4777-4.
Ebani VV, Mancianti F. Potential role of birds in the epidemiology of Coxiella burnetii, Coxiella-like agents and Hepatozoon spp. Pathogens. 2022;11:298. https://doi.org/10.3390/pathogens11030298.
Allen KE, Johnson EM, Little SE. Hepatozoon spp Infections in the United States. Vet Clim North Am Small Anim Pract. 2011;41:1221–38. https://doi.org/10.1016/j.cvsm.2011.08.006.
Allen KE, Yabsley MJ, Johnson EM, Reichard MV, Panciera RJ, Ewing SA, et al. Novel Hepatozoon in vertebrates from the southern United States. J Parasitol. 2011;97:648–53. https://doi.org/10.1645/GE-2672.1.
Olavo E. Ciclo evolutivo do Hepatozoon triatomae (Sporozoa, Haemogregarinidae) parasita de triatomíneos. Rev Saúde Públ. 1975;9:91. https://doi.org/10.1590/S0034-89101975000300011.
Ferguson LV, Smith TG. Fecundity reduction in the second gonotrophic cycle of Culex pipiens infected with the apicomplexan blood parasite. Hepatozoon sipedon J Parasitol. 2014;100:442–6. https://doi.org/10.1645/13-378.1.
Murata T, Taura Y, Nakama S, Inoue M, Abe H, Fujisaki K. Detection of Hepatozoon canis oocyst from ticks collected from the infected dogs. J Vet Med Sci. 1995;57:111–2. https://doi.org/10.1292/jvms.57.111.
Ewing SA, Mathew JS, Panciera RJ. Transmission of Hepatozoon americanum (Apicomplexa: Adeleorina) by ixodids (Acari: Ixodidae). J Med Entomol. 2002;39:631–4. https://doi.org/10.1603/0022-2585-39.4.631.
Watkins RA, Moshier SE, Pinter AJ. The flea, Megabothris abantis: An invertebrate host of Hepatozoon sp. and a likely definitive host in Hepatozoon infections of the montane vole Microtus montanus. J Wildl Dis. 2006;42:386–90. https://doi.org/10.7589/0090-3558-42.2.386.
Rigó K, Majoros G, Szekeres S, Molnár I, Jablonszky M, Majláthová V, et al. Identification of Hepatozoon erhardovae Krampitz, 1964 from bank voles (Myodes glareolus) and fleas in Southern Hungary. Parasitol Res. 2016;115:2409–13. https://doi.org/10.1007/s00436-016-4992-7.
Baneth G, Mathew JS, Shkap V, Macintire DK, Barta JR, Ewing SA. Canine hepatozoonosis: two disease syndromes caused by separate Hepatozoon spp. Trends Parasitol. 2003;19:27–31. https://doi.org/10.1016/S1471-4922(02)00016-8.
Criado-Fornelio A, Ruas JL, Casado N, Farias NAR, Soares MP, Müller G, et al. New molecular data on mammalian Hepatozoon species (Apicomplexa: Adeleorina) from Brazil and Spain. J Parasitol. 2006;92:93–9. https://doi.org/10.1645/ge-464r.1.
Baneth G, Sheiner A, Eyal O, Hahn S, Beaufils JP, Anug Y, et al. Redescription of Hepatozoon felis (Apicomplexa: Hepatozoidae) based on phylogenetic analysis, tissue and blood form morphology, and possible transplacental transmission. Parasit Vectors. 2013;6:1–10. https://doi.org/10.1186/1756-3305-6-102.
Zinsstag J, Schelling E, Waltner-Toews D, Tanner M. From, “one medicine” to “one health” and systemic approaches to health and well-being. Prev Vet Med. 2011;101:148–56. https://doi.org/10.1016/j.prevetmed.2010.07.003.
Dupré J. (Some) species are processes. In: Wilkins JS, Pavlinov I, Pavlinov I, editors. Species problems and beyond: contemporary issues in philosophy and practice. 1st ed. New York: CRC Press; 2022. p. 284–6.
Arrais RC, Paula RC, Martins TF, Nieri-Bastos FA, Marcili A, Labruna MB. Survey of ticks and tick-borne agents in maned wolves (Chrysocyon brachyurus) from a natural landscape in Brazil. Ticks Tick Borne Dis. 2021;12:101639. https://doi.org/10.1016/j.ttbdis.2020.101639.
Thompson AT, White SA, Shaw D, Garrett KB, Wyckoff ST, Doub EE, et al. A multi-seasonal study investigating the phenology, host and habitat associations, and pathogens of Haemaphysalis longicornis in Virginia, USA. Ticks Tick Borne Dis. 2021;12:101773. https://doi.org/10.1016/j.ttbdis.2021.101773.
Hornok S, Tánczos B, Fernández de Mera IG, de la Fuente J, Hofmann-Lehmann R, Farkas R. High prevalence of Hepatozoon-infection among shepherd dogs in a region considered to be free of Rhipicephalus sanguineus. Vet Parasitol. 2013;196:189–93. https://doi.org/10.1016/j.vetpar.2013.02.009.
Sumrandee C, Baimai V, Trinachartvanit W, Ahantarig A. Hepatozoon and Theileria species detected in ticks collected from mammals and snakes in Thailand. Ticks Tick Borne Dis. 2015;6:309–15. https://doi.org/10.1016/j.ttbdis.2015.02.003.
Merino S, Vásquez RA, Martínez J, Celis-Diez JL, GutiÉrrez-JimÉnez L, Ippi S, et al. Molecular characterization of an ancient Hepatozoon species parasitizing the ‘living fossil’ marsupial ‘Monito del Monte’ Dromiciops gliroides from Chile. Biol J Linn Soc. 2009;98:76. https://doi.org/10.1111/j.1095-8312.2009.01302.x.
Najm NA, Meyer-Kayser E, Hoffmann L, Pfister K, Silaghi C. Hepatozoon canis in German red foxes (Vulpes vulpes) and their ticks: Molecular characterization and the phylogenetic relationship to other Hepatozoon spp. Parasitol Res. 2014;113:2979–85. https://doi.org/10.1007/s00436-014-3923-8.
Hamšíková Z, Silaghi C, Rudolf I, Venclíková K, Mahríková L, Slovák M, et al. Molecular detection and phylogenetic analysis of Hepatozoon spp in questing Ixodes ricinus ticks and rodents from Slovakia and Czech Republic. Parasitol Res. 2016;115:3897–904. https://doi.org/10.1007/s00436-016-5156-5.
Card LR, McShea WJ, Fleischer RC, Maldonado JE, Stewardson K, Campana MG, et al. Tick burdens in a small-mammal community in Virginia. Northeast Nat. 2019;26:641–55. https://doi.org/10.1656/045.026.0317.
Muñoz-Leal S, Lopes MG, Marcili A, Martins TF, González-Acuña D, Labruna MB. Anaplasmataceae, Borrelia and Hepatozoon agents in ticks (Acari: Argasidae, Ixodidae) from Chile. Acta Trop. 2019;192:91–103. https://doi.org/10.1016/j.actatropica.2019.02.002.
de Miranda RL, de Castro JR, Olegário MMM, Beletti ME, Mundim AV, O’Dwyer LH, et al. Oocysts of Hepatozoon canis in Rhipicephalus (Boophilus) microplus collected from a naturally infected dog. Vet Parasitol. 2011;177:392–6. https://doi.org/10.1016/j.vetpar.2011.01.044.
Forlano M, Scofield A, Elisei C, Fernandes KR, Ewing SA, Massard CL. Diagnosis of Hepatozoon spp in Amblyomma ovale and its experimental transmission in domestic dogs in Brazil. Vet Parasitol. 2005;134:1–7. https://doi.org/10.1016/j.vetpar.2005.05.066.
Rubini AS, Paduan KS, Martins TF, Labruna MB, O’Dwyer LH. Acquisition and transmission of Hepatozoon canis (Apicomplexa: Hepatozoidae) by the tick Amblyomma ovale (Acari: Ixodidae). Vet Parasitol. 2009;164:324–7. https://doi.org/10.1016/j.vetpar.2009.05.009.
Baneth G, Shkap V, Samish M, Pipano E, Savitsky I. Antibody response to Hepatozoon canis in experimentally infected dogs. Vet Parasitol. 1998;74:299–305. https://doi.org/10.1016/s0304-4017(97)00160-x.
Vincent-Johnson NA, Macintire DK, Lindsay DS, Lenz SD, Baneth G, Snkap V, et al. A new Hepatozoon species from dogs: Description of the causative agent of canine hepatozoonosis in north America. J Parasitol. 1997;83:1165–72. https://doi.org/10.2307/3284379.
Mathew JS, Ewing SA, Panciera RJ, Woods JP, et al. Experimental transmission of Hepatozoon americanum Vincent-Johnson to dogs by the Gulf Coast tick, Amblyomma maculatum Koch. Vet Parasitol. 1997;1998:1–14. https://doi.org/10.1016/s0304-4017(98)00189-7.
Mathew JS, Ewing SA, Panciera RJ, Kocan KM. Sporogonic development of Hepatozoon americanum (Apicomplexa) in its definitive host, Amblyomma maculatum (Acarina). J Parasitol. 1999;85:1023–31. https://doi.org/10.2307/3285663.
Kocan AA, Breshears M, Cummings C, Panciera RJ, Ewing SA, Barker RW. Naturally occurring hepatozoonosis in Coyotes from Oklahoma. J Wildl Dis. 1999;35:86–9. https://doi.org/10.7589/0090-3558-35.1.86.
Di Cataldo S, Cevidanes A, Ulloa Contreras C, Cabello J, Gambino D, Gargano V, et al. Large-scale survey for canine vector-borne parasites in free-ranging dogs and foxes from six diverse bioclimatic regions of Chile. Vet Parasitol Reg Stud Rep. 2022;30:100721. https://doi.org/10.1016/j.vprsr.2022.100721.
Perles L, de Macedo GC, Barreto WTG, Francisco GV, Herrera HM, Barros-Battesti DM, et al. Longitudinal dynamics and health impact of Hepatozoon procyonis (Apicomplexa: Hepatozoidae) on naturally infected ring-tailed coatis Nasua nasua (Carnivora: Procyonidae) from Midwestern Brazil. Ticks Tick Borne Dis. 2022;13:101982. https://doi.org/10.1016/j.ttbdis.2022.101982.
Blanco CM, Teixeira BR, da Silva AG, de Oliveira RC, Strecht L, Ogrzewalska M, et al. Microorganisms in ticks (Acari: Ixodidae) collected on marsupials and rodents from Santa Catarina, Paraná and Mato Grosso do Sul states. Brazil Ticks Tick Borne Dis. 2017;8:90–8. https://doi.org/10.1016/j.ttbdis.2016.10.003.
Laakkonen J, Henttonen H, Hastriter MW, Niemimaa J, Jarrell GH. Hemoparasites and fleas of shrews and rodents from Alaska. Acta Parasitol. 2002;47:255–7.
Desser SS, Hong H, Martin DS. The life history, ultrastructure, and experimental transmission of Hepatozoon catesbianae n comb., an apicomplexan parasite of the bullfrog, Rana catesbeiana and the mosquito, Culex territans in Algonquin Park. Ontario J Parasitol. 1995;81:212–22. https://doi.org/10.2307/3283922.
Baneth G. Tick-borne infections of animals and humans: a common ground. Int J Parasitol. 2014;44:591–6. https://doi.org/10.1016/j.ijpara.2014.03.011.
Fava NMN, Alves TS, Lopes MG, Labruna MB, Santos AQ, Cury MC. Occurrence and molecular identification of hemoparasites in wild mammals kept in rehabilitation Centers in Brazil. Acta Parasitol. 2022;67:476–86. https://doi.org/10.1007/s11686-021-00492-3.
Silveira JAG, D’Elia ML, de Oliveira AI, de Almeida LR, dos Santos HA, de Magalhães Soares DF, et al. Rangelia vitalii in a free-ranging maned wolf (Chrysocyon brachyurus) and co-infections. Int J Parasitol Parasites Wildl. 2016;5:280–5.
da Silva MRL, Mattoso CRS, Costa A, Saito ME, Tchaicka L, O’Dwyer LH. Rangelia vitalii and Hepatozoon canis coinfection in pampas fox Lycalopex gymnocercus from Santa Catarina State. Brazil Rev Bras Parasitol Vet. 2018;27:377–83. https://doi.org/10.1590/S1984-296120180018.
Almeida AP, Souza TD, Marcili A, Labruna MB. Novel Ehrlichia and Hepatozoon agents infecting the Crab-Eating fox (Cerdocyon thous) in Southeastern Brazil. J Med Entomol. 2013;50:640–6. https://doi.org/10.1603/ME12272.
Santos FM, de Sousa KCM, Sano NY, Nantes WAG, Liberal SC, Machado RZ, et al. Relationships between vector-borne parasites and free-living mammals at the Brazilian Pantanal. Parasitol Res. 2021;120:1003–10. https://doi.org/10.1007/s00436-020-07028-0.
O’Dell WD, Watkins RA, Moshier SE, Pinter AJ. Giardia and other parasites of small mammals in Grand Teton National Park. UW-National Park Service Res Station Ann Rep. 1990;14:91–3. https://doi.org/10.1301/uwnpsrc.1990.2893.
Soares HS, Marcili A, Barbieri ARM, Minervino AHH, Moreira TR, Gennari SM, et al. Novel piroplasmid and Hepatozoon organisms infecting the wildlife of two regions of the Brazilian Amazon. Int J Parasitol Parasites Wildl. 2017;6:115. https://doi.org/10.1016/j.ijppaw.2017.05.002.
Crandall KE, Kerr JT, Millien V. Emerging tick-borne pathogens in Central Canada: Recent detections of Babesia odocoilei and Rickettsia rickettsii. Vector Borne Zoonotic Dis. 2022;22:535–44. https://doi.org/10.1089/vbz.2022.0036.
Santodomingo AM, Thomas RS, Quintero-Galvis JF, Echeverry-Berrio DM, la Fuente MCS, de, Moreno-Salas L, et al. Apicomplexans in small mammals from Chile, with the first report of the Babesia microti group in South American rodents. Parasitol Res. 2022;121:1009–20. https://doi.org/10.1007/s00436-022-07452-4.
Modarelli JJ, Westrich BJ, Milholland M, Tietjen M, Castro-Arellano I, Medina RF, et al. Prevalence of protozoan parasites in small and medium mammals in Texas, USA. Int J Parasitol Parasites Wildl. 2020;11:229–34. https://doi.org/10.1016/j.ijppaw.2020.02.005.
Richards CS. Hepatozoon procyonis, n sp, from the raccoon. J Protozool. 1961;8:360–2. https://doi.org/10.1111/j.1550-7408.1961.tb01229.x.
Schaffer GD, Hanson WL, Davidson WR, Nettles VF. Hematotropic parasites of translocated raccoons in the southeast. J Am Vet Med Assoc. 1978;173:1148–51.
Pietrzak SM, Pung OJ. Trypanosomiasis in raccoons from Georgia. J Wildl Dis. 1998;34:132–6. https://doi.org/10.7589/0090-3558-34.1.132.
Colle AC, De Mendonça RFB, Maia MO, da Freitas L, et al. Molecular survey of tick-borne pathogens in small mammals from Brazilian Amazonia. Rev Bras Parasitol Vet. 2019;28:592–604. https://doi.org/10.1590/S1984-29612019086.
Perles L, Ikeda P, Francisco G de V, Torres JM, de Oliveira CE, Lourenço EC, et al. Molecular detection of Hepatozoon spp. in non-hematophagous bats in Brazil. Ticks Tick Borne Dis. 2020;11:101401. https://doi.org/10.1016/j.ttbdis.2020.101401.
Perles L, Roque ALR, D’Andrea PS, Lemos ERS, Santos AF, Morales AC, et al. Genetic diversity of Hepatozoon spp. in rodents from Brazil. Sci Rep. 2019;9:10122. https://doi.org/10.1038/s41598-019-46662-2.
Cardoso L, Cortes HCE, Eyal O, Reis A, Lopes AP, Vila-Viçosa MJ, et al. Molecular and histopathological detection of Hepatozoon canis in red foxes (Vulpes vulpes) from Portugal. Parasit Vectors. 2014;7:1–6. https://doi.org/10.1186/1756-3305-7-113.
Nordgren RM, Craig TM. Experimental transmission of the Texas strain of Hepatozoon canis. Vet Parasitol. 1984;16:207–14. https://doi.org/10.1016/0304-4017(84)90038-4.
Karagenc TI, Pasa S, Kirli G, Hosgor M, Bilgic HB, Ozon YH, et al. A parasitological, molecular and serological survey of Hepatozoon canis infection in dogs around the Aegean coast of Turkey. Vet Parasitol. 2006;135:113–9. https://doi.org/10.1016/j.vetpar.2005.08.007.
Rubini AS, Paduan KDS, Lopes VVA, O’Dwyer LH. Molecular and parasitological survey of Hepatozoon canis (Apicomplexa: Hepatozoidae) in dogs from rural area of Sao Paulo state. Brazil Parasitol Res. 2008;102:895–9. https://doi.org/10.1007/s00436-007-0846-7.
Criado-Fornelio A, Buling A, Cunha-Filho NA, Ruas JL, Farias NAR, Rey-Valeiron C, et al. Development and evaluation of a quantitative PCR assay for detection of Hepatozoon sp. Vet Parasitol. 2007;150:352–6. https://doi.org/10.1016/j.vetpar.2007.09.025.
Grillini M, Beraldo P, Frangipane di Regalbono A, Dotto G, Tessarin C, Franzo G, et al. Molecular survey of Cytauxzoon spp and Hepatozoon spp in felids using a novel real-time PCR approach. Front Vet Sci. 2023;10:1113681. https://doi.org/10.3389/fvets.2023.1113681.
Gardner MJ, Bishop R, Shah T, De Villiers EP, Carlton JM, Hall N, et al. Genome sequence of Theileria parva, a bovine pathogen that transforms lymphocytes. Science. 1979;2005:134–7. https://doi.org/10.1126/science.1110439.
Hikosaka K, Watanabe Y, Kobayashi F, Waki S, Kita K, Tanabe K. Highly conserved gene arrangement of the mitochondrial genomes of 23 Plasmodium species. Parasitol Int. 2011;60:80. https://doi.org/10.1016/j.parint.2011.02.001.
Cornillot E, Hadj-Kaddour K, Dassouli A, Noel B, Ranwez V, Vacherie B, et al. Sequencing of the smallest Apicomplexan genome from the human pathogen Babesia microti†. Nucleic Acids Res. 2012;40:9102–14. https://doi.org/10.1093/nar/gks700.
Lang-Unnasch N, Reith ME, Munholland J, Barta JR. Plastids are widespread and ancient in parasites of the phylum Apicomplexa. Int J Parasitol. 1998;28:1743–54. https://doi.org/10.1016/s0020-7519(98)00136-2.
Kim B, Smith TG, Desser SS. The life history and host specificity of Hepatozoon clamatae (Apicomplexa: Adeleorina) and ITS-1 nucleotide sequence variation of Hepatozoon species of frogs and mosquitoes from Ontario. J Parasitol. 1998;84:789–97. https://doi.org/10.2307/3284589.
Boulianne B, Evans RC, Smith TG. Phylogenetic analysis of Hepatozoon species (Apicomplexa: Adeleorina) infecting frogs of Nova Scotia, Canada, determined by ITS-1 sequences. J Parasitol. 2007;93:1435–41. https://doi.org/10.1645/ge-1041.1.
Carreno RA, Martin DS, Barta JR. Cryptosporidium is more closely related to the gregarines than to coccidia as shown by phylogenetic analysis of apicomplexan parasites inferred using small-subunit ribosomal RNA gene sequences. Parasitol Res. 1999;85:899–904. https://doi.org/10.1007/s004360050655.
Barta JR, Ogedengbe JD, Martin DS, Smith TG. Phylogenetic position of the adeleorinid Coccidia (Myzozoa, Apicomplexa, Coccidia, Eucoccidiorida, Adeleorina) inferred using 18S rDNA sequences. J Eukaryot Microbiol. 2012;59:171–80. https://doi.org/10.1111/j.1550-7408.2011.00607.x.
Maia JP, Carranza S, Harris DJ. Comments on the systematic revision of adeleid Haemogregarines: are more data needed? J Parasitol. 2016;102:549–52. https://doi.org/10.1645/15-930.
Léveillé AN, El Skhawy N, Barta JR. Multilocus sequencing of Hepatozoon cf griseisciuri infections in Ontario eastern gray squirrels (Sciurus carolinensis) uncovers two genotypically distinct sympatric parasite species. Parasitol Res. 2020;119:713–24. https://doi.org/10.1007/s00436-019-06583-5.
Hrazdilová K, Červená B, Blanvillain C, Foronda P, Modrý D. Quest for the type species of the genus Hepatozoon – phylogenetic position of hemogregarines of rats and consequences for taxonomy. Syst Biodivers. 2021;19:622–31. https://doi.org/10.1080/14772000.2021.1903616.
Wilson RJ, Williamson DH. Extrachromosomal DNA in the Apicomplexa. Microbiol Mol Biol Rev. 1997;61:1–16. https://doi.org/10.1128/mmbr.61.1.1-16.1997.
Léveillé AN, Baneth G, Barta JR. Next generation sequencing from Hepatozoon canis (Apicomplexa: Coccidia: Adeleorina): complete apicoplast genome and multiple mitochondrion-associated sequences. Int J Parasitol. 2019;49:375–87. https://doi.org/10.1016/j.ijpara.2018.12.001.
Leveille AN, Ogedengbe ME, Hafeez MA, Tu HH, Barta JR. The complete mitochondrial genome sequence of Hepatozoon catesbianae (Apicomplexa: Coccidia: Adeleorina), a blood parasite of the Green frog, Lithobates (Formerly Rana) clamitans. J Parasitol. 2014;100:651–6. https://doi.org/10.1645/13-449.1.
. Karadjian G, Chavatte JM, Landau I. Systematic revision of the adeleid haemogregarines, with creation of Bartazoon n. g., reassignment of Hepatozoon argantis Garnham, 1954 to Hemolivia, and molecular data on Hemolivia stellata. Parasite. 2015;22:31. https://doi.org/10.1051/parasite/2015031.
Hadziavdic K, Lekang K, Lanzen A, Jonassen I, Thompson EM, Troedsson C. Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PLoS ONE. 2014;9:e87624. https://doi.org/10.1371/journal.pone.0087624.
Alabí AS, Monti G, Otth C, Sepulveda-García P, Perles L, Machado RZ, et al. Genetic diversity of Hepatozoon in rodents from Chile. Rev Bras Parasitol Vet. 2021;30:e012721. https://doi.org/10.1590/s1984-29612021082.
Gutiérrez-Liberato GA, Lotta-Arévalo IA, Rodríguez-Almonacid CC, Vargas-Ramírez M, Matta NE. Molecular and morphological description of the first Hepatozoon (Apicomplexa: Hepatozoidae) species infecting a neotropical turtle, with an approach to its phylogenetic relationships. Parasitology. 2021;148:747–59. https://doi.org/10.1017/s0031182021000184.
Megía-Palma R, Martínez J, Fitze PS, Cuervo JJ, Belliure J, Jiménez-Robles O, et al. Genetic diversity, phylogenetic position, and co-phylogenetic relationships of Karyolysus, a common blood parasite of lizards in the western Mediterranean. Int J Parasitol. 2023;53:185–96. https://doi.org/10.1016/j.ijpara.2022.12.006.
Bazzano V, Félix ML, Parodi P, Carvalho LA, Nava S, Armúa-Fernández MT, et al. Phylogenetic analysis of Hepatozoon spp (Apicomplexa: Hepatozoidae) infecting Philodryas patagoniensis (Serpentes: Dipsadidae) in Uruguay. Parasitol Res. 2020;119:1093–100. https://doi.org/10.1007/s00436-020-06605-7.
Weck BC, Serpa MCA, Ramos VN, Luz HR, Costa FB, Ramirez DG, et al. Novel genotypes of Hepatozoon spp. in small mammals, Brazil. Parasit Vectors. 2022;15:87. https://doi.org/10.1186/s13071-022-05216-8.
Varela O, Cormenzana-Méndez A, Krapovickas L, Bucher EH. Seasonal diet of the pampas fox (Lycalopex gymnocercus) in the chaco dry woodland Northwestern. Argentina J Mammal. 2008;89:1012–9. https://doi.org/10.1644/07-MAMM-A-125.1.
de Carvalho RF, Passos DC, Lessa LG. Diet variations in Short-tailed opossum Monodelphis domestica (Didelphimorphia, Didelphidae) due to seasonal and intersexual factors. Mastozool Neotrop. 2019;26:340–8. https://doi.org/10.31687/saremMN.19.26.2.0.14.
Mignino J, Martinez S, Luengos Vidal E, Lucherini M. Actualistic taphonomy of pampas fox (Lycalopex gymnocercus) scat-derived bone accumulations from central Argentina: contributions to archaeological and palaeontological studies. Hist Biol. 2023:1-13 https://doi.org/10.1080/08912963.2023.2183858.
Dutra-Vieira FM, Silva MS, Vieira GS, Carvalho AS, Schimming BC. Diet of crab-eating fox (Cerdocyon thous) in two conservation units of the Amazon rainforest. Brazil Br J Biol. 2024;84:e252093. https://doi.org/10.1590/1519-6984.252093.
Farias AA, Kittlein MJ. Small-scale spatial variability in the diet of pampas foxes (Pseudalopex gymnocercus) and human-induced changes in prey base. Ecol Res. 2008;23:543–50. https://doi.org/10.1007/s11284-007-0407-7.
Sloboda M, Kamler M, Bulantová J, Votýpka J, Modrý D. Rodents as intermediate hosts of Hepatozoon ayorgbor (Apicomplexa: Adeleina: Hepatozoidae) from the African ball python, Python regius? Folia Parasitol. 2008;55:13–6. https://doi.org/10.14411/fp.2008.003.
Goebel LGA, Longo GR, Oliveira MA, Fermiano EC, da Silva DJ, Ignácio ÁRA, et al. Registro de depredación de Rhinella marina (Linnaeus, 1758) (Anura: Bufonidae) por Cerdocyon thous (Carnivora: Canidae) usando cámara trampa en Vale do Paraíso, Rondonia Brasil. Mammalogy Notes. 2023;9:362. https://doi.org/10.47603/mano.v9n1.362.
de Castro L, Demoner NM, Magro MR, da Silva L, Azevedo JM, et al. Hepatozoon spp infections in wild rodents in an area of endemic canine hepatozoonosis in southeastern Brazil. Ticks Tick Borne Dis. 2016;7:64. https://doi.org/10.1016/j.ttbdis.2016.04.002.
de Sousa KCM, Fernandes MP, Herrera HM, Benevenute JL, Santos FM, Rocha FL, et al. Molecular detection of Hepatozoon spp in domestic dogs and wild mammals in southern Pantanal, Brazil with implications in the transmission route. Vet Parasitol. 2017;237:37–46. https://doi.org/10.1016/j.vetpar.2017.02.023.
de Andreazzi CS, Rademaker V, Gentile R, Herrera HM, Jansen AM, D’Andrea PS. Population ecology of small rodents and marsupials in a semi-deciduous tropical forest of the southeast Pantanal, Brazil. Zoologia. 2011;28:762–70. https://doi.org/10.1590/S1984-46702011000600009.
Maia JP, Álvares F, Boratyński Z, Brito JC, Leite JV, Harris DJ. Molecular assessment of Hepatozoon (Apicomplexa: Adeleorina) infections in wild canids and rodents from North Africa, with implications for transmission dynamics across taxonomic groups. J Wildl Dis. 2014;50:837–48. https://doi.org/10.7589/2013-10-280.
Criado-Fornelio A, Buling A, Casado N, Gimenez C, Ruas J, Wendt L, et al. Molecular characterization of arthropod-borne hematozoans in wild mammals from Brazil. Venezuela Spain Acta Parasitol. 2009;54:187–93. https://doi.org/10.2478/s11686-009-0031-5.
Lucas MR, da Silva F, Fornazari L. Didelphis albiventris naturally infected with Hepatozoon canis in southeastern Brazil. Ticks Tick Borne Dis. 2017;8:81. https://doi.org/10.1016/j.ttbdis.2017.07.005.
de Azevedo GL, Moraes LA, Figueira Aguiar DC, Tavares Dias HL, Sardinha AS, et al. Genetic diversity of Hepatozoon spp in Hydrochoerus hydrochaeris and Pecari tajacu from eastern Amazon. Ticks Tick Borne Dis. 2018;9:314–8. https://doi.org/10.1016/j.ttbdis.2017.11.005.
Carey JR. Insect biodemography. Annu Rev Entomol. 2001;46:79–110. https://doi.org/10.1146/annurev.ento.46.1.79.
Stumpf MPH. Haplotype diversity and SNP frequency dependence in the description of genetic variation. Eur J Human Genet. 2004;12:469–77. https://doi.org/10.1038/sj.ejhg.5201179.
Baker E, Jensen A, Miller D, Garrett KB, Cleveland CA, Brown J, et al. Hepatozoon spp infection in wild canids in the eastern United States. Parasit Vectors. 2023;16:1–10. https://doi.org/10.1186/s13071-023-05968-x.
Tomé B, Maia JPMC, Harris DJ. Hepatozoon infection prevalence in four snake genera: Influence of diet, prey parasitemia levels, or parasite type? J Parasitol. 2012;98:913–7. https://doi.org/10.1645/ge-3111.1.
Beerntsen BT, James AA, Christensen BM. Genetics of mosquito vector competence. Microbiol Mol Biol. 2000;64:115–37. https://doi.org/10.1128/mmbr.64.1.115-137.2000.
Dantas-Torres F, Chomel BB, Otranto D. Ticks and tick-borne diseases: a one health perspective. Trends Parasitol. 2012;28:437–46. https://doi.org/10.1016/j.pt.2012.07.003.
Medeiros MCI, Hamer GL, Ricklefs RE. Host compatibility rather than vector–host-encounter rate determines the host range of avian Plasmodium parasites. Proc Biol Sci. 2013. https://doi.org/10.1098/rspb.2012.2947.
de la Fuente J, Antunes S, Bonnet S, Cabezas-Cruz A, Domingos AG, Estrada-Peña A, et al. Tick-pathogen interactions and vector competence: Identification of molecular drivers for tick-borne diseases. Front Cell Infect Microbiol. 2017;7:114. https://doi.org/10.3389/fcimb.2017.00114.
Davidson WR, Calpin JP. Hepatozoon griseisciuri infection in gray squirrels of the southeastern United States. J Wildl Dis. 1976;12:72–6. https://doi.org/10.7589/0090-3558-12.1.72.
Simpson VR, Birtles RJ, Bown KJ, Panciera RJ, Butler H, Davison N. Hepatozoon species infection in wild red squirrels (Sciurus vulgaris) on the Isle of Wight. Vet Rec. 2006;159:202–5. https://doi.org/10.1136/vr.159.7.202.
Whiteman NK, Matson KD, Bollmer JL, Parker PG. Disease ecology in the Galpagos Hawk (Buteo galapagoensis): host genetic diversity, parasite load and natural antibodies. Proc Biol Sci. 2006;273:797.
Dick CW, Patterson BD. Against all odds: Explaining high host specificity in dispersal-prone parasites. Int J Parasitol. 2007;37:871–6. https://doi.org/10.1016/j.ijpara.2007.02.004.
Wolf RW, Aragona M, Muñoz-Leal S, Pinto LB, Melo ALT, Braga IA, et al. Novel Babesia and Hepatozoon agents infecting non-volant small mammals in the Brazilian Pantanal, with the first record of the tick Ornithodoros guaporensis in Brazil. Ticks Tick Borne Dis. 2016;7:449–56. https://doi.org/10.1016/j.ttbdis.2016.01.005.
Desser SS. The haemogregarinidae and lankesterellidae Parasitic Protozoa. Elsevier: Academic Press; 1993.
Hoberg EP, Brooks DR. Beyond vicariance: integrating taxon pulses, ecological fitting, and oscillation in evolution and historical biogeography. In: Morand S, Krasnov BR, editors. The biogeography of host-parasite interactions. Oxford: Oxford University Press; 2010. p. 7–20.
Goater TM, Goater CP, Esch GW. Parasite biogeography and phylogeography parasitism: the diversity and ecology of animal parasites. 2nd ed. New York: Cambridge University Press; 2013.
Keesing F, Belden LK, Daszak P, Dobson A, Harvell CD, Holt RD, et al. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature. 2010;468:647–52. https://doi.org/10.1038/nature09575.
Keesing F, Ostfeld RS. Impacts of biodiversity and biodiversity loss on zoonotic diseases. PNAS. 2021;118:e2023540118. https://doi.org/10.1073/pnas.2023540118.
Thompson RCA, Lymbery AJ, Smith A. Parasites, emerging disease and wildlife conservation. Int J Parasitol. 2010;40:1163–70. https://doi.org/10.1016/j.ijpara.2010.04.009.
Allen KE, Li Y, Kaltenboeck B, Johnson EM, Reichard MV, Panciera RJ, et al. Diversity of Hepatozoon species in naturally infected dogs in the southern United States. Vet Parasitol. 2008;154:220–5. https://doi.org/10.1016/j.vetpar.2008.03.027.
Carvalho L, Félix ML, Bazzano V, da Costa A, Armúa-Fernández MT, Muñoz-Leal S, et al. An Hepatozoon americanum-like protozoan in crab-eating (Cerdocyon thous) and grey pampean (Lycalopex gymnocercus) foxes from Uruguay. Parasitol Res. 2021;120:3587–93. https://doi.org/10.1007/s00436-021-07305-6.
Pedó E, Tomazzoni AC, Hartz SM, Christoff AU. Diet of crab-eating fox, Cerdocyon thous (Linnaeus) (Carnivora, Canidae), in a suburban area of southern Brazil. Rev Bras Zool. 2006;23:637–41. https://doi.org/10.1590/S0101-81752006000300005.
Sábato MAL, de Melo LFB, Magni EMV, Young RJ, Coelho CM. A note on the effect of the full moon on the activity of wild maned wolves, Chrysocyon Brachyurus. Behav Process. 2006;73:228–30. https://doi.org/10.1016/j.beproc.2006.05.012.
Weckel M, Giuliano W, Silver S. Jaguar (Panthera onca) feeding ecology: distribution of predator and prey through time and space. J Zool. 2006;270:25–30. https://doi.org/10.1111/j.1469-7998.2006.00106.x.
Gilbert-Norton LB, Leaver LA, Shivik JA. The effect of randomly altering the time and location of feeding on the behaviour of captive coyotes (Canis latrans). Appl Anim Behav Sci. 2009;120:179–85. https://doi.org/10.1016/j.applanim.2009.06.007.
Wallach AD, Dekker AH, Lurgi M, Montoya JM, Fordham DA, Ritchie EG. Trophic cascades in 3D: network analysis reveals how apex predators structure ecosystems. Methods Ecol Evol. 2017;8:135–42. https://doi.org/10.1111/2041-210X.12663.
Marinho PH, Bezerra D, Antongiovanni M, Fonseca CR, Venticinque EM. Activity patterns of the threatened northern tiger cat Leopardus tigrinus and its potential prey in a Brazilian dry tropical forest. Mamm Biol. 2018;89:30–6. https://doi.org/10.1016/j.mambio.2017.12.004.
Hody AW, Moreno R, Meyer NFV, Pacifici K, Kays R. Canid collision—expanding populations of coyotes (Canis latrans) and crab-eating foxes (Cerdocyon thous) meet up in Panama. J Mammal. 2019;100:1819–30. https://doi.org/10.1093/jmammal/gyz158.
Szynwelski BE, Kretschmer R, Matzenbacher CA, Ferrari F, Alievi MM, de Freitas TRO. Hybridization in canids—A case study of Pampas fox (Lycalopex gymnocercus) and domestic dog (Canis lupus familiaris) hybrid. Animals. 2023;13:2505. https://doi.org/10.3390/ani13152505.
Otranto D, Cantacessi C, Pfeffer M, Dantas-Torres F, Brianti E, Deplazes P, et al. The role of wild canids and felids in spreading parasites to dogs and cats in Europe: Part I: Protozoa and tick-borne agents. Vet Parasitol. 2015;213:12–23. https://doi.org/10.1016/j.vetpar.2015.04.022.
Davis DS, Robinson RM, Craig TM. Naturally occurring hepatozoonosis in a Coyote. J Wildl Dis. 1978;14:244–6. https://doi.org/10.7589/0090-3558-14.2.244.
Mercer SH, Jones LP, Rappole JH, Twedt D, Lack LL, Craig TM. Hepatozoon sp in wild carnivores in Texas. J Wildl Dis. 1988;24:574–6. https://doi.org/10.7589/0090-3558-24.3.574.
Kocan AA, Cummings CA, Panciera RJ, Mathew JS, Ewing SA, Barker RW. Naturally occurring and experimentally transmitted Hepatozoon americanum in Coyotes from Oklahoma. J Wildl Dis. 2000;36:149–53. https://doi.org/10.7589/0090-3558-36.1.149.
Starkey LA, Panciera RJ, Paras K, Allen KE, Reiskind MH, Reichard MV, et al. Genetic diversity of Hepatozoon spp in Coyotes from the South-Central United States. J Parasitol. 2013;99:375–8. https://doi.org/10.1645/ge-3104.1.
Alencar NX, Kohayagawa A, Santarém VA. Hepatozoon canis infection of wild carnivores in Brazil. Vet Parasitol. 1997;70:279–82. https://doi.org/10.1016/s0304-4017(96)01119-3.
André MR, Adania CH, Teixeira RHF, Vargas GH, Falcade M, Sousa L, et al. Molecular detection of Hepatozoon spp in Brazilian and exotic wild carnivores. Vet Parasitol. 2010;173:134–8. https://doi.org/10.1016/j.vetpar.2010.06.014.
Santos EM de S, Cunha RC de SC da, Farias MPO, Fonseca CF da, Oliveira JB de, Carvalho RRN de, et al. View of ticks and fleas in Crab-eating fox (Cerdocyon thous) of Pernambuco state, Brazil. Braz J Vet Res Anim Sci São Paulo. 2016;53:295–9. https://doi.org/10.11606/issn.1678-4456.bjvras.2016.110634
Han BA, Schmidt JP, Bowden SE, Drake JM. Rodent reservoirs of future zoonotic diseases. Proc Natl Acad Sci USA. 2015;112:7039–44. https://doi.org/10.1073/pnas.1501598112.
Pagel MD, May RM, Collie AR. Ecological aspects of the geographical distribution and diversity of mammalian species. Am Nat. 1991;137:791–815.
Hudson PJ, Dobson AP, Lafferty KD. Is a healthy ecosystem one that is rich in parasites? Trends Ecol Evol. 2006;21:381–5. https://doi.org/10.1016/j.tree.2006.04.007.
Mitchell GF. Co-evolution of parasites and adaptive immune responses. Immunol Today. 1991;7:2–5. https://doi.org/10.1016/s0167-5699(05)80002-7.
Metzger B, dos Santos K, Paduan AS, Rubini TG, de Oliveira C, Pereira LH. The first report of Hepatozoon sp (Apicomplexa: Hepatozoidae) in neotropical felids from Brazil. Vet Parasitol. 2008;152:33. https://doi.org/10.1016/j.vetpar.2007.12.006.
Kubo M, Miyoshi N, Yasuda N. Hepatozoonosis in two species of Japanese wild cat. J Vet Med Sci. 2006;68:833–7. https://doi.org/10.1292/jvms.68.833.
Rojas A, Rojas D, Montenegro V, Gutiérrez R, Yasur-Landau D, Baneth G. Vector-borne pathogens in dogs from Costa Rica: first molecular description of Babesia vogeli and Hepatozoon canis infections with a high prevalence of monocytic ehrlichiosis and the manifestations of co-infection. Vet Parasitol. 2014;199:121–8. https://doi.org/10.1016/j.vetpar.2013.10.027.
Alić A, Šupić J, Goletić T, Rešidbegović E, Lutvikadić I, Hodžić A. A unique case of fatal coinfection caused by Leptospira spp and Hepatozoon canis in a Red fox cub (Vulpes vulpes). Pathogens. 2021;11:11. https://doi.org/10.3390/pathogens11010011.
Pessôa SB, de Biasi P, Puorto G. Transferência do Hepatozoon tupinambis, parasita do lagarto Tupinambis teguixin, para a serpente cascavel (Crotalus durissus terrificus), por intermédio do mosquito Culex fatigans. Mem Inst Oswaldo Cruz. 1974;72:295–9. https://doi.org/10.1590/S0074-02761974000200013.
Wozniak EJ, Telford SR. The fate of Hepatozoon species naturally infecting Florida black racers and watersnakes in potential mosquito and soft tick vectors, and histological evidence of pathogenicity in unnatural host species. Int J Parasitol. 1991;21:511–6. https://doi.org/10.1016/0020-7519(91)90055-c.
Simonato G, Franco V, Salvatore G, Manzocchi S, Dotto G, Morelli S, et al. First autochthonous clinical case of Hepatozoon silvestris in a domestic cat in Italy with unusual presentation. Parasit Vectors. 2022;15:1–7. https://doi.org/10.1186/s13071-022-05534-x.
Garrett JJ, Kocan AA, Reichard MV, Panciera RJ, Bahr RJ, Ewing SA. Experimental infection of adult and juvenile coyotes with domestic dog and wild coyote isolates of Hepatozoon americanum (Apicomplexa: Adeleorina). J Wildl Dis. 2005;41:588–92. https://doi.org/10.7589/0090-3558-41.3.588.
East ML, Wibbelt G, Lieckfeldt D, Ludwig A, Goller K, Wilhelm K, et al. A Hepatozoon species genetically distinct from H canis infecting spotted hyenas in the Serengeti ecosystem Tanzania. J Wildl Dis. 2008;44:45–52. https://doi.org/10.7589/0090-3558-44.1.45.
Basson PA, McCully M, Bigalke RD, van Niekerk JW. Observations on a Hepatozoon-like parasite in the Impala. Vet Med Ass. 1967;38:12–4.
Craig TM. Tissue-Inhabiting protozoans: Hepatozoon spp. and hepatozoonosis. In: Samuel WM, Margo JP, Kocan AA, editors. Iowa State University Press; 2001. p. 462–8. https://doi.org/10.1002/9780470377000.ch17b.
Miller WW. Hepatozoon perniciosum a haemogregarine pathogenic for white rats; with a description of the sexual cycle in the intermediate host, a mite (Lelaps echidninus). 46th ed. Washington: Government Printing Office; 1908.
Porter A. Leucocytozoön musculi, sp n, a parasitic protozoon from the blood of white mice. Proc Zool Soc Lond. 1908;78:703–16. https://doi.org/10.1111/j.1469-7998.1908.tb07403.x.
Hoogstraal H. The life cycle and incidence of Hepatozoon balfouri (Laveran, 1905) in Egyptian jerboas (Jaculus spp) and mites (Haemolaelaps aegyptius Keegan, 1956). J Protozool. 1961;8:231–48. https://doi.org/10.1111/j.1550-7408.1961.tb01209.x.
Krampitz HE. Development of Hepatozoon erhardovae krampitz, 1964 (Protozoa: Haemogregarinidae) in experimental mammalian and arthropod hosts. II. Sexual development in fleas and sporozoite indices in xenodiagnosis. Trans R Soc Trop Med Hyg. 1981;75:155–7. https://doi.org/10.1016/0035-9203(81)90052-3.
Johnson EM, Allen KE, Breshears MA, Panciera RJ, Little SE, Ewing SA. Experimental transmission of Hepatozoon americanum to rodents. Vet Parasitol. 2008;151:164–9. https://doi.org/10.1016/j.vetpar.2007.10.017.
Ferrari G, Girardi M, Cagnacci F, Devineau O, Tagliapietra V. First record of Hepatozoon spp in alpine wild rodents Implications and perspectives for transmission dynamics across the food web. Microorganisms. 2022;10:712. https://doi.org/10.3390/microorganisms10040712.
Dahmana H, Granjon L, Diagne C, Davoust B, Fenollar F, Mediannikov O. Rodents as hosts of pathogens and related zoonotic disease risk. Pathogens. 2020;9:202. https://doi.org/10.3390/pathogens9030202.
IUCN. IUCN Red List of Threatened Species. 2023. https://www.iucnredlist.org/about/citationinfo
Dantas-Torres F. Climate change, biodiversity, ticks and tick-borne diseases: the butterfly effect. Int J Parasitol Parasites Wildl. 2015;4:452–61. https://doi.org/10.1016/j.ijppaw.2015.07.001.
Fayer R, Dubey JP, Lindsay DS. Zoonotic protozoa: From land to sea. Trends Parasitol. 2004;20:531–6. https://doi.org/10.1016/j.pt.2004.08.008.
Huggins LG, Koehler AV, Ng-Nguyen D, Wilcox S, Schunack B, Inpankaew T, et al. A novel metabarcoding diagnostic tool to explore protozoan haemoparasite diversity in mammals: a proof-of-concept study using canines from the tropics. Sci Rep. 2019;9:1–10. https://doi.org/10.1038/s41598-019-49118-9.
Acknowledgements
We dedicate this work to the memory of Daniel González Acuña, an extraordinary person who contributed significantly to studying parasites and wildlife conservation in Chile. He always demonstrated unwavering confidence towards our work, and we sincerely thank him for his trust and guidance.
Funding
This study was funded by the “Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT)” No. 11220177 and by the ANID BECAS/Scholarship Program/DOCTORADO NACIONAL/2019–21190078 and 2020–21200182. Funders had no role in the study design, data collection, analysis, preparation of the manuscript, and decision to publish.
Author information
Authors and Affiliations
Contributions
RT and AS conceived and designed the study and analyzed the data. RT coordinated the preparation and drafted the manuscript. LS-A, JFQ-G, LM, JEU, and SM-L verified data analyses and commented on initial versions of the manuscript. JEU and SM-L critically reviewed the manuscript for publication. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the review was conducted in the absence of any commercial or financial relationships that could be construed as a potential competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Wild mammals with reported Hepatozoon spp. in the Americas.Table S2. Primer details and thermal conditions used for the molecular diagnosis and genotyping of Hepatozoon in wild mammals of the Americas.Table S3. 18S rDNA Hepatozoon sequences (≥387 bp) listed in GenBank database until December 2022, flanking one or more of the nine SSU rRNA regions.Table S4. Genetic variants of Hepatozoon spp. circulating among wild mammals in the Americas.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Thomas, R., Santodomingo, A., Saboya-Acosta, L. et al. Hepatozoon (Eucoccidiorida: Hepatozoidae) in wild mammals of the Americas: a systematic review. Parasites Vectors 17, 108 (2024). https://doi.org/10.1186/s13071-024-06154-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-024-06154-3