
Abstract
Background
Bats (Mammalia: Chiroptera) serve as natural reservoirs for many zoonotic pathogens worldwide, including vector-borne pathogens. However, bat-associated parasitic arthropods and their microbiota are thus far not thoroughly described in many regions across the globe, nor is their role in the spillover of pathogens to other vertebrate species well understood. Basic epidemiological research is needed to disentangle the complex ecological interactions among bats, their specific ectoparasites and microorganisms they harbor. Some countries, such as Ukraine, are particularly data-deficient in this respect as the ectoparasitic fauna is poorly documented there and has never been screened for the presence of medically important microorganisms. Therefore, the aims of this study were to provide first data on this topic.
Methods
A total of 239 arthropod specimens were collected from bats. They belonged to several major groups of external parasites, including soft ticks, fleas, and nycteribiid flies from six chiropteran species in Northeastern Ukraine. The ectoparasites were individually screened for the presence of DNA of Rickettsia spp., Anaplasma/Ehrlichia spp., Bartonella spp., Borrelia spp., and Babesia spp. with conventional PCRs. Positive samples were amplified at several loci, sequenced for species identification, and subjected to phylogenetic analysis.
Results
Rickettsia DNA was detected exclusively in specimens of the soft tick, Carios vespertilionis (7 out of 43 or 16.3%). Sequencing and phylogenetic analysis revealed high similarity to sequences from Rickettsia parkeri and several other Rickettsia species. Bacteria from the family Anaplasmataceae were detected in all groups of the ectoparasites (51%, 122/239 samples), belonging to the genera Anaplasma, Ehrlichia, and Wolbachia. The detection of Bartonella spp. was successful only in fleas (Nycteridopsylla eusarca) and bat flies (Nycteribia koleantii, N. pedicularia), representing 12.1% (29/239) of the collected ectoparasites. No DNA of Babesia or Borrelia species was identified in the samples.
Conclusions
We report for the first time in Ukraine the molecular detection of several bacterial agents in bat ectoparasites collected from six species of bats. The data presented extend the knowledge on the distribution of ectoparasite species in bats and their involvement in potentially circulating agents pathogenic for humans and vertebrate animals.
Graphical Abstract

Similar content being viewed by others

Bats (Mammalia: Chiroptera) represent the second-most diverse order of mammals after rodents [1]. The multitude of their ecological interactions with other animals and their shared physical environment puts bats in close contact with a large variety of viruses, bacteria, fungi, and parasites [2, 3]. Currently, bat microbiota are poorly documented and understood although during the last 2 decades bats received increased research attention as a natural source of well-known and potentially zoonotic pathogens, especially viruses [4]. While the role of bats in circulation and spillover of zoonotic viruses such as lyssaviruses, filoviruses, henipaviruses, and coronaviruses is relatively well established [5, 6], limited knowledge exists regarding their role as reservoirs for arthropod-borne pathogens, which represent a substantial proportion of zoonoses worldwide [7].
Previous research mostly focused on detection of vector-borne bacteria from the genera Bartonella, Rickettsia, and Borrelia in samples of bat tissues, excreta, and their ectoparasites [8,9,10,11,12]. More research efforts are needed to determine the relevance of these findings to the circulation of zoonotic vector-borne pathogens or their significance for human and animal health.
Many areas, especially in Eastern Europe, are thus far lacking this type of research. For example, very little is known about bat ectoparasites in Ukraine, and the vector-borne bacteria they might carry have never been surveyed in the country [13,14,15]. The Kharkiv oblast (synonym with region), Northeastern Ukraine, is the most intensively studied region in the country, with 20 years of bat-related stationary research and monitoring activities, where the bat diversity reaches 15–16 species [16]. However, bat ectoparasites have thus far not been the main research focus, and there is only one publication dedicated to bat ectoparasites in the region, which was published in a local journal [15]. The authors identified eight species of ectoparasites, including mesostigmatid mites (genera Spinturnix: 3 spp.; Macronyssus: 1 sp.; Steatonyssus: 1 sp.), fleas (genus Ischnopsyllus: 2 spp.), and a bat fly (Nycteribia koleantii) occurring in five bat taxa. Moreover, they examined only a small number of opportunistically collected samples (total of 142 specimens), and the ectoparasites were not screened for microorganisms [15]. Thus, our present study is aimed at filling the gap by conducting molecular screening of selected vector-borne microorganisms in recent samples of ectoparasitic arthropods collected from bats in Kharkiv oblast.
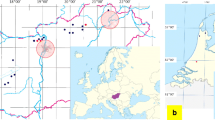
The field survey was conducted in five localities divided among the three habitat types: urban (November 2019), natural (August–September 2019), and rural (July 2020), representing main types of land use in the region (Additional file 1: Table S1). Bats were mist-netted in autumn swarming and wintering sites or hand captured from two breeding colonies that roosted in private houses in a countryside area. Each individual was identified to the species level, its sex, age, and reproductive status were noted, and forearm length and body mass were measured (for details see: [17]). After taking the measurements from a bat, the body, coat, ears, wing, and uropatagium membranes of each animal were examined for ectoparasites in daylight or using a headlamp. All detected arthropods were collected with tweezers and cotton swabs and placed in individually labeled tubes with 96% ethanol. From the total number of collected ectoparasites (~ 1000), so far 239 specimens have been morphologically identified to the species level using the Nikon SMZ800 stereomicroscope and taxonomic keys [18,19,20]. After the morphological identification, these specimens were transferred to 70% ethanol and sent to the Institute of Infectology, Friedrich-Loeffler-Institut, Germany, for further molecular screening.
The samples were processed individually, each ectoparasite being homogenized in sterile phosphate-buffered saline (PBS) with steel beads using the Tissue Lyser II (Qiagen, Hilden, Germany). The DNA extraction was performed from 100 μl aliquots using NucleoMag® VET kit (Macherey–Nagel, Düren, Germany) and the King Fisher® Flex Purification system (ThermoFisher, Darmstadt, Germany), according to the manufacturer’s instructions. Total DNA was eluted in 100 μl elution buffer and then stored at −80 °C until further analysis.
Four species of ectoparasites collected from different bat species (Table 1) were PCR screened for the presence of Babesia spp., Rickettsia spp., Bartonella spp., and Anaplasma/Ehrlichia spp., while only Carios vespertilionis ticks were additionally screened for DNA of Borrelia spp. The screening was done using specific primers listed in Additional file 1: Table S2 and PCR conditions described in the publications cited therein. The reaction products from samples with successfully amplified target genes were purified with NucleoSEQ® kit (Mackerey Nagel, Düren, Germany) following manufacturer’s instructions and Sanger sequenced in the Laboratory for Applied Bioinformatics and Sequencing of Viral Genomes and Transcriptomes, Institute of Diagnostic Virology, Friedrich-Loeffler-Institut.
Among the tested samples, DNA of Rickettsia spp. was identified in 2.9% (7/239) of ectoparasites (Table 2). All positive samples were C. vespertilionis ticks, six specimens collected from Pipistrellus pygmaeus and one from P. kuhlii bat species. Sequence analysis of the rickettsial gltA gene did not successfully differentiate the species, with all detected sequences showing 100% similarity to several Rickettsia spp.: Rickettsia parkeri (GenBank access. no.: MK814825), R. africae (MH938655), R. sibirica (KU310587), or uncultured Rickettsia sp. (MG228263). PCR based on the ompA gene followed by sequencing of positive C. vespertilionis ticks for Rickettsia indicated 100% similarity of all samples with R. parkeri (MK962698) and Rickettsia sp. (KX137902).
The BLASTn analysis for the Rickettsia ompB sequences also showed 100% similarity of the samples from this study with R. parkeri (CP040325), uncultured Rickettsia sp. (MK405417), and Rickettsia sp. (AF123720).
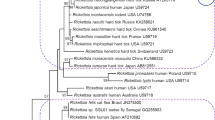
The phylogenetic analysis (Mega X [21]) was done using the gltA, ompA, and ompB concatenated Rickettsia sequences and sequences from representative Rickettsia species available in GenBank. The analysis demonstrates that all Rickettsia sequences detected in C. vespertilionis in this study are phylogenetically closely related to R. parkeri and R. africae (Fig. 1a).

Phylogenetic analysis of sequences within the Rickettsiales order. a Phylogenetic tree of Rickettsia sequences based on concatenated gltA, ompA, and ompB genes using the maximum likelihood analysis and Tamura three parameter with discrete gamma distribution; b phylogenetic analysis of Ehrlichia detected in C. vespertilionis based on the 16S rRNA loci, using the maximum likelihood analysis and Hasegawa-Kishino-Yano; c phylogenetic analysis of Anaplasma detected in C. vespertilionis based on the 16S rRNA loci, using the maximum likelihood analysis and Tamura three parameter. Bootstrap values are indicated at the nodes. The red dot preceding the sample names indicates the sequences obtained in this study
The detection of R. parkeri-like sequences in bat-associated soft ticks from Europe represents a noteworthy finding as this alphaproteobacterium is known to be associated primarily with hard ticks in the genus Amblyomma occurring in the Americas [22]. However, a recent study ostensibly identified R. parkeri sequences in tissues of Pipistrellus pipistrellus bats from China [23], which would significantly expand the geography and host range of the species. While some of the strains in R. parkeri sensu lato complex are well-established human pathogens, little is known about their natural reservoir hosts [22].
Moreover, neither pathogenicity in vertebrates nor transmissibility by argasid ticks is known for the R. parkeri-like species presently detected in Eurasia. Further research efforts, therefore, should focus on isolating and establishing the identity of the bacterium as well as elucidating its enzootic cycle. This would also be of public health relevance, keeping in mind that C. vespertilionis may occasionally bite humans in situations when their bat hosts are no longer available [24].
Regarding DNA of Anaplasma/Ehrlichia spp., 51% (122/239) of the samples tested positive: 4.7% (2/43) of C. vespertilionis, 56% (56/100) of Nycteridopsylla eusarca, 69.2% (54/78) of Nycteribia kolenatii, and 55.6% (10/18) of N. pedicularia (Table 2). The two C. vespertilionis samples positive for Anaplasma/Ehrlichia spp. after the initial 16S rRNA PCR were further amplified by hemi-nested PCR targeting a 16S rRNA fragment then sequenced. The DNA sequence analysis showed that one sequence was 98.5% similar to Candidatus Ehrlichia shimanensis (AB074459) while the other sample had 99.7% similarity to uncultured Anaplasma sp. clone Erz1600 (MT601947). The phylogenetic analysis based on the 16S rRNA partial sequence indicates that the Ehrlichia sequence detected in C. vespertilionis clusters in a clade that includes Candidatus Ehrlichia shimanensis, uncultured Ehrlichia: E. minasensis or E. canis (Fig. 1b). The phylogenetic tree based on the 16S rRNA partial sequence shows that the Anaplasma sequence was detected in C. vespertilionis clusters in a clade that includes uncultured Anaplasma and Anaplasma ovis (Fig. 1c). Eight N. eusarca samples positive for Anaplasma/Ehrlichia spp. were sequenced, two having 100% identity to uncultured bacterium clone layman_j06 (DQ980970), and six sequences were 99.6–100% similar to Wolbachia endosymbiont (MH618381). Four N. pedicularia and six N. kolenatii flies positive for Anaplasma/Ehlichia spp. were also sequenced, having 99.3–100% similarity to Wolbachia endosymbiont (MH618380).
While zoonotic Anaplasmataceae such as Anaplasma phagocytophilum have been previously reported from insectivorous bats and their ticks in Europe [12], it remains unclear whether bats play any role in the epidemiology of granulocytic anaplasmosis. Most of the hits in the genera Ehrlichia and Anaplasma from the present study are clustering with poorly characterized species (e.g. E. minasensis) or with microorganisms not commonly associated with bats, such as E. canis or A. ovis. This implies that either a much broader range of vertebrate reservoirs and arthropod vectors support circulation of these pathogens in nature or, more parsimoniously, that the molecular markers selected for species identification have poor discriminatory capacity at this level. On the other hand, the finding of such endosymbionts as Wolbachia sp. is not surprising as these bacteria are almost universally present in many groups of arthropods and in filarial nematodes and have been reported previously in multiple bat fly species [25].
The detection of Bartonella spp. was successful in 12.1% (29/239) ectoparasites collected from bats. All bat ticks tested negative, while N. eusarca, N. kolenatii, and N. pedicularia showed prevalence rates that varied from 7 to 21.8% and 27.8%, respectively (Table 2). The analysis based on the gltA gene of DNA sequences from N. eusarca revealed 99–100% similarity to uncultured Bartonella sp. clone 198T155 (MK140218) detected in C. vespertilionis from The Netherlands (n = 3 isolates) and uncultured Bartonella sp. isolate M451 (AJ871615) found in the blood of Nyctalus noctula from UK (n = 4 isolates). Eight N. kolenatii- and two N. pedicularia-positive samples for Bartonella 16S-23S rRNA were also sequenced. Sequence analysis indicated that three sequences from N. kolenatii had 94.5% similarity to Bartonella sp. strain 44601 (MF288119) obtained from Myotis blythii, and two sequences matched 98.7% and 99%, respectively, to Bartonella sp. strain 44718 (MF288128) from Pipistrellus pygmaeus. The other five sequences (three from N. kolenatii and two from N. pedicularia) had 96.7–97.4% similarity to uncultured Bartonella sp. clone 137 (KX420735) found in Rhinolophus ferrumequinum bat species.
Bartonella gltA and 16S-23S rRNA sequences were further used to construct phylogenetic trees (Fig. 2a, b). The phylogenetic analysis based on the gltA partial sequence shows that three Bartonella sequences detected in N. eusarca cluster in a clade that includes uncultured Bartonella (MK140218) and Bartonella washoensis (AF050108). The additional four obtained sequences cluster in a separate clade. The analysis based on the partial 16S-23S rRNA sequence shows that Bartonella sequences detected in N. kolenatii and N. pedicularia cluster close to uncultured Bartonella (KX420735) and Bartonella sp. (MF288119 and MF288128). All sequence data and accession nos. are shown in Additional file 1: Table S3.

Phylogenetic analysis of Bartonella spp. isolates obtained in this study. a Phylogenetic tree based on citrate synthase (gltA) partial gene using the maximum likelihood analysis and Tamura three-parameter model with a discrete gamma distribution; b phylogenetic analysis of Bartonella spp. based on 16S-23S rRNA sequence using the maximum likelihood analysis and Tamura three parameter with a discrete gamma distribution. Bootstrap values are indicated at the nodes. The red dots highlight the sequences of this study. Brucella abortus (X95889) was used as outgroup
Various Bartonella sequences have been detected in many bat species and in their ectoparasites (fleas, ticks, and flies) across the world [12, 26]. While some of those findings are thought to represent zoonotic bacteria [27], others cluster with sequences detected only in bats, bats and Nycteribiidae flies, or solely in bat flies, with no known vertebrate host association [28, 29]. It has been hypothesized that pathogenic bartonellae evolved from insect-specific ancestors through their association with hematophagous vectors, which allowed them to adapt to mammalian blood [30]. While currently pathogenic Bartonella spp. are believed to be highly host/vector specific, the remarkable diversity of sequences belonging to this genus found in bats and their ectoparasites suggests the ancient nature and evolutionary importance of this association [31].
All ectoparasite samples were negative for Babesia spp., while only C. vespertilionis were screened for Borrelia spp. and were positive in 4.7% (2/43) using the 16S-23S IGS specific primers [32]. The following sequencing attempts for the locus were unsuccessful, suggesting non-specific amplification and leaving the samples without further identification.
This study offers first glimpses on the microbial diversity found in ectoparasites collected from several species of insectivorous bats in Northeast Ukraine. Our research effort creates the impetus for disentangling the vector-host-pathogen interactions among bats and their ectoparasites in an understudied part of Europe. Further studies employing larger sample sizes, greater diversity of the host and parasite species, and variable methods, including next generation sequencing, should reveal a complete and more complex picture. Given the globally changing patterns of bat distribution, their increasing proximity to humans, and the high rates of the infectious disease emergence in wildlife, domestic animals, and human populations, this basic research is important from a public health perspective as well as for conservation biology.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. Newly generated sequences used for the phylogenetic analysis were submitted to the GenBank database. All sequence data and accession nos. are shown in Table S3.
References
Burgin CJ, Colella JP, Kahn PL, Upham NS. How many species of mammals are there? J Mammal. 2018;99:1–14.
Ingala MR, Simmons NB, Perkins SL, McMahon K. Bats are an untapped system for understanding microbiome evolution in mammals. mSphere. 2018;3:e00397-18. https://doi.org/10.1128/mSphere.00397-18.
Farina LL, Lankton JS. Chiroptera. In: Terio KA, McAloose D, St. leger J, editors. Pathology of wildlife and zoo animals. Cambridge: Academic Press; 2018. p. 607–33.
Van Brussel K, Holmes EC. Zoonotic disease and virome diversity in bats. Curr Opin Virol. 2022;52:192–202. https://doi.org/10.1016/j.coviro.2021.12.008.
Klein A, Calvelage S, Schlottau K, Hoffmann B, Eggerbauer E, Müller T, et al. Retrospective enhanced bat lyssavirus surveillance in Germany between 2018–2020. Viruses. 2021;13:1538.
Fagre AC, Kading RC. Can bats serve as reservoirs for arboviruses? Viruses. 2019;11:215.
Loh EH, Zambrana-Torrelio C, Olival KJ, Bogich TL, Johnson CK, Mazet JAK, et al. Targeting transmission pathways for emerging zoonotic disease surveillance and control. Vector Borne Zoonotic Dis. 2015;15:432–7. https://doi.org/10.1089/vbz.2013.1563.
Dietrich M, Kearney T, Seamark EC, Markotter W. The excreted microbiota of bats: evidence of niche specialisation based on multiple body habitats. FEMS Microbiol Lett. 2017;364:fnw284.
Sándor AD, Földvári M, Krawczyk AI, Sprong H, Corduneanu A, Barti L, et al. Eco-epidemiology of novel Bartonella genotypes from parasitic flies of insectivorous bats. Microb Ecol. 2018;76:1076–88.
Evans NJ, Bown K, Timofte D, Simpson VR, Birtles RJ. Fatal borreliosis in bat caused by relapsing fever spirochete, United Kingdom. Emerg Infect Dis. 2009;15:1331.
Jaenson TG, Wilhelmsson P. First Record of a Suspected Human-Pathogenic Borrelia Species in Populations of the Bat Tick Carios vespertilionis in Sweden. Microorganisms. 2021;9:1100.
Hornok S, Szőke K, Meli ML, Sándor AD, Görföl T, Estók P, et al. Molecular detection of vector-borne bacteria in bat ticks (Acari: Ixodidae, Argasidae) from eight countries of the Old and New Worlds. Parasit Vectors. 2019;12:1–7.
Bobkova O. Mites and ticks (Acari) as bats’(Chiroptera) ectoparasites of eastern part of Ukraine. Vestn Zool. 2005;39:73–8.
Bobkova O. Distribution of Ticks (Ixodoidea, Parasitiformes)–Ectoparasites of Bats (Chiroptera) in Ukraine. Vestn Zool. 2003;37:23–8.
Naglov VA, Tkach GE. 2002. Fauna of ectoparasites of bats in Eastern Ukraine. Plecotus et al. 120–3.
Vlaschenko A, Kravchenko K, Yatsiuk Y, Hukov V, Kramer-Schadt S, Radchuk V. Bat assemblages are shaped by land cover types and forest age: a case study from Eastern Ukraine. Forests. 2022;13:1732.
Kravchenko K, Vlaschenko A, Prylutska A, Rodenko O, Hukov V, Shuvaev V. Year-round monitoring of bat records in an urban area: Kharkiv (NE Ukraine), 2013, as a case study. Turk J Zool. 2017;41:530–48.
Theodor O, Moscona A. On the bat parasites in Palestine I. Nycteribiidae, Streblidae, Hemiptera Siphonaptera. Parasitology. 1954;44:157–245.
Medvedev S. Fleas of the family Ischnopsyllidae (Siphonaptera) of the fauna of Russia and adjacent countries. Entomol Rev. 1996;76:480–93.
Filippova N. 1966. Argasid ticks (Argasidae). Fauna SSSR. Paukoobraznye 4. vol. 4. Moscow: Nauka;
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9. https://doi.org/10.1093/molbev/msy096.
Allerdice ME, Paddock CD, Hecht JA, Goddard J, Karpathy SE. Phylogenetic differentiation of Rickettsia parkeri reveals broad dispersal and distinct clustering within North American strains. Microbiol Spectr. 2021;9:e01417-e1421.
Zhao S, Yang M, Liu G, Hornok S, Zhao S, Sang C, et al. Rickettsiae in the common pipistrelle Pipistrellus pipistrellus (Chiroptera: Vespertilionidae) and the bat soft tick Argas vespertilionis (Ixodida: Argasidae). Parasit Vectors. 2020;13:10. https://doi.org/10.1186/s13071-020-3885-x.
Jaenson TG, TäLleklint L, Lundqvist L, Olsen B, Chirico J, Mejlon H. Geographical distribution, host associations, and vector roles of ticks (Acari: Ixodidae, Argasidae) in Sweden. J Med Entomol. 1994;31:240–56.
Wilkinson DA, Duron O, Cordonin C, Gomard Y, Ramasindrazana B, Mavingui P, et al. The Bacteriome of bat flies (Nycteribiidae) from the Malagasy region: a community shaped by host ecology, bacterial transmission mode, and host-vector specificity. Appl Environ Microbiol. 2016;82:1778–88. https://doi.org/10.1128/AEM.03505-15.
Reeves WK, Rogers TE, Durden LA, Dasch GA. Association of Bartonella with the fleas (Siphonaptera) of rodents and bats using molecular techniques. J Vector Ecol. 2007;32:118–22.
Veikkolainen V, Vesterinen EJ, Lilley TM, Pulliainen AT. Bats as reservoir hosts of human bacterial pathogen Bartonella mayotimonensis. Emerg Infect Dis. 2014;20:960–7. https://doi.org/10.3201/eid2006.130956.
Qiu Y, Kajihara M, Nakao R, Mulenga E, Harima H, Hang’ombe BM, et al. Isolation of Candidatus Bartonella rousetti and other bat-associated Bartonellae from bats and their flies in Zambia. Pathogens. 2020;9:469.
Nabeshima K, Sato S, Kabeya H, Komine N, Nanashima R, Takano A, et al. Detection and phylogenetic analysis of Bartonella species from bat flies on eastern bent-wing bats (Miniopterus fuliginosus) in Japan. Comp Immunol Microbiol Infect Dis. 2020;73:101570. https://doi.org/10.1016/j.cimid.2020.101570.
Segers FH, Kešnerová L, Kosoy M, Engel P. Genomic changes associated with the evolutionary transition of an insect gut symbiont into a blood-borne pathogen. ISME J. 2017;11:1232–44. https://doi.org/10.1038/ismej.2016.201.
Lei BR, Olival KJ. Contrasting patterns in mammal-bacteria coevolution: Bartonella and Leptospira in bats and rodents. PLoS Negl Trop Dis. 2014;8:e2738. https://doi.org/10.1371/journal.pntd.0002738.
Bunikis J, Garpmo U, Tsao J, Berglund J, Fish D, Barbour AG. Sequence typing reveals extensive strain diversity of the Lyme borreliosis agents Borrelia burgdorferi in North America and Borrelia afzelii in Europe. Microbiology. 2004;150:1741–55. https://doi.org/10.1099/mic.0.26944-0.
Acknowledgements
The authors dedicate this work to the truth, which will never die in a free world with free and free-thinking people. Furthermore, the authors thank Dr. Alona Prylutska, Oleksii Parfilov, Vitaliy Hukov, Marharyta Moiseienko, and other employees and volunteers of The Bat Rehabilitation Center of Feldman Ecopark for field and laboratory assistance in this study. A special thank you goes to Natalie Shanyuk, illustrator of Bat Rehabilitation Center, Kharkiv, Ukraine, for providing the visual abstract to this article.
Funding
This research was partly supported (2019) by grant of The Youth Activity Fund of The Explorers Club “Where worlds encounter—searching for zoonotic pathogen exchange between bats and other vertebrates,” project leader Olena Rodenko. The regular activities of The Bat Rehabilitation Center of Feldman Ecopark were supported by the International Charitable Foundation “Oleksandr Feldman Foundation.”
Author information
Authors and Affiliations
Contributions
AV, OR, VB, and IT did bat catching and collection of ectoparasites. VB, OR, and SF provided the identification of ectoparasites. CS and CR designed the molecular analysis of the samples and analysed the data. CR and OT performed the molecular screening. AV and DM provided the general management of the project from Ukrainian side. AV, CR, and SF drafted the manuscript. AV, OR, VB, and IT partly contributed to the first version of the manuscript. CS and SF critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The Bat Rehabilitation Center of Feldman Ecopark works under the general permission of the Kharkiv Oblast Authority of Ecology and Natural Resources.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Locations and roosting sites of bats collected for the purpose of this study. Table S2. Primers used for pathogen screening of ectoparasites. Table S3. GenBank accession numbers for sequences obtained in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Vlaschenko, A., Răileanu, C., Tauchmann, O. et al. First data on bacteria associated with bat ectoparasites collected in Kharkiv oblast, Northeastern Ukraine. Parasites Vectors 15, 443 (2022). https://doi.org/10.1186/s13071-022-05582-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05582-3