
Abstract
Background
In Mongolia, the taiga tick Ixodes persulcatus is the major vector of tick-borne pathogens. Knowledge about co-infections of these pathogens in ticks is necessary both for understanding their persistence in nature and for diagnosing and treating tick-borne diseases.
Methods
The prevalence of seven tick-borne infections in 346 I. persulcatus collected from the Selenge and Bulgan provinces of Mongolia was evaluated using real-time PCR. Quantification of Borrelia spp. was performed using multiplex quantitative PCR targeting the 16S rRNA gene. Genetic analysis of Borrelia spp. in 11 ticks infected with Borrelia miyamotoi, including six ticks co-infected with Borrelia burgdorferi sensu lato (s.l.), was performed by high-throughput sequencing of the flaB gene fragment.
Results
Six ticks (1.7%) were infected with tick-borne encephalitis virus (TBEV); 171 (49.4%), with B. burgdorferi sensu lato; 17 (4.9%), with B. miyamotoi; 47 (13.6%), with Anaplasma phagocytophilum; and 56 (16.2%), with Ehrlichia sp. Neither Rickettsia sibirica nor R. heilongjiangensis were detected. Borrelia burgdorferi s.l. occurred as co-infection in 55 (32.2%) of all infected ticks. The other pathogens co-infected ticks in 58.8–70.2% of cases. No pairwise associations between co-infecting pathogens were observed, with the exception of a positive association between A. phagocytophilum and Ehrlichia sp. infections. The spirochete loads of B. miyamotoi were significantly higher than those of B. burgdorferi s.l. (mean: 5.2 vs 4.0 log10 genome copies/tick, respectively). Ten isolates of B. miyamotoi belonged to the Siberian lineage. Borrelia burgdorferi s.l was represented by nine isolates of B. afzelii, B. bavariensis and B. garinii.
Conclusions
In populations of I. persulcatus inhabiting the Selenge and Bulgan provinces of Mongolia, five vector-borne pathogens, i.e. TBEV, B. burgdorferi s.l., B. miyamotoi, A. phagocytophilum and Ehrlichia sp., persist independently from each other, with the exception of A. phagocytophilum and Ehrlichia sp. which seem to share the circulation mode. The discrepancies in B. burgdorferi s.l. and B. miyamotoi prevalence and spirochete load per tick suggest that different ecological niches are occupied by Lyme disease and relapsing fever agents. High-throughput sequencing allows genetic identification of borreliae species in co-infected ticks.
Graphical Abstract

Similar content being viewed by others

Background
In Mongolia, tick-borne infections have been recognized as a significant threat to human health since the 1990s. A significant increase in the incidence, geographic range and fatality rates of tick-borne infections has been recorded [1, 2], and the seroprevalence of tick-borne pathogens may reach 15% [3]. About 30.5% of local nomadic herders report a history of tick bites and exposure to some tick-borne pathogens, particularly Anaplasma spp. and Rickettsia spp., comprising 46.3% of all tick bites [4]. Among the major administrative provinces (hereinafter referred to as aimags) of Mongolia, the northern Selenge and Bulgan aimags are characterized by an especially high risk of tick-borne infections. The landscape of these two adjacent aimags is formed by slopes of the Khentii Ridge and the Selenga River Basin, which provide one of the most humid climate conditions in Mongolia [5]. This results in a wide spread of mixed boreal subalpine forests in the region, with about 33% of land area in Selenge and 29% in Bulgan covered with forests in comparasion to the Mongolian national average of 7.7% [5]. Due to these suitable ecological conditions, the taiga tick Ixodes persulcatus (Schulze, 1930) is widely distributed in Selenge and Bulgan aimags where it is the most epidemiologically important vector tick species [6,7,8]. A number of studies have indicated the presence of such pathogens as tick-borne encephalitis virus (TBEV), Borrelia burgdorferi sensu lato (s.l.), B. miyamotoi, Rickettsia sibirica, Anaplasma phagocytophilum and Ehrlichia muris in Mongolia ([9], [10], reviewed in [11], [12]). The causative agents of North Asian tick-borne spotted fever, R. sibirica and Rickettsia heilongjiangensis, are also well recognized human pathogens that are usually associated with Dermacentor nuttalli (Olenev, 1928) and Haemaphysalis concinna (Koch, 1884) ticks, respectively [13, 14]. However, the infection of I. persulcatus with R. sibirica and R. heilongjiangensis has also been described [15]. In Mongolia, around 4% of Dermacentor sp. ticks are infected with R. sibirica [12, 16], but no infection of I. persulcatus with R. sibirica or R. heilongjiangensis has been detected so far.
In spite of the high prevalence of tick-borne pathogens in ticks, a relatively small number of disease cases are reported in Mongolia. Thus, the morbidity of Lyme disease in the northern aimags is approximately twofold lower than that in neighboring Russian regions [11]. Also, discrepancies between the number of reported tick bites, symptoms and antibody seroprevalence to tick-borne infections among nomadic Mongolian herders is reported [4]. These discrepancies suggest that ecological and social drivers of tick-borne diseases in Mongolia are still not understood completely. A better characterization of the spatial and temporal distribution of ticks and associated pathogens, long-term surveillance and knowledge of the qualitative characteristics of tick-borne infections are necessary in Mongolia, especially in the northern aimags [4, 11, 12].
The ability of pathogens to infect the same tick lead to multiple co-infections, i.e. simultaneous infection of the same tick specimen with two or more microorganisms. For I. persulcatus, co-infections with multiple pathogens have been studied in different parts of the distribution range, including Japan [17,18,19], Russia [15, 20,21,22] and Finland [23]. It has been shown that in different geographic locations co-infecting microorganisms can either have no effect on each other, such as TBEV, B. burgdorferi s.l. and A. phagocytophilum in Western Siberia [24], or there may have positive and negative interactions [22, 25]. In addition, co-infection with some pathogens has an epidemiological importance as it may facilitate the emergence of some zoonoses (e.g. babesiosis in co-infection with he Lyme disease agent) and affect disease severity [25, 26]. For Borrelia sp. co-infections are an even more complicated problem due to the significant diversity of these bacteria. Tick-borne borreliae are currently divided into two major groups: the Lyme borreliosis (LB) group and relapsing fever (RF) group [27]. The LB group comprises > 20 species belonging to the B. burgdorferi s.l.complex, of which four i.e. B. burgdorferi sensu stricto (s.s.), B. afzelii, B. garinii and B. bavariensis are recognized as human pathogens [28]. The RF group is mainly associated with soft (argasid) ticks, although one species, B. miyamotoi, is associated with hard (ixodid) ticks and is widely spread across the Northern hemisphere [29]. The regional populations of this pathogen are highly clonal and cluster into three evolutionary lineages designated as Siberian, American and European [30]. While B. burgdorferi s.l. and B. miyamotoi can infect the same range of tick and vertebrate hosts, co-infections with these pathogens are usual in ticks [30,31,32]. In Mongolia, the LB group is represented by the genetically diverse B. afzelii, B. garinii and B. bavariensis isolates [33, 34], whereas all known isolates of B. miyamotoi are very similar and belong to the Siberian lineage [18]. However, due to high genetic similarity of B. garinii, B. afzelii, B. bavariensis and B. miyamotoi, their molecular identification and analysis in co-infected samples is complicated by mixed DNA/RNA templates, especially when the conserved genome regions are investigated [18]. Generally, the information on co-infections of I. persulcatus with different pathogens in tick populations in Mongolia is insufficient.
To better understand the ecology of tick-borne infections in Mongolia, we evaluated the contingency of several epidemiologically important infections in the Selenge and Bulgan aimags using co-infection study, quantitatively characterized the infection with Borrelia sp. in questing I. persulcatus ticks using quantitative PCR (qPCR) and assessed the genetic diversity of Borrelia spp. in five ticks infected with B. miyamotoi and six ticks co-infected with B. miyamotoi and B. burgdorferi s.l. using the high-throughput sequencing approach.
Methods
Tick collection and processing
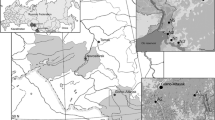
Questing adult I. persulcatus ticks were collected from vegetation using flannel flags at eight sampling sites in the Selenge and Bulgan aimags of Mongolia between 30 May 2019 and 2 February 2019. Geographical coordinates of the collection sites were determined using a “Garmin 60CSx” GPS device (Garmin Ltd., Olathe, KS, USA) at the beginning of the tick collection routes and mapped using Google Earth Pro 7.3 software (Fig. 1). Ticks were delivered to the laboratory alive and processed immediately. The ticks were first identified to the species level using a morphological key guide [35]. Afterwards, individual ticks were washed twice in 70% alcohol, once in sterile phosphate buffered saline (PBS, pH = 7.4), crushed with a sterile RNase/DNase-free pestle and resuspended in 100 μl of PBS.

Sampling sites (yellow marks) where questing Ixodes persulcatus were collected, in the Selenge and Bulgan aimags of Mongolia. The mapping of sampling sites and the image generation were performed using Google EarthPro 7.3 software (accessed September 2019)
Extraction of nucleic acids
Total RNA/DNA was isolated from an entire tick suspension using the RealBest Extraction100 kit (VektorBest, Novosibirsk, Russia) according to the manufacturer’s instructions. The final DNA/RNA sample was dissolved in 200 μl of the elution buffer. To prevent any carryover of amplified PCR fragments, tick processing, extraction of nucleic acids from ticks, reverse transcription and preparation of the PCR reaction, and PCR and subsequent electrophoresis were conducted in four separate rooms.
Reverse transcription, PCR and qPCR
The RNA of TBEV was transcribed with Superscript IV reverse transcriptase (Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA) and random hexanucleotides according to the manufacturer’s instruction. The primers and probes used in this work are listed in Table 1. Real-time PCR was performed according to the protocol published by Schwaiger and Casinotti [36] with minor modifications. Briefly, the reaction was carried out in a volume of 25 μl that contained 1 U of Taq polymerase HSTaq (Eurogen, Moscow, Russian Federation), 2.5 μl of an optimized reaction buffer (Eurogen), 0.25 mM of each dNTP, the primers F-TBE1 and R-TBE1 at concentration of 50 and 300 nM, respectively, and the probe TBE-WT at concentration of 200 nM (Sintol, Moscow, Russian Federation). The cycling program included preheating at 95 °C for 10 min, followed by 45 cycles of 95 °C for 15 s and 60 °C for 1 min. The results were recorded on the FAM channel at 60 °C.
Taqman quantitative real-time PCR (qPCR) targeting the 16S ribosomal RNA (rRNA) gene of Borrelia sp. was used to detect, differentiate and quantify B. burgdorferi s.l. and B. miyamotoi as described previously [29, 30], with minor modifications. The standard DNA samples of B. miyamotoi and B. burgdorferi s.l. were prepared according to Takano et al. [30]. The quantity of spirochetes in the specimens was assessed using serial tenfold dilutions of standard DNA samples of corresponding Borrelia sp. and expressed as log10 of genome copies per tick. The PCR was performed in a reaction volume of 25 μl containing 1 U of Taq polymerase HSTaq (Eurogen), 2.5 μl of DNA template, primers BspF-16s and BspR-16s at a concentration of 900 nM each and the probes FAM-LD and VIC-RF at a concentration of 200 nM each. The PCR conditions were an initial cycle at 50° C for 2 min, then a cycle at 95 °C for 2 min, followed by 45 cycles of 95 °C for 15 s and 63 °C for 60 s, with reading of the fluorescence.
All samples that were positive for B. miyamotoi were subjected to PCR that targeted the flagellin B gene (flaB), with primers Bfla-PAD and Bfla-PDU, as described previously [37]. Target PCR fragments of approximately 450 nucleotides were purified by agarose gel electrophoresis and used for high-throughput DNA sequencing (next-generation sequencing [NGS]) library preparation.
The nucleic acids of A. phagocytophilum, E. muris and/or E. chaffeensis (hereafter, Ehrlichia sp.) R. sibirica and R. heilongjiangensis were detected using commercial multiplex real-time PCR kits (VektorBest) according to the manual of the supplier.
All PCR experiments were performed on a CFX Touch T1000 thermal cycler (Bio-Rad Laboratories Inc., Hercules, CA, USA) and processed with Bio-Rad CFX Manager 3.1 software.
Sequencing
The flaB PCR amplicons, purified by agarose gel electrophoresis and the QIAquick Gel Extraction kit (Qiagen, Hilden, Germany), were subjected to NGS analysis on NextSeq 550 sequencer (Illumina Inc., San Diego, CA, USA). Libraries were prepared using the Nextera DNA Flex Library Prep kit, and sequencing was performed using the NextSeq 500/550 Mid Output Kit v2.5 (Illumina) with 300 cycles.
The reads were processed and quality was assessed using the FastQ and FastQC programs implemented in the BaseSpace Sequence Hub (Illumina; https://basespace.illumina.com/dashboard). The sequencing data were then uploaded to the Galaxy web platform, and this public server (https://usegalaxy.org/) was used to analyze the data [38]. The de-novo assembly was performed using the Shovill assembly platform with the Megahit sequence assembler [39]. Resulting contigs were used to generate the consensus sequences, following which the PCR primer sequences were trimmed and obtained sequences were deposited in GenBank under the accession numbers OL580771—OL580777. Sequences were processed and multiple alignments were composed using BioEdit software [40]. The evolutionary distances were computed using the Tamura 3-parameter model [41], which was optimal according to the Bayesian information criterion scores. The evolutionary history was inferred using the maximum likelihood method. The reliability of the trees was evaluated by 1000 bootstrap resampling, and those nodes with bootstrap support > 70% were assumed to be reliable. The phylogenetic analysis was performed using MEGA X sequence analysis software [42].
Data analysis
The gender ratio and the prevalence of infections was estimated as the proportion of infected ticks and expressed in percentage with 95% confidence intervals (95% CI). The gender rates in different geographic locations and prevalence rates in the tick groups from different geographic locations and genders were compared using the unpaired Chi-square (χ2) test for proportions. Borrelia genome copy number quantification data are presented as mean values with the 95% CI. Differences in quantities of B. burgdorferi s.l. and B. miyamotoi were analyzed using the unpaired Mann–Whitney test.
To assess the associations between microorganism and frequency of co-infection with any other microorganism, the 2 × 5 contingency table was composed with single-infection and co-infection states of each microorganism determining the binary condition. To assess the associations between TBEV, B.burgdorferi s.l., B. miyamotoi, A. phagocytophilum and Ehrlichia sp. infections in ticks, the 2 × 2 contingency tables were constructed for each pair of microorganisms [43], with infected and non-infected states of ticks determining the binary conditions. For each contingency table, the observed values were compared with values expected mathematically under the assumption that the pathogens are distributed in tick populations by chance only. The χ2 test was used for this comparison, using the Yates correction for tables containing cells with ≤ 5 counts.
The statistical analyses were performed using MS Excel (Microsoft Corp., Redmond, WA, USA), the MaxStat Light free statistical program and R version 4.0.2 (R Foundation, Vienna, Austria) software. Significance was set at 0.95; all significance tests were two-tailed. The differences were considered statistically significant when P-values were below 0.05.
Results
Tick population
In total, 138 and 208 I. persulcatus ticks were sampled from the Bulgan aimag and Selenge aimag, respectively. The proportion of females was 58% (n = 80, 95% CI: 49.2–66.2) and 56% (n = 114, 95% CI: 48.8–62.8), respectively, and there was no significant difference in gender ratio between geographic locations (χ2 = 0.07357, df = 1, P = 0.7862). Four nymphs (1.9%) were collected in the Selenge aimag only.
Prevalence of infections
Neither R. sibirica nor R. heilongjiangensis were detected in the entire collected sample and, therefore, these bacteria were not analyzed further. The prevalence of other infections is summarized for each infection separately in Table 2.
There were no significant differences in the prevalence of infections between different geographic locations or between the groups of male and female ticks, with the exception of A. phagocytophilum, which was more prevalent in ticks from the Selenge aimag than in those from the Bulgan aimag (16.8% vs 8.7%, respectively; χ2 = 4.673, df = 1, P = 0.0306). This difference was probably due to lower rate of anaplasma infection among females in the Bulgan aimag, and was significant both for females from the Selenge aimag (3.7% vs 21.9%; χ2 = 11.152, df = 1, P = 0.0008) and males from the Bulgan aimag (3.7% vs 15.5%; χ2 = 4.476, df = 1, P = 0.0344).
Based on these results we assumed that ticks in both the Selenge and Bulgan samples belong to the same general population. We therefore combined the data for further analyses. In the combined sample, the most prevalent pathogen was the B. burgdorferi s.l., which was detected in 49% of ticks, followed by Ehrlichia sp. (16.0%), A. phagocytophilum (13.5%), B. miyamotoi (4.9%) and TBEV (1.7%). In total, 64.2% of ticks were infected with at least one pathogen (Table 2).
Co-infection rate
The data on co-infection by tick-borne pathogens are summarized in Table 3. The majority of infected ticks (70.7%) harbored a single pathogen; however, only B. burgdorferi s.l. occurred as single infection (116 ticks), and at an infection rate that was higher than statistically expected (90 ticks). Indeed, 67.0% of all the ticks infected with B. burgdorferi s.l. were not infected with any other pathogen. Other pathogens occurred as single infections in 29.8–41.2% of ticks (Table 3), which was less than statistically expected for TBEV (observed single infections vs. single infections: 2 vs. 3 ticks), B. miyamotoi (7 vs 9), A. phagocytophilum (14 vs 25) and Ehrlichia sp. (18 vs 30). Observed correlations were statistically significant (χ2 = 36.928, df = 4, P = 0.0001).
Among the co-infected ticks, the most prevalent combinations were B. burgdorferi s.l./Ehrlichia sp. and B. burgdorferi s.l./A. phagocytophilum; these combinations were observed in 30.8% and 26.2% of all co-infected ticks, respectively. The rarest combination was co-infection with B. miyamotoi and A. phagocytophilum, which was detected in 1.5% of co-infections. Notably, there were no cases of multiple (> 2 pathogens) co-infections involving B. miyamotoi. Also, of all B. burgdorferi s.l.-positive ticks, six (9.2%) were co-infected with B. miyamotoi, and the total prevalence of B. burgdorferi s.l. + B. miyamotoi co-infection in the tick population was 1.7%. No other infections were detected in ticks simultaneously infected with B. burgdorferi s.l. and B. miyamotoi. The only significant positive association between pairs of infections was co-infection with A. phagocytophilum and Ehrlichia sp.; this co-infection was detected in 14 ticks, whereas only 7.6 cases were statistically expected if these pathogens were distributed in tick populations by chance only (χ2 = 6.303, df = 1, P = 0.0121). Other infections, including every combination that involved B. burgdorferi s.l. and B. miyamotoi, were not associated with each other, suggesting independent modes of transmission and/or different ecological niches.
Quantification of B. miyamotoi and B. burgdorferi s.l.
In I. persulcatus ticks, the mean concentration (± 95% CI) of B. miyamotoi was 5.2 ± 0.9 log10 genome copies/tick; in comparison, the mean concentration of B. burgdorferi s.l. was 4.0 ± 0.2 log10 genome copies/tick. This difference was statistically significant (difference between median values: 2.0 log10 genome copies/tick; U = 2110.5, P = 0.0021), indicating that the mean load of B. miyamotoi was approximately 28-fold higher than that of B. burgdorferi s.l. To verify that these results are not affected by artifacts of the qPCR test (e.g. by non-specific probe hydrolysis at the late stages of PCR), we compared those ticks with a Borrelia spp. load of > 10 genome copies per reaction. We found that the mean spirochete load was 6.25 ± 0.4 and 4.3 ± 0.4 log10 genome copies/tick for B. miyamotoi and B. burgdorferi s.l., respectively (difference between median values: 2.05 log10 genome copies/tick; U = 1677, P = 0.0001), thereby supporting the significance of an approximately 30-fold higher mean spirochete load of B. miyamotoi in comparison to B. burgdorferi s.l.
In five co-infected ticks, the load of B. miyamotoi was approximately 10- to 50-fold higher than that of B. burgdorferi s.l., with mean quantities of 4.5 ± 0.8 and 3.4 ± 0.4 log10 genome copies per tick, respectively (difference in median values: 1.363 log10 genome copies/tick; U = 23, P = 0.028). In the sixth co-infected tick, the concentration of B. miyamotoi was lower than that of the B. burgdorferi s.l.; however this quantification of B. miyamotoi was not confirmed by flaB sequencing (Table 4).
The distribution of the log-transformed spirochete amounts in the infected ticks significantly differed between B. burgdorferi s.l. and B. miyamotoi. The amounts of B. burgdorferi s.l. were normally distributed in general, with a slight tail to the low values (Fig. 2a). However, the distribution of amounts of B. miyamotoi had a bimodal shape: the load of the first group of ticks was between 2.5 and 3 log10 genome copies/tick, and that of the second group of ticks was– between 5.5 and 7 log10 genome copies/tick (Fig. 2b).

Histograms of the frequency distribution of spirochete loads of Borrelia burgdorferi sensu lato, n = 171 (a) and Borrelia miyamotoi, n = 17 (b) in infected Ixodes persulcatus ticks. Trend lines were constructed using a polynomial regression model
Genotyping of Borrelia sp. in co-infected samples
All 17 samples infected with B. miyamotoi were subjected to PCR targeting flaB gene fragment. Of these, five failed to produce any PCR product; four of these flaB-negative samples contained low amounts of B. miyamotoi DNA (2.9–7.5 genome copies/tick), while the fifth sample had a high bacterial load of 28250 B. miyamotoi genome copies/tick according to 16S rRNA qPCR. Of the other 12 samples, one did not produce sufficient amount of PCR product to perform further research. The remaining 11 amplicons, including six co-infected with B. burgdorferi s.l., were successfully analyzed using NGS. The primer sequences were trimmed from the final gene fragments, and 10 sequences of B. miyamotoi, five sequences of B. bavariensis and four sequences of B. afzelii 397-bp flaB gene fragments were assembled (Table 4).
Five samples were infected solely with B. miyamotoi, which was consistent with the qPCR data. Co-infected sample Mng_B19-16 did not contain any sequences of B. miyamotoi flaB, and only the flaB fragment of B. afzelii was detected in this sample. Of the remaining five co-infected samples, two were double infections with B. miyamotoi and B. bavariensis, two were triple infections with B. miyamotoi, B. bavariensis and B. afzelii and one was a triple infection with B. miyamotoi, B. garinii and B. afzelii. In total, of the 11 analyzed samples, five were infected with B. miyamotoi only, one was infected with B. afzelii only, two were co-infected with two pathogens and three were co-infected with three Borrelia species (Table 4). The Megahit assembly coverage varied from 5 to 16,591 reads per base and, on average, comprised 2236 reads per base for B. miyamotoi, 264 reads per base for B. bavariensis, 162 reads per base for B. afzelii and 43 reads per base for B. garinii (Table 4).
All samples of B. miyamotoi, both single- and co-infecting I. persulcatus, had the identical flaB sequences belonging to the Siberian lineage. All of these sequences were identical to the reference strain B. miyamotoi HT31, isolated from I. persulcatus, as well as with previously described isolates from the Irkutsk Region and Mongolia (Fig. 3a).

Phylogenetic relationships of B. miyamotoi (a) and B. burgdorferi sensu lato (b) co-infecting I. persulcatus ticks. The isolates obtained in this work are indicated by inverted triangles. The evolutionary history was inferred using the maximum likelihood method. The evolutionary distances were computed using the Tamura 3-parameter model TN92. The scale indicates the number of base substitutions per site. The bootstrap support (in percentage, from 1000 replicates) is shown next to the branches; bootstrap values < 70% are hidden. Evolutionary analyses were conducted in MEGA X software
The B. bavariensis nucleotide sequences could be divided into three distinct genotypes. Sample Bb_Mng_B19-11 was closely related to the BgVir strain from the Tomsk Region; two identical samples, Bb_Mng_A19-58 and Bb_Mng_C19-45, clustered with the well-described strain NT29; and Bb_Mng_A19-7 clustered with the reference strain of B. bavariensis PBi (Fig. 3b). At the amino acid level, all studied samples were identical to B. garinii 20047 and B. bavariensis NT29, but not to the B. bavariensis PBi that contained mutation A203S (the residue numbering of B. burgdorferi s.s. B31, accession NC_001318).
The sample Bg_Mng_C19-111 was identical to B. garinii strain JEM3 whose flaB nucleotide sequence was unique in the NCBI BLAST database. The amino acid sequence of Bg_Mng_C19-111 was identical to both B. garinii 20047 and B. bavariensis NT29.
The B. afzelii samples were represented by two genotypes. Three samples (i.e. Ba_Mng_A19-7, Ba_Mng_C19-45 and Ba_Mng_C19-111) were most closely related to the strain Mp6 that was isolated from I. persulcatus in the European part of Russia [44]. Sample Ba_Mng_B19-16 was identical to the B. afzelii PKo strain isolated from the skin of patient in Germany [45], as well as to a number of Siberian and European B. afzelii isolates from different sources. At the amino acid level, samples Ba_Mng_A19-7, Ba_Mng_C19-45 and Ba_Mng_C19-11 had the unique mutation A209S in comparison to the B. afzelii PKo strain (in the residue numbering of NC_001318). No identical mutations were found in other B. afzelii isolates in the NCBI database using BLASTp service.
Discussion
The prevalence of infection
The overall prevalence of TBEV in I. persulcatus in 2019 comprised 1.7% (95% CI: 0.7–4.0), with no significant difference between the Selenge (1.4%, 95% CI: 0.4–5.0) and Bulgan (2.2%, 95% CI: 0.6–7.0) aimags. TBEV prevalence in previous studies was mainly reported for the Selenge aimag and recorded in different years as being 5.5% [46], 1.6% [47], 1.3% [48] and 1.9% [49], with an average estimation of 3.2% [11]. In the Bulgan aimag, the prevalence of TBEV has been reported to vary from 1.5% [11] to 4.4% [46]. Thus, our results, in general, correspond to the most recent evaluations. A slight trend towards a decrease in the overall prevalence of TBEV in I. persulcatus may be assumed. However, a larger sample of ticks and a longer observation period are necessary to precisely evaluate the dynamics of TBEV infection in Mongolian populations of I. persulcatus.
The prevalence of B. burgdorferi s.l. in Mongolian populations of I. persulcatus in different years was reported to be 32.8–36.1% [9], 22–24.5% [34], 55% [19] and 45% [18]. In 2019, the prevalence of the Lyme disease agent was 49.4%, which is close to the highest values registered during the entire period of our surveillance in Mongolia, indicating the high risk of human cases of Lyme disease.
The relapsing fever agent B. miyamotoi from the questing ticks collected in the 2013–2015 periodwas reported in Mongolia in 2017 for the first time [18]. In that period, the prevalence of the infection in I. persulcatus was the same in the Selenge, Bulgan and Huvsugul aimags, averaging 4.5% [18]. This value is very similar to our observations of the pathogen 4–6 years later (4.9%). In other populations of I. persulcatus, the prevalence of the pathogen varied, being 1.6–2% in Japan [18, 30], 2.6–5% in neighboring Inner Mongolia, China [50, 51], 1.7–2.9% in the Irkutsk Region [37, 52], up to 6.3% in Western Siberia [15], 1.8% in the European parts of Russia [53] and about 4% in Finland [54]. Thus, the mean prevalence of B. miyamotoi in Mongolian taiga ticks seems to be relatively high in comparison with other geographic locations, which may increase the risk of relapsing fever disease in the Selenge and Bulgan aimags.
In the Selenge aimag, A. phagocytophilum was previously reported in 6.2% of ticks [55], whereas Ehrlichia muris was only detected in 0.1–2.5% of I. persulcatus ticks [10]. In our samples, these microorganisms were two- to fivefold more prevalent (13.6% vs 16.2%, respectively), which is unusual, not only for Mongolia but also for other geographic locations. There have been several cases of R. sibirica isolation from I. presulcatus in Western Siberia [56]; these previous results allowed us to assume the vector competence of the taiga tick for this pathogen. In addition, Rar and colleagues, who studied 334 adult ticks of I. persulcatus from Western Siberia, reported the prevalence of R. sibirica and R. heilongjiangensis in those ticks to be 2.4% and 0.3%, respectively [15]. In contrast, we did not observe either of these two rickettsia species in taiga ticks despite having a comparable sample size of 346 specimens. In our opinion, the vector ability of I. persulcatus for these bacteria is minimal (if any), whereas the reported cases are sporadic and most likely caused by accidental pathogen “leakage” from sympatric vertebrate and invertebrate reservoir hosts.
The observed variability in prevalence of tick-borne infections at the same locations of Mongolia during the time may be caused not only by natural processes (e.g. if local ecosystems became more suitable for particular pathogen) but by technical biases as well (e.g. by differences in pathogen detection techniques, variable amount of sampling sites, etc.). Further prolonged observations and intense surveillance using unified methodology would help to elucidate the dynamics of tick-borne infections in Mongolian populations of I. persulcatus.
Co-infections
In this work we studied the co-infection of I. persulcatus in Mongolia with seven epidemiologically significant pathogens; this is the first report of a study using this approach. However, the study is restricted by our assumption that B. bavariensis, B. afzelii and B. garinii act as a single pathogen of the B. burgdorferi s.l. complex. However, we propose that our results in this study case are valid from epidemiological perspective because each of these borrelias is a well-recognized agent of Lyme disease, and no other species from the B. burgdorferi s.l. complex has ever been detected in Mongolia or neighboring territories [18, 33, 34, 57, 58].
The abundance of co-infections appeared to be quite high, with about one third of all infected ticks being co-infected with two or more pathogens. The vast majority of co-infections were caused by two pathogens, and no ticks were infected with more than three pathogens. All co-infections involved different combinations of TBEV, B. burgdorferi s.l., A. phagocytophilum and Ehrlichia sp. (most likely E. muris). Interestingly, B. miyamotoi was present at the same prevalence as TBEV; however, the only co-infection observed for this pathogen was for the pair B. burgdorferi s.l./B. miyamotoi, with no statistically significant contingency between these spirochetes. In contrast, TBEV was observed in co-infection with B. burgdorferi s.l., A. phagocytophilum and Ehrlichia sp.
In Mongolia, co-infections with varying combinations of tick-borne pathogens have been occasionally reported in previous studies. For the pair B. burgdorferi s.l./A. phagocytophilum, the prevalence of co-infections was determined to be 4.7% in the Selenge aimag [19]. Co-infection with B. miyamotoi and B. burgdorferi s.l. was observed in 1.2% of ticks from the Selenge, Bulgan and Huvsgol aimags [18]. In our study, corresponding rates were very similar, being 4.9% and 1.7%, respectively.
The independent distribution of pathogens in ticks suggest differences in vertebrate hosts, mechanisms of transmission and, probably, different tissue tropism in vertebrates and ticks.
Quantitative characteristics of Borrelia sp. in I. persulcatus
The quantitative characteristics of Borrelia sp. in I. persulcatus found in the present study are in very good agreement with previously published data on I. scapularis in the USA [29], especially in terms of the almost identical shape of the frequency distribution curves (Fig. 2). However, in our study, the mean spirochete loads in I. persulcatus were approximately 10- to 100-fold higher higher for both species. Thus, for B. burgdorferi s.l., mean counts in I. scapularis versus I. persulcatus were 3155 (or 3.5 log10) and 4.0 log10 spirochetes/infected tick, respectively. For B. miyamotoi, the difference in spirochete loads was even more notable, comprising 4246 (or 3.6 log10) and 5.9 log10 genome copies, respectively. There are two major considerations that may explain these differences. First, differences in the species and life stage studied: in the present study, we studied I. persulcatus adults, while Barbour et al. [29] studied I. scapularis nymphs. Both the species of tick and the life stage may affect the amount of spirochetes harbored by an infected tick. Secondly, our DNA extraction protocol differed from that used by Barbour et al. [29], possibly increasing the detection limit. The synergy of these two considerations may also have occurred. It should be noted, however, that under laboratory conditions, the concentration of B. burgdorferi s.s. in infected nymphs of I. scapularis was found to be 4–5 log10 genome copies/tick [59], which is is very similar to values found in natural populations of I. persulcatus.
In contrast, in the study of questing adult I. persulcatus in the Far East of Russia, the average load of B. burgdorferi s.l. was evaluated to be 5.6 × 107 (or 7.7 log10) genome equivalents/tick [60], which is approximately 1000-fold higher than the values observed in our survey (4.0 log10). The data on B. miyamotoi quantification are also contradictive: 9.52 × 103 (or 4.0 log10) versus to 5.2 log10, respectively. This discrepancy, however, could be explained by technical biases. Indeed, Pukhovskaya and colleagues used a different quantification approach that initially had been developed for B. burgdorferi s.l. solely [61], and B. burgdorferi s.s. B31 was used as universal standard to quantify both B. burgdorferi s.l. and B. miyamotoi [60]. We used the same approach as Barbour and colleagues [29] and quantified both borrelia species simultaneously in a one-tube multiplex qPCR test using separate sets of DNA copy number standards for B. burgdorferi s.l. and B. miyamotoi. As well, our quantification results are supported by two independent methods, namely the qPCR assay of the 16S rRNA gene and NGS analysis of the flaB gene fragment. Therefore, we hypothesize that B. burgdorferi s.l. and B. miyamotoi co-exist in the ecosystems with I. persulcatus as a competent vector using the mechanism of partitioning of ecological niches, similar to that discovered for B. burgdorferi s.s. and B. miyamotoi in ecosystems with I. scapularis being the competent vector [29].
Genetic diversity of Borrelia spp. in co-infected ticks
To identify the species and perform phylogenetic analysis of Borrelia spp. in six ticks co-infected with B. burdorferi s.l. and B. miyamotoi and in five ticks with single infection of B. miyamotoi, we used the high-throughput massive parallel sequencing of the flaB gene fragment. flaB is a chromosomal gene encoding flagellin B, a core protein in the spirochete periplasmic flagella [62]. This gene is localized in a chromosomal part of the genome and is highly conserved among different species of the genus Borrelia, which makes it a popular target for universal PCR detection systems as it allows the detection and quantification of borrelia infections irrespective of the causative species and plasmid strain [63, 64]. Despite its high level of conservation, there are numerous minor differences in the nucleotide sequences of flaB, and these enable both the species of borreliae to be identied as well as reconstruction of the phylogenetic relationships of the borreliae [19, 65, 66].
Using this approach, we successfuly characterized Borrelia sp. diversity in six co-infected ticks. It appeared that up to three different species, i.e. B. miyamotoi, B. afzelii and B. bavariensis or B. garinii, may infect a single tick simultaneously. The prevalence of triple borrelial infections was quite high, about 1% of the entire I. persulcatus population, however no quadruple infection was detected. Previously, only dual co-infections of B. garinii with B. afzelii (7.8%) or B. bavariensis (6.2%) were reported [19].
The Mng_B19-16 sample contained both B. miyamotoi and B. burgdorferi s.l. DNA according to the qPCR targeting the 16S rRNA gene. However, only B. afzelii was detected in the sample when the flaB amplicon was analyzed. In this sample, the concentration of B. miyamotoi was approximately 10,000-fold lower than that of B. afzelii (2.5 vs 6.5 log10 copies/tick, respectively), whereas in other co-infected samples the difference in concentrations did not exceed 100-fold (Table 4). Most likely, the less abundant flaB template of B. miyamotoi failed to compete with the much more abundant B. afzelii template. In this case, for successful resolution of closely related pathogens in co-infected ticks, the difference in concentrations of the microorganisms should not exceed 2–3 log10 genome copies per sample. Another explanation could be false-positive signal for B. miyamotoi during qPCR targeting the 16S rRNA gene due to, for example, non-specific Taqman probe binding and hydrolysis on the template of B. afzelii or some other bacteria in that particular tick. In this case, every sample with a low concentration of B. miyamotoi should be assumed as suspicious. Notably, every single-infected sample with a very low (below 10 copies per tick) concentration of B. miyamotoi failed to produce the flaB PCR fragment. To resolve this issue, the detailed study of ticks with low concentrations of Borrelia sp. is necessary.
There was no diversity in B. miyamotoi detected in Mongolia. All characterized isolates were highly homogeneous and belonged to the Siberian lineage that is associated with I. persulcatus and widely distributed from Japan to Europe [18, 67]. In contrast, the remarkable diversity of Lyme disease agents was described. Thus, it had been shown previously that 26.3% of B. burgdorferi s.l. belong to B. garinii 20047, followed by 7.8% of B. afzelii and 7.0% of B. garinii NT29 type (nowadays B. bavariensis) [19]. In another study of 91 borreliae-positive ticks, 62% were infected with the B. bavariensis, 13% were infected with B. afzelii and 15% were co-infected with both species [34]. Generally, the genetic diversity of Borrelia sp. in co-infected samples in our study is rather high and corresponds to previously published data [34]. No strict geographic associations in the phylogeny of Borrelia sp. were detected for studied isolates from Mongolia, which could be explained by an intense exchange of the isolates between different populations of I. persulcatus and with closely related I. ricinus ticks. These results also characterize the NGS as a powerful tool that is very effective in studies of tick-borne co-infections.
Conclusion
At least five vector-borne pathogens, i.e. TBEV, B. burgdorferi s.l., B. miyamotoi, A. phagocytophilum and Ehrlichia sp. (most likely, E. muris), simultaneously circulate among I. persulcatus ticks in the Selenge and Bulgan aimags of Mongolia, and > 64% of ticks are infected with at least one pathogen. The analysis of contingency tables of infections indicates that these microorganisms persist independently of each other and, probably, occupy different ecological niches. The only exception is the pair A. phagocytophilum and Ehrlichia sp., which occur significantly more often as co-infection than expected for independent distribution, suggesting a common mode of transmission, shared range of vertebrate hosts and tissue tropism. Among the I. persulcatus ticks, B. burgdorferi s.l. had a tenfold higher prevalence of infection than B. miyamotoi, but the spirochete loads of B. burgdorferi s.l. in infected ticks were, vice versa, 28- to 30-fold lower than concentration of B. miyamotoi. From the healthcare perspective, this result could mean that in spite of a lower abundance of relapsing fever agent, the risk of infection with this pathogen is similar to that of Lyme disease due to the higher initial dose of pathogen in each case of infection. The analysis of the flaB gene fragment of Borrelia sp. in five single-infected and six co-infected samples revealed that all 10 B. miyamotoi isolates belong to the Siberian evolutionary lineage. The Borrelia burgdorferi s.l. was represented by B. afzelii (four isolates), B. bavariensis (four isolates) and B. garinii (one isolate). The most diverse group was B. bavariensis, consisting of three genotypes, and B. afzelii, divided into two genotypes; in contrast, B. garinii was represented by single isolate only. A unique amino acid mutation of alanine to serine was discovered in residue 209 of the flaB gene of Mongolian isolates of B. afzelii. Introduction of quantitative analysis of the kinetics of tick-borne infections in vectors, vertebrate hosts and human patients would enhance our understanding of the persistence of these pathogens in nature and help to improve the preventive measures, diagnostics and treatment of tick-borne diseases.
Availability of data and materials
The data generated in this study are available from the corresponding author upon request.
References
Baasandavga U, Badrakh B, Burged N, Davaajav O, Khurelsukh T, Barnes A, et al. A case series of fatal meningoencephalitis in Mongolia: epidemiological and molecular characteristics of tick-borne encephalitis virus. West Pac Surveill Response J WPSAR. 2019;10:25–31.
von Fricken ME, Rolomjav L, Illar M, Altantogtokh D, Hogan KM, Uyanga B, et al. Geographic range of Lyme Borreliosis in Mongolia. Vector Borne Zoonotic Dis. 2019;19:658–61.
Walder G, Lkhamsuren E, Shagdar A, Bataa J, Batmunkh T, Orth D, et al. Serological evidence for tick-borne encephalitis, borreliosis, and human granulocytic anaplasmosis in Mongolia. Int J Med Microbiol. 2006;296:69–75.
Lkhagvatseren S, Hogan KM, Boldbaatar B, von Fricken ME, Anderson BD, Pulscher LA, et al. Discrepancies between self-reported tick bites and evidence of tick-borne disease exposure among nomadic Mongolian herders. Zoonoses Public Health. 2019;66:480–6.
Xopoo Yндэcний C. Mongolian Statistical Information Service. Yндэcний Cтaтиcтикийн Xopoo. 2022. https://1212.mn. Accessed 20 Apr 2022.
Dash M, Biambaa B, Neronov VM. The ixodid tick fauna of the Mongolian People’s Republic. I. The species distribution [Fauna iksodovykh kleshcheǐ MNR. Soobshchenie I. Rasprostranenie vidov]. Meditsinskaya Parazitol Parazit Bolezni. 1988;3:37–42.
Danchinova GA, Khasnatinov MA, Abmed D, Bataa J, Nyamdavaa P, Tserennorov D, et al. Fauna and ecology of ixodid ticks in Mongolia. Acta Biomed Sci East Sib Biomed J. 2007;S3:90–3.
Narankhajid M, Yeruult C, Gurbadam A, Battsetseg J, Aberle SW, Bayartogtokh B, et al. Some aspects on tick species in Mongolia and their potential role in the transmission of equine piroplasms, Anaplasma phagocytophilum and Borrelia burgdorferi L. Parasitol Res. 2018;117:3557–66.
Danchinova GA, Khasnatinov MA, Zlobin VI, Kozlova IV, Verkhozina MM, Sountsova OV, et al. Ixodid ticks in Southern part of Eastern Siberia and Mongolia and their spontaneous infectiveness by infectious agents. Bull Sib Med. 2006;5:137–43.
von Fricken ME, Qurollo BA, Boldbaatar B, Wang Y-W, Jiang R-R, Lkhagvatseren S, et al. Genetic diversity of Anaplasma and Ehrlichia bacteria found in Dermacentor and Ixodes ticks in Mongolia. Ticks Tick-Borne Dis. 2020;11:101316.
Černý J, Buyannemekh B, Needham T, Gankhuyag G, Oyuntsetseg D. Hard ticks and tick-borne pathogens in Mongolia—a review. Ticks Tick-Borne Dis. 2019;10:101268.
von Fricken ME, Voorhees MA, Koehler JW, Asbun C, Lam B, Qurollo B, et al. Molecular characteristics of rickettsia in ticks collected along the southern border of Mongolia. Pathogens. 2020;9:943.
Socolovschi C, Mediannikov O, Raoult D, Parola P. The relationship between spotted fever group Rickettsiae and ixodid ticks. Vet Res. 2009;40:34.
Igolkina Y, Rar V, Vysochina N, Ivanov L, Tikunov A, Pukhovskaya N, et al. Genetic variability of Rickettsia spp in Dermacentor and Haemaphysalis ticks from the Russian Far East. Ticks Tick-Borne Dis. 2018;9:1594–603.
Rar V, Livanova N, Sabitova Y, Igolkina Y, Tkachev S, Tikunov A, et al. Ixodes persulcatus/pavlovskyi natural hybrids in Siberia: Occurrence in sympatric areas and infection by a wide range of tick-transmitted agents. Ticks Tick-Borne Dis. 2019;10:101254.
Speck S, Derschum H, Damdindorj T, Dashdavaa O, Jiang J, Kaysser P, et al. Rickettsia raoultii, the predominant Rickettsia found in Mongolian Dermacentor nuttalli. Ticks Tick-Borne Dis. 2012;3:227–31.
Inokuma H, Ohashi M, Tanabe S, Miyahara K. Prevalence of tick-borne rickettsia and ehrlichia in Ixodes persulcatus and Ixodes ovatus in Tokachi District, Eastern Hokkaido, Japan. J Vet Med Sci. 2007;69:661–4.
Iwabu-Itoh Y, Bazartseren B, Naranbaatar O, Yondonjamts E, Furuno K, Lee K, et al. Tick surveillance for Borrelia miyamotoi and phylogenetic analysis of isolates in Mongolia and Japan. Ticks Tick-Borne Dis. 2017;8:850–7.
Masuzawa T, Masuda S, Fukui T, Okamoto Y, Bataa J, Oikawa Y, et al. PCR detection of Anaplasma phagocytophilum and Borrelia burgdorferi in Ixodes persulcatus ticks in Mongolia. Jpn J Infect Dis. 2014;67:47–9.
Korenberg EI, Shcherbakov SV, Bannova GG, Levin ML. Karavanov AS [The infectiousness of Ixodes persulcatus ticks with the causative agents of Lyme disease and tick-borne encephalitis simultaneously]. Parazitologiia. 1990;24:102–5.
Alekseev AN, Dubinina HV, Jushkova OV. First report on the coexistence and compatibility of seven tickborne pathogens in unfed adult Ixodes persulcatus Schulze (Acarina: Ixodidae). Int J Med Microbiol Suppl. 2004;293:104–8.
Movila A, Dubinina HV, Sitnicova N, Bespyatova L, Uspenskaia I, Efremova G, et al. Comparison of tick-borne microorganism communities in Ixodes spp. of the Ixodes ricinus species complex at distinct geographical regions. Exp Appl Acarol. 2014;63:65–76.
Sormunen JJ, Andersson T, Aspi J, Bäck J, Cederberg T, Haavisto N, et al. Monitoring of ticks and tick-borne pathogens through a nationwide research station network in Finland. Ticks Tick-Borne Dis. 2020;11:101449.
Morozova OV, Dobrotvorsky AK, Livanova NN, Tkachev SE, Bakhvalova VN, Beklemishev AB, et al. PCR Detection of Borrelia burgdorferi sensu lato, tick-borne encephalitis virus, and the human granulocytic ehrlichiosis agent in Ixodes persulcatus ticks from Western Siberia, Russia. J Clin Microbiol. 2002;40:3802–4.
Ginsberg HS. Potential effects of mixed infections in ticks on transmission dynamics of pathogens: comparative analysis of published records. Exp Appl Acarol. 2008;46:29–41.
Diuk-Wasser MA, Vannier E, Krause PJ. Coinfection by the tick-borne pathogens Babesia microti and Borrelia burgdorferi: ecological, epidemiological and clinical consequences. Trends Parasitol. 2016;32:30–42.
Margos G, Gofton A, Wibberg D, Dangel A, Marosevic D, Loh S-M, et al. The genus Borrelia reloaded. PLoS ONE. 2018;13:e0208432.
Stanek G, Strle F. Lyme borreliosis—from tick bite to diagnosis and treatment. FEMS Microbiol Rev. 2018;42:233–58.
Barbour AG, Bunikis J, Travinsky B, Hoen AG, Diuk-Wasser MA, Fish D, et al. Niche partitioning of Borrelia burgdorferi and Borrelia miyamotoi in the same tick vector and mammalian reservoir species. Am J Trop Med Hyg. 2009;81:1120–31.
Takano A, Toyomane K, Konnai S, Ohashi K, Nakao M, Ito T, et al. Tick surveillance for relapsing fever spirochete Borrelia miyamotoi in Hokkaido, Japan. PLoS ONE. 2014;9:e104532.
Hamšíková Z, Coipan C, Mahríková L, Minichová L, Sprong H, Kazimírová M. Borrelia miyamotoi and co-infection with Borrelia afzelii in Ixodes ricinus ticks and rodents from Slovakia. Microb Ecol. 2017;73:1000–8.
Raileanu C, Moutailler S, Pavel I, Porea D, Mihalca AD, Savuta G, et al. Borrelia diversity and co-infection with other tick borne pathogens in ticks. Front Cell Infect Microbiol. 2017;7:36.
Sabitova Y, Fomenko N, Tikunov A, Stronin O, Khasnatinov M, Abmed D, et al. Multilocus sequence analysis of Borrelia burgdorferi sensu lato isolates from Western Siberia, Russia and Northern Mongolia. Infect Genet Evol. 2018;62:160–9.
Scholz HC, Margos G, Derschum H, Speck S, Tserennorov D, Erdenebat N, et al. High prevalence of genetically diverse Borrelia bavariensis-like strains in Ixodes persulcatus from Selenge aimag. Mongolia Ticks Tick-Borne Dis. 2013;4:89–92.
Filippova NA. Ixodid ticks of the subfamily Ixodinae. Fauna of USSR. Arachnida. Leningrad: Nauka; 1977.
Schwaiger M, Cassinotti P. Development of a quantitative real-time RT-PCR assay with internal control for the laboratory detection of tick borne encephalitis virus (TBEV) RNA. J Clin Virol. 2003;27:136–45.
Khasnatinov MA, Danchinova GA, Takano A, Kawabata H, Ohashi N, Masuzawa T. Prevalence of Borrelia miyamotoi in Ixodes persulcatus in Irkutsk City and its neighboring territories. Russia Ticks Tick-Borne Dis. 2016;7:394–7.
Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Čech M, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–44.
Seemann T. Shovill: Faster SPAdes assembly of Illumina reads. 2017. https://github.com/tseemann/shovill. Accessed 20 Apr 2022.
Hall TA. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Tamura K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol Biol Evol. 1992;9:678–87.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Mina MJ, Burke RM, Klugman KP. Estimating the prevalence of coinfection with influenza virus and the atypical bacteria Bordetella pertussis, Chlamydophila pneumoniae, and Mycoplasma pneumoniae. Eur J Clin Microbiol Infect Dis. 2014;33:1585–9.
Masuzawa T, Kharitonenkov IG, Kadosaka T, Hashimoto N, Kudeken M, Takada N, et al. Characterization of Borrelia burgdorferi sensu lato isolated in Moscow province–a sympatric region for Ixodes ricinus and Ixodes persulcatus. Int J Med Microbiol. 2005;294:455–64.
Wilske B, Preac-Mursic V, Göbel UB, Graf B, Jauris S, Soutschek E, et al. An OspA serotyping system for Borrelia burgdorferi based on reactivity with monoclonal antibodies and OspA sequence analysis. J Clin Microbiol. 1993;31:340–50.
Khasnatinov M, Tserennorov D, Nymadavaa P, Tchaporgina EA, Glushenkova T, Arbatskaya E, et al. Tick-borne encephalitis virus in Mongolia. Int J Infect Dis. 2010;14:e372–3.
Frey S, Mossbrugger I, Altantuul D, Battsetseg J, Davaadorj R, Tserennorov D, et al. Isolation, preliminary characterization, and full-genome analyses of tick-borne encephalitis virus from Mongolia. Virus Genes. 2012;45:413–25.
Muto M, Bazartseren B, Tsevel B, Dashzevge E, Yoshii K, Kariwa H. Isolation and characterization of tick-borne encephalitis virus from Ixodes persulcatus in Mongolia in 2012. Ticks Tick-Borne Dis. 2015;6:623–9.
Boldbaatar B, Jiang R-R, von Fricken ME, Lkhagvatseren S, Nymadawa P, Baigalmaa B, et al. Distribution and molecular characteristics of rickettsiae found in ticks across Central Mongolia. Parasit Vectors. 2017;10:61.
Sato K, Liu D, Cui Y, Yin X, et al. Surveillance of Borrelia miyamotoi-carrying ticks and genomic analysis of isolates in Inner Mongolia, China. Parasit Vectors. 2021;14:368.
Gao Y, Lv X-L, Han S-Z, Wang W, Liu Q, Song M. First detection of Borrelia miyamotoi infections in ticks and humans from the northeast of Inner Mongolia. China Acta Trop. 2021;217:105857.
Fomenko NV, Livanova NN, Borgoiakov VI, Kozlova IV, Shulaĭkina IV, Pukhovskaia NM, et al. Detection of Borrelia miyamotoi in ticks Ixodes persulcatus from Russia. Parazitologiia. 2010;44:201–11.
Masuzawa T, Sakakibara K, Suzuki K, Sato H, Yasuda S. Detection of Asian-type Borrelia miyamotoi from Ixodes ricinus inhabiting Tver Province (Russia): a sympatric region for I. ricinus and Ixodes persulcatus. Vector-Borne Zoonotic Dis. 2020;20:921–3.
Pakanen V-M, Sormunen JJ, Sippola E, Blomqvist D, Kallio ER. Questing abundance of adult taiga ticks Ixodes persulcatus and their Borrelia prevalence at the north-western part of their distribution. Parasit Vectors. 2020;13:384.
Karnath C, Obiegala A, Speck S, Essbauer S, Derschum H, Scholz H, et al. Detection of Babesia venatorum, Anaplasma phagocytophilum and Candidatus Neoehrlichia mikurensis in Ixodes persulcatus ticks from Mongolia. Ticks Tick-Borne Dis. 2016;7:357–60.
Shpynov SN, Fournier P-E, Rudakov NV, Samoilenko IE, Reshetnikova TA, Yastrebov VK, et al. Molecular identification of a collection of spotted fever group rickettsiae obtained from patients and ticks from Russia. Am J Trop Med Hyg. 2006;74:440–3.
Ni X-B, Jia N, Jiang B-G, Sun T, Zheng Y-C, Huo Q-B, et al. Lyme borreliosis caused by diverse genospecies of Borrelia burgdorferi sensu lato in northeastern China. Clin Microbiol Infect. 2014;20:808–14.
Ostapchuk YO, Perfilyeva YV, Zhigailov AV, Maltseva ER, Neupokoyeva AS, Bissenbay AO, et al. Monitoring of pathogenic Borrelia burgdorferi sensu lato in the Almaty oblast. Kazakhstan Ticks Tick-Borne Dis. 2021;12:101725.
Hodzic E, Feng S, Freet KJ, Borjesson DL, Barthold SW. Borrelia burgdorferi population kinetics and selected gene expression at the host-vector interface. Infect Immun. 2002;70:3382–8.
Pukhovskaya NM, Morozova OV, Vysochina NP, Belozerova NB, Ivanov LI. Prevalence of Borrelia burgdorferi sensu lato and Borrelia miyamotoi in ixodid ticks in the Far East of Russia. Int J Parasitol Parasites Wildl. 2019;8:192–202.
Morozova OV, Grishechkin AE, Konkova-Reidman AB. Quantitations of Borrelia and Bartonella DNA and tick-borne encephalitis virus RNA in Ixodes persulcatus ticks collected in Chelyabinsk region. Mol Genet Microbiol Virol. 2011;26:41–5.
Ge Y, Li C, Corum L, Slaughter CA, Charon NW. Structure and expression of the FlaA periplasmic flagellar protein of Borrelia burgdorferi. J Bacteriol. 1998;180:2418–25.
de Leeuw BHCGM, Maraha B, Hollemans L, Sprong H, Brandenburg AH, Westenend PJ, et al. Evaluation of Borrelia real time PCR DNA targeting OspA, FlaB and 5S–23S IGS and Borrelia 16S rRNA RT-qPCR. J Microbiol Methods. 2014;107:41–6.
Nunes M, Parreira R, Carreira T, Inácio J, Vieira ML. Development and evaluation of a two-step multiplex TaqMan real-time PCR assay for detection/quantification of different genospecies of Borrelia burgdorferi sensu lato. Ticks Tick-Borne Dis. 2018;9:176–82.
Fukunaga M, Okada K, Nakao M, Konishi T, Sato Y. Phylogenetic analysis of Borrelia species based on flagellin gene sequences and its application for molecular typing of Lyme disease borreliae. Int J Syst Bacteriol. 1996;46:898–905.
Wodecka B. flaB gene as a molecular marker for distinct identification of Borrelia species in environmental samples by the PCR-restriction fragment length polymorphism method. Appl Environ Microbiol. 2011;77:7088–92.
Nakao R, Kasama K, Boldbaatar B, Ogura Y, Kawabata H, Toyoda A, et al. The evolution of hard tick-borne relapsing fever borreliae is correlated with vector species rather than geographical distance. BMC Ecol Evol. 2021;21:105.
Acknowledgements
High-throughput sequencing was performed on a Illumina NextSeq 550 sequencer of the Collective Research Center “Center for the development of progressive personalized technologies for health” SC FHHRP, Irkutsk. All authors thank Viacheslav V. Sinkov (SC FHHRP, Irkutsk) for technical assistance in sequencing.
Funding
This research was funded by the budget of FSPSI Scientific Centre for family health and human reproduction problems (SC FHHRP), Irkutsk, Russian Federation.
Author information
Authors and Affiliations
Contributions
All authors have agreed to be personally accountable for the author's own contributions. All authors ensure that questions related to the accuracy or integrity of any part of the work, even ones for which the author was not personally involved, are appropriately investigated and resolved, and the resolution documented in the literature.
Conception and design of the work: MAK, GAD, NE and DA. Acquisition of data: EKL, MAK, NAL, DT, GO, DN, NE, DA, KS and HK. Analysis and interpretation of data: MAK, EKL, DA, GAD and HK. MAK, HK and EKL drafted the work and substantively revised it. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. This research did not involve humans, client-owned animals, vertebrates of regulated invertebrates, as well as human embryos, gametes and stem cells.
Consent for publication
Not applicable. This research did not include details, images, or videos relating to an individual person.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lagunova, E.K., Liapunova, N.A., Tuul, D. et al. Co-infections with multiple pathogens in natural populations of Ixodes persulcatus ticks in Mongolia. Parasites Vectors 15, 236 (2022). https://doi.org/10.1186/s13071-022-05356-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05356-x