
Abstract
Background
Avian haemosporidia infect both domestic and wild birds, causing anemia, acute tissue degeneration, and depopulation in wild birds. Poultry and wild birds have been reported as common reservoirs of haemosporidia, but limited information is available for red junglefowl (Gallus gallus) in China. The present study investigated the prevalence and molecular characterization of haemosporidia in red junglefowl.
Methods
Blood samples were collected from 234 red junglefowl from Jinghong City of Yunnan Province, and genomic DNA was extracted from these samples. The prevalence of haemosporidia was determined by nested PCR targeting the mitochondrial cytochrome b (cytb) gene. Molecular characterization was investigated based on phylogenetic analysis of cytb sequences, and associated risk factors were analyzed using the Chi-square (χ2) test.
Results
The overall prevalence of haemosporidia was 74.8% (175/234), and three species were identified, namely Haemoproteus enucleator, Leucocytozoon californicus, and Plasmodium juxtanucleare. The prevalence of haemosporidia in adult fowl (81.1%, 107/132) was significantly higher (χ2 = 6.32, df = 1, P = 0.012) than that in juveniles (66.7%, 68/102). Three novel haemosporidian lineages were revealed.
Conclusions
This study examined the prevalence and identified species of avian haemosporidians in red junglefowl, providing new information on the molecular epidemiology and geographical distribution of haemosporidian parasites. Our results indicated high prevalence and diverse species distribution of these haemosporidians in red junglefowl. To the best of our knowledge, this is the first record of haemosporidian infection in red junglefowl in China.
Graphical Abstract

Similar content being viewed by others

Background
Avian haemosporidia are a whole group of organisms containing hundreds of species [1]. The group has been used for decades as a model to study the mechanisms of disease transmission and interspecific co-evolution [2]. Haemosporidia of the genera Plasmodium, Haemoproteus, and Leucocytozoon are diverse groups of vector-transmitted blood parasites that are abundant in most avian families and can cause disease [2,3,4]. At present, there are more than 4000 lineages defined based on the barcode sequence of the mitochondrial cytochrome b gene (cytb). Approximately 2000 bird species can be infected by haemosporidia, and these parasites have been found in all regions of the world except Antarctica, posing a serious threat to the health and even survival of infected poultry and birds [5].
Over the many centuries since the domestication of chickens, they have been respected by different cultures all over the world. Compared with sheep, cattle, pigs, and other livestock, chicken is the preferred source of animal protein. Red junglefowl (Gallus gallus) has been identified as the wild ancestor of domestic chicken (Gallus gallus domesticus) [6]. Due to the warm and humid tropical rain forest climate, Xishuangbanna is rich in biodiversity, which is highly suitable for domestic chickens and their insect vectors. Avian haemosporidia are mainly transmitted by dipteran-blood sucking insects such as mosquitoes, biting midges, and black flies [7, 8]. In poultry, haemosporidiosis can lead to clinical manifestations such as multiple organ injury, anemia, and weight loss, which seriously affects the economic benefits of poultry breeding [9, 10]. Failure to provide timely preventive treatment will lead to higher rates of infection and mortality [11, 12].
Information about patterns of distribution of haemosporidia in poultry contributes to better prevention, control, and treatment of avian haemosporidiosis. However, to date, there are limited studies on haemosporidian infection in red junglefowl. Therefore, the main objectives of the present study were to investigate the prevalence, molecular characterization, and associated risk factors of haemosporidia in red junglefowl using molecular biology and high-throughput sequencing, evaluating the factors associated with haemosporidian infection in red junglefowl using cross-sectional analysis.
Methods
Sample collection
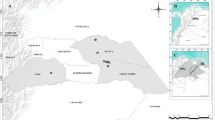
The red junglefowl is a tropical member of the pheasant family and the direct ancestor of the domestic chicken. With the help of the staff of Yunnan Province Center for Animal Disease Control and Prevention, Xishuangbanna Dai Autonomous Prefecture Technical Extension Station for Animal Husbandry and Veterinary Medicine, from November 2020 to May 2021, a total of 234 blood specimens were collected from red junglefowl in a tea plantation habitat in Jinghong City (21°27′ ~ 22°36′N, 100°25′ ~ 101°31′E), Yunnan Province, southwestern China. These domestic chickens were divided into two age groups: juveniles and adults. Samples were divided into three groups according to body weight: < 0.5 kg, 0.5–1.0 kg, and > 1 kg. Each fresh blood specimen was randomly obtained from the inferior pterygoid vein of each apparently healthy fowl using a vacuum blood collection tube with anticlotting agents including ethylenediaminetetraacetic acid (EDTA). The vacuum blood collection tubes containing approximately 2–4 ml individual animal blood samples were then labeled with sex, weight, age, sampling site, and sampling time, and immediately kept on ice packs at −80 °C during transport.
Molecular analysis
The genomic DNA of each blood sample was extracted using a commercial DNA kit (Tiangen Bio-tech Co., Ltd, Beijing, China) according to the manufacturer’s instructions. The extracted genomic DNA was stored at −20 °C for further polymerase chain reaction (PCR) analysis. Avian haemosporidian infection in red junglefowl was detected by nested PCR amplification of a 479-base-pair fragment of the mitochondrial cytb gene using primers and procedures described previously [13]. For the first PCR, the primers HaemNFI (5′-CATATATTAAGAGAAITATGGAG-3′) and HaemNR3 (5′-ATAGAAAGATAAGAAATACCATTC-3′) were used. In the second PCR, two primer pairs were applied: the primers HaemNF (5′-ATGGTGCTTTCGATATATGCATG-3′) and HaemNR2 (5′-GCATTATCTGGATGTGATAATGGT-3′), and HaemNFL (5′-ATGGTGTTTTAGATACTTACATT-3′) and HaemNR2L (5′-CATTATCTGGATGAGATAATGGIGC-3′). Amplification products were tested by running 2 μl of the second PCR product on 1.5% agarose gel stained with SYBR Green I and visualized with UVP GelStudio DNA Gel Documentation Imaging Systems (Analytik Jena Company, US, https://www.laboratory-equipment.com/uvp-gelstudio-dna-gel-documentation-systems-analytik-jena.html). One negative control (nuclease-free water) and three positive controls were used to determine possible false amplifications.
Bioinformatics, lineage identification, and phylogenetic analysis
All positive secondary PCR products were purified and sequenced by Kunming Sangon Biotech (Shanghai) Co., Ltd. Sequences obtained were firstly proofread with their DNA peak-form graph using Chromas 2.6. Using MEGA X (Version 10.2.6, https://www.megasoftware.net/), the sequences of amplification products were aligned with the most similar lineages according to the BLAST result in the MalAvi database (http://130.235.244.92/Malavi/blast.html) [5, 14]. Haplotypes were defined as new lineages if they differed by one base pair from lineages deposited in the MalAvi database (http://mbio-serv2.mbioe kol.lu.se/Malavi). The phylogenetic analysis was performed using the neighbor-joining (NJ) method with MEGA X; the Kimura 2-parameter model was selected, and 1000 bootstrap replicates were applied in this study. The numbers at the nodes indicate the bootstrap support obtained by repeating the analysis 1000 times, and values above 50% are shown.
Statistical analysis
The prevalence of avian haemosporidian parasites among different red junglefowl groups according to sex, age, weight, and sampling season were calculated by Chi-square (χ2) tests using SPSS 22.0 (IBM Corporation, https://www.ibm.com/cn-zh), and were considered statistically significant if P < 0.05. The odds ratios (ORs) and their 95% confidence intervals (95% CIs) were calculated and analyzed by using GraphPad Prism Version 8.02 for Windows (GraphPad Software, Inc., https://www.graphpad.com/).
Results
Prevalence of avian haemosporidia in red junglefowl
Haemosporidia belonging to the genera Haemoproteus, Plasmodium, and Leucocytozoon were detected in red junglefowl (Tables 1 and 2, Fig. 1). As shown in Table 1, 175 out of 234 DNA samples were positive for avian haemosporidia, representing a 74.8% overall prevalence.

Phylogenetic tree of avian haemosporidia (Plasmodium, Haemoproteus, and Leucocytozoon) based on cytb sequences. One lineage of Hepatocystis spp. was used as an outgroup. Parasite species names and GenBank accession numbers are provided in the tree. The parasite lineages reported in this study are marked by blue squares, green dots, and yellow triangles, respectively. The bootstrap value is shown when the value is greater than 50%
Among samples positive for haemosporidian infection, 107 were in adult fowls, with an infection rate of 81.1% (107/132), while the infection rate in juveniles was 66.7% (68/102). A significant difference was observed between the two age groups (χ2 = 6.32, df = 1, P = 0.012). The positive rate of blood samples collected in summer (80.9%, 106/131) was higher than that in winter (67.0%, 69/103). According to Chi-square tests, we identified the risk factors for the prevalence of haemosporidia in fowls as age (OR 0.47, 95% CI 0.26–0.58, P 0.012) and season (OD 2.09, 95% CI 1.15–3.80, P 0.015).
We found single and mixed haemosporidian infections in red junglefowl (Table 2). Of 175 blood samples that tested positive by the PCR technique, 153 (153/175, 87.4%) samples were single pathogen infections, of which seven samples were Haemoproteus infections, 32 were Leucocytozoon infections, and 114 were Plasmodium infections. In addition, there were 22 (22/175, 12.6%) samples which were mixed infections, with 16 samples infected with two pathogens and six samples infected with three pathogens.
Molecular characterization of avian haemosporidia
Molecular analysis revealed parasites belonging to three different genera: Haemoproteus, Plasmodium, and Leucocytozoon (Fig. 1). The three lineages of haemosporidia clustered with their genetically most similar lineages within the corresponding parasite genera. Our three representative lineages hGALGAL01, lGALGAL01, and pGALGAL01 were more similar to H. enucleator, L. californicus, and P. juxtanucleare, respectively (Fig. 1).
Discussion
The global prevalence of haemosporidia in red junglefowl was 74.8% (175/234), which is much higher than that of fighting cocks from Thailand (20.8%, 52/250) [15], but lower than that in domestic chickens from Nan, Prachinburi, and Chachoengsao provinces of Thailand (79.6%, 125/157) [16] and in indigenous chickens from the north central part of Nigeria (75.0%, 81/108) [17]. The reason for this may be the abundance of vegetation in tropical areas, with species of Culicoides and avian haemosporidia transmitted by biting midges and other insect vectors [18, 19]. In addition, the reason for the variation in prevalence is complicated, and many factors will affect the detection rate, such as sampling time, age group, sampling number, and geographical conditions [20]. In addition, similar to previous studies, the proportion of single infection was much higher than that of mixed infections [21, 22], and mixed infections showed multiple combinations [23, 24].
Avian haemosporidia were detected in juvenile and adult fowls with infection rates of 66.7% (68/102) and 81.1% (107/132) (P 0.012), respectively. Previous studies showed that infection rates were higher in young birds relative to adults, possibly due to the lower immune resistance in young birds [25, 26]. The greater area of bare skin of young domestic chickens makes them more easily accessible to the pathogen vectors [27]. The weight of red junglefowl did not appear to contribute significantly to Haemoproteus spp. infection. It is true that many studies have shown that different host traits and abiotic factors are important determinants in a host–parasite interaction [28, 29]. Factors such as plant richness, vector species, temperature, and humidity in wild bird habitats contribute significantly to the prevalence and diversity of Haemoproteus spp. [30,31,- 32].
Avian haemosporidia in birds is genetically diverse [33, 34]. The representative Haemoproteus gene (accession no. OM965002) is closely related to Haemoproteus spp. in birds from India (99–100% similarity) [34]. The lineage detected in the present study is new and may be a novel lineage from red junglefowl. We revealed that the known and novel lineages found in this study have biological transmission in China and can be transmitted to other birds.
Conclusion
Using a PCR-based molecular approach, the present study revealed the high prevalence (74.8%) and species of avian haemosporidians in red junglefowl of different sex and age from Yunnan Province, southwestern China. Three species (H. enucleator, L. californicus, and P. juxtanucleare) were identified. This is the first record of avian haemosporidian infection in red junglefowl in China, which extends the host range and genetic diversity of avian haemosporidians and has implications for the control of avian haemosporidia infection in red junglefowl.
Availability of data and materials
The datasets supporting the findings of this article are included within the article. Representative nucleotide sequences obtained in this study were deposited in the GenBank under accession numbers OM965002–OM965004.
Abbreviations
- cytb :
-
Cytochrome b gene
- ORs:
-
Odds ratios
- CIs:
-
Confidence intervals
- PCR:
-
Polymerase chain reaction
References
Galen SC, Borner J, Martinsen ES, Schaer J, Austin CC, West CJ, et al. The polyphyly of Plasmodium: comprehensive phylogenetic analyses of the malaria parasites (order Haemosporida) reveal widespread taxonomic conflict. R Soc Open Sci. 2018;5:171780.
Rivero A, Gandon S. Evolutionary ecology of avian malaria: past to present. Trends Parasitol. 2018;34:712–26.
Clark NJ, Clegg SM, Lima MR. A review of global diversity in avian haemosporidians (Plasmodium and Haemoproteus: Haemosporida): new insights from molecular data. Int J Parasitol. 2014;44:329–38.
Dunn JC, Outlaw DC. Flying into the future: avian haemosporidians and the advancement of understanding host-parasite systems. Parasitology. 2019;146:1487–9.
Bensch S, Hellgren O, Perez-Tris J. MalAvi: a public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol Ecol Resour. 2009;9:1353–8.
Wang MS, Thakur M, Peng MS, Jiang Y, Frantz LAF, Li M, et al. 863 genomes reveal the origin and domestication of chicken. Cell Res. 2020;30:693–701.
Valkiunas G. Avian malaria parasites and other haemosporidia. Boca Raton: CRC Press; 2005.
Pramual P, Thaijarern J, Tangkawanit U, Wongpakam K. Molecular identification of blood meal sources in black flies (Diptera: Simuliidae) suspected as leucocytozoon vectors. Acta Trop. 2020;205:105383.
Lee HR, Koo BS, Jeon EO, Han MS, Min KCh, Lee SB, et al. Pathology and molecular characterization of recent Leucocytozoon caulleryi cases in layer flocks. J Biomed Res. 2016;30:517–24.
Galosi L, Scaglione FE, Magi GE, Cork SC, Peirce MA, Ferraro S, et al. Fatal Leucocytozoon Infection in a captive Grey-headed Parrot (Poicephalus robustus suahelicus). J Avian Med Surg. 2019;33:179–83.
Naqvi MA, Khan MK, Iqbal Z, Rizwan HM, Khan MN, Naqvi SZ, et al. Prevalence and associated risk factors of haemoparasites, and their effects on hematological profile in domesticated chickens in District Layyah, Punjab, Pakistan. Prev Vet Med. 2017;143:49–53.
Miranda Paez A, Chalkowski K, Zohdy S, Willoughby JR. Management of avian malaria in populations of high conservation concern. Parasit Vectors. 2022;15:208.
Hellgren O, Waldenström J, Bensch S. A new PCR assay for simultaneous studies of Leucocytozoon, Plasmodium, and Haemoproteus from avian blood. J Parasitol. 2004;90:797–802.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Piratae S, Vaisusuk K, Chatan W. Prevalence and molecular identification of Leucocytozoon spp. in fighting cocks (Gallus gallus) in Thailand. Parasitol Res. 2021;120:2149–55.
Xuan MNT, Kaewlamun W, Saiwichai T, Thanee S, Poofery J, Tiawsirisup S, et al. Development and application of a novel multiplex PCR assay for the differentiation of four haemosporidian parasites in the chicken Gallus gallus domesticus. Vet Parasitol. 2021;293:109431.
Mohammed BR, Ojo AA, Opara MN, Jegede OC, Agbede RIS. Haemo- and endoparasites of indigenous chickens reared in Gwagwalada area council, Abuja, Nigeria. Ann Parasitol. 2019;65:293–6.
Žiegytė R, Platonova E, Kinderis E, Mukhin A, Palinauskas V, Bernotienė R. Culicoides biting midges involved in transmission of haemoproteids. Parasit Vectors. 2021;14:27.
Bernotienė R, Žiegytė R, Vaitkutė G, Valkiūnas G. Identification of a new vector species of avian haemoproteids, with a description of methodology for the determination of natural vectors of haemosporidian parasites. Parasit Vectors. 2019;12:307.
Fecchio A, Ellis VA, Bell JA, Andretti CB, D’Horta FM, Silva AM, et al. Avian malaria, ecological host traits and mosquito abundance in southeastern Amazonia. Parasitology. 2017;144:1117–32.
Hanel J, Doležalová J, Stehlíková Š, Modrý D, Chudoba J, Synek P, et al. Blood parasites in northern goshawk (Accipiter gentilis) with an emphasis to Leucocytozoon toddi. Parasitol Res. 2016;115:263–70.
Ivanova K, Zehtindjiev P, Mariaux J, Georgiev BB. Genetic diversity of avian haemosporidians in Malaysia: cytochrome b lineages of the genera Plasmodium and Haemoproteus (Haemosporida) from Selangor. Infect Genet Evol. 2015;31:33–9.
Marzal A, Ibáñez A, González-Blázquez M, López P, Martín J. Prevalence and genetic diversity of blood parasite mixed infections in Spanish terrapins Mauremys leprosa. Parasitology. 2017;144:1449–57.
Bernotienė R, Palinauskas V, Iezhova T, Murauskaitė D, Valkiūnas G. Avian haemosporidian parasites (Haemosporida): a comparative analysis of different polymerase chain reaction assays in detection of mixed infections. Exp Parasitol. 2016;163:31–7.
Jia T, Huang X, Valkiūnas G, Yang M, Zheng C, Pu T, et al. Malaria parasites and related haemosporidians cause mortality in cranes: a study on the parasites diversity, prevalence and distribution in Beijing Zoo. Malar J. 2018;17:234.
Thul JE, Forrester DJ, Greiner EC. Hematozoa of wood ducks (Aix spons) in the Atlantic flyway. J Wildl Dis. 1980;16:383–90.
Imura T, Suzuki Y, Ejiri H, Sato Y, Ishida K, Sumiyama D, et al. Prevalence of avian haematozoa in wild birds in a high-altitude forest in Japan. Vet Parasitol. 2012;183:244–8.
Rodrigues RA, Felix GMF, Pichorim M, Moreira PA, Braga EM. Host migration and environmental temperature influence avian haemosporidians prevalence: a molecular survey in a Brazilian Atlantic rainforest. PeerJ. 2021;9:e11555.
Eastwood JR, Peacock L, Hall ML, Roast M, Murphy SA, Gonçalves da Silva A, et al. Persistent low avian malaria in a tropical species despite high community prevalence. Int J Parasitol Parasites Wildl. 2019;8:88–93.
Castaño-Vázquez F, Merino S. Differential effects of environmental climatic variables on parasite abundances in blue tit nests during a decade. Integr Zool. 2022;17:511–29.
Castaño-Vázquez F, Schumm YR, Bentele A, Quillfeldt P, Merino S. Experimental manipulation of cavity temperature produces differential effects on parasite abundances in blue tit nests at two different latitudes. Int J Parasitol Parasites Wildl. 2021;14:287–97.
Fecchio A, Lima MR, Bell JA, Schunck F, Corrêa AH, Beco R, et al. Loss of forest cover and host functional diversity increases prevalence of avian malaria parasites in the Atlantic forest. Int J Parasitol. 2021;51:719–28.
Padilla DP, Illera JC, Gonzalez-Quevedo C, Villalba M, Richardson DS. Factors affecting the distribution of haemosporidian parasites within an oceanic island. Int J Parasitol. 2017;47:225–35.
Ishtiaq F, Gering E, Rappole JH, Rahmani AR, Jhala YV, Dove CJ, et al. Prevalence and diversity of avian hematozoan parasites in Asia: a regional survey. J Wildl Dis. 2007;43:382–98.
Acknowledgements
We would like to thank Sangon Biotech (Shanghai) for technical assistance.
Funding
This work was supported by the Yunnan Fundamental Research Projects (Grant No. 202101AU070064, 202201AT070067), the Scientific research fund project of Yunnan Provincial Department of Education (Grant No. 2022J0011), the Veterinary Public Health Innovation Team of Yunnan Province (Grant No. 202105AE160014), and the Fund for Shanxi “1331 Project” (Grant No. 20211331–13). The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Author information
Authors and Affiliations
Contributions
ZL and QSL conceived and designed the study, and JJH and XQZ critically revised the manuscript. ZL, XXR, YJZ and LTY performed the experiment, ZL analyzed the data and drafted the manuscript. ZL, BFD and NYH conducted the sample collection. JJH, FCZ, XQZ and QSL helped in the implementation of the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The protocol of the present study was reviewed and approved by the Animal Ethical and Welfare Committee of Yunnan University. All blood samples were collected from red junglefowl after obtaining permission from the poultry farmers and functional management departments without other authorities, and all procedures were performed in strict accordance with the legal requirements of the Animal Ethics Procedures and Guidelines of the People’s Republic of China. All efforts were made to minimize suffering of fowls.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. The co-author Prof. Xing-Quan Zhu serves as the Subject Editor for the section “Parasite genetics, genomics and proteomics” of Parasites & Vectors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Z., Ren, XX., Zhao, YJ. et al. First report of haemosporidia and associated risk factors in red junglefowl (Gallus gallus) in China. Parasites Vectors 15, 275 (2022). https://doi.org/10.1186/s13071-022-05389-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05389-2