
Abstract
Background
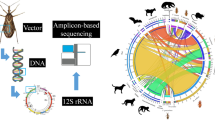
Tsetse flies can transmit various Trypanosoma spp. that cause trypanosomiasis in humans, wild animals, and domestic animals. Amplicon deep sequencing of the 12S ribosomal RNA (rRNA) gene can be used to detect mammalian tsetse hosts, and the 18S rRNA gene can be used to detect all associated eukaryotic pathogens, including Trypanosoma spp.
Methods
Tsetse flies were collected from the Serengeti National Park (n = 48), Maswa Game Reserve (n = 42), and Tarangire National Park (n = 49) in Tanzania in 2012–13. Amplicon deep sequencing targeting mammal-specific 12S rRNA and 18S rRNA genes was performed to screen the blood-feeding sources of tsetse flies and eukaryotic parasites in tsetse flies, respectively.
Results
12S rRNA gene deep sequencing revealed that various mammals were blood-feeding sources of the tsetse flies, including humans, common warthogs, African buffalos, mice, giraffes, African elephants, waterbucks, and lions. Genes of humans were less frequently detected in Serengeti (P = 0.0024), whereas African buffaloes were detected more frequently as a blood-feeding source (P = 0.0010). 18S rRNA gene deep sequencing showed that six tsetse samples harbored the Trypanosoma gene, which was identified as Trypanosoma godfreyi and Trypanosoma simiae in subsequent ITS1 gene sequencing.
Conclusions
Through amplicon deep sequencing targeting the 12S rRNA and 18S rRNA genes, various mammalian animals were identified as blood-meal sources, and two Trypanosoma species were detected in tsetse flies collected from the Maswa Game Reserve, Serengeti National Park, and Tarangire National Park in Tanzania. This study illustrates the patterns of parasitism of tsetse fly, wild animals targeted by the fly, and Trypanosoma spp. carried by the fly in Tanzania. It may provide essential data for formulating better strategies to control African trypanosomes.
Graphical Abstract

Similar content being viewed by others
Background
The tsetse fly is a vector of Trypanosoma brucei gambiense and T.b. rhodesiense, both of which can cause African human trypanosomiasis (HAT) (also known as sleeping sickness). The tsetse fly is also a vector of T. vivax, T. simiae, T.b. brucei, and T. congolense, all of which can cause trypanosomiasis in wild and domestic animals (AAT) [1]. HAT and AAT are severe and sometimes fatal diseases affecting the central nervous system in humans and the nervous system and muscles in animals, respectively. The spread of AAT consequently puts a significant constraint on the development of animal husbandry in Africa while HAT is a severe public health problem. As adult tsetse flies are frequently found on cattle and horses, among other animals, and feed on animal blood, they can carry parasites that cause HAT and AAT. There is no cure for either disease, and the only method of prevention involves controlling tsetse flies [2, 3].
Preference of tsetse flies for blood-feeding hosts can vary significantly depending on the species, wildlife, and geographic location [4, 5]. Tsetse flies feed on various wild and domestic mammals [6]. For example, humans, cattle, dogs, bush pigs, African buffaloes, warthogs, greater kudus, rats, and bats were confirmed as the blood-meal sources of tsetse flies in Zambia, using 12S ribosomal RNA (rRNA) gene deep sequencing [7]. Analysis of tsetse flies using vertebrate cytochrome c oxidase I (COX1) and cytochrome b gene polymerase chain reaction (PCR) revealed that humans are the most common vertebrate hosts [8]. This also indicated that other wild species, such as hippopotamuses, African buffaloes, African savannah elephants, and giraffes, may be involved in trypanosomiasis transmission. Blood meal collection and identification are essential for determining the hosts of tsetse flies for epidemiological studies and controlling their population. A study, in which the blood of vertebrates was analyzed in tsetse flies, revealed that the changes in the environment, fauna, and host availability can affect tsetse feeding patterns [9].
The internal transcribed spacer (ITS) region of ribosomal DNA (rDNA) is commonly used to detect Trypanosoma spp. in tsetse flies because of the highly conserved flanking regions and size variability between Trypanosoma spp. and their subgroups [10,11,12]. Recently, attempts have been made to detect Trypanosoma spp. by using the 18S rRNA region [6, 13]. The 18S rRNA region in eukaryotes is highly conserved across species and allows the detection of a variety of eukaryotic organisms [14,15,16].
The recent development of high-throughput sequencing allows the use of a metagenomic approach to detect all prokaryotic and eukaryotic species in a sample in a single sequencing run at a low cost [17,18,19]. In this study, we used amplicon deep sequencing of the 12S rRNA and 18S rRNA genes to identify the mammalian hosts of the tsetse fly and associated eukaryotic pathogens (including Trypanosoma spp.), respectively. This efficient, inexpensive, and sensitive method for monitoring biodiversity may provide essential information for formulating new strategies to control tsetse flies in Africa.
Methods
Sample collection and identification of tsetse flies
We collected tsetse flies between January 2012 and February 2013 from the Serengeti National Park (n = 48), Maswa Game Reserve (n = 42), and Tarangire National Park (n = 49) in Tanzania. The samples were collected within a 5-km radius of our accommodation (Serengeti National Park-2.434974741607467, 34.85272334722886; Maswa Game Reserve-3.2568282434634996, 34.595773504540574; Tarangire National Park-3.991658476578967, 35.96541568649041). The tsetse flies were caught using a net mounted on the back of a moving vehicle and preserved in absolute ethanol. DNA was extracted from each tsetse fly using a Nucleospin Tissue Kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions and stored in a deep freezer until testing. Molecular identification of tsetse flies was performed by ITS2 gene amplification and sequencing [20].
Illumina sequencing and bioinformatics
The 18S rRNA V9 region was identified as 1391f (5'-TCGTCGGCAGCGTCAGATG TGTATAAGAGACAG GTACACACCGCCCGTC-3') and EukBr (5'-GTCTCGTGGG CTCGGAGATGTGTATAAGAGACAGTGATCCTTCTGCAGGTTCACCTAC-3') [17]. The 12S rRNA genes were identified as L1085 (5'-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCCAAACTGGGATTAGATAACCC-3') and H1259 (5'-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGTTTGCTGAAGATGGCGGTA-3') [18]. A limited-cycle (eight cycles) amplification step was performed to add multiplexing indices and Illumina sequencing adapters. Mixed amplicons were pooled and sequenced on an Illumina iSeq 100 sequencing system using the Illumina iSeq™ 100 i1 Reagent v2 kit (San Diego, CA, USA) according to the manufacturer’s instructions.
Geneious Prime® 2022.0.2 (Biomatters Ltd., Auckland, New Zealand) was used to process and assemble raw 18S V9 and 12S rRNA reads in the following steps [21, 22]. Sequences < 100 bp were deleted, and 151 bp regions were amplified. The forward and reverse reads were merged to produce a single consensus sequence. Closely related sequences were clustered into separate contigs using de novo assembly. We used the default setting according to the online manual (https://www.geneious.com/tutorials/metagenomic-analysis/), using ‘Minimum Overlap Identity’ as 98%. Operational taxonomic units (OTUs) were defined via sequence clustering using Basic Local Alignment Search Tool (BLAST) on NCBI “nt” GenBank database (November 2021 version). A curated database was created for taxonomic classification. BLAST hits were used to create a sequence classification database. Lastly, the extracted BLAST hits were assigned the name of the source organism.
Polymerase chain reaction and sequencing analysis
PCR was performed to identify Trypanosoma spp. using the following primer sets: ITS1 CF (5'-CCGGAAGTTCACCGATATTG-3') and ITS1 BR (5'-TTGCTGCGTTCTTCAACGAA-3') [10]. Sequencing of positive PCR amplicons was performed by Bionics Co., Ltd. (Seoul, Korea). A BLAST search was used.
To compare the sequence of the obtained ITS1 gene with the series available in GenBank, the obtained sequence was compared to the line deposited in GenBank using BLAST. Gene sequences, except for the primer regions, were aligned using the Multisequence Alignment Program (Geneious).
Results
Among the 139 tsetse flies collected from the Maswa Game Reserve (n = 48), Serengeti National Park (n = 42), and Tarangire National Park (n = 49), 2 tsetse flies in Tarangire National Park (T51 and T52) were identified as Glossina morsitans, and the remaining samples were identified as Glossina swynnertoni.
Amplicon deep sequencing targeting a mammalian-specific 12S rRNA gene was performed to determine the blood-meal sources of collected tsetse flies. Only 100 samples (41 from Maswa, 29 from Serengeti, and 30 from Tarangire) successfully underwent sequencing and bioinformatics analysis. Various mammalian genes were detected, including those of humans, common warthogs, African buffaloes, mice, giraffes, African elephants, waterbucks, domestic pigs, Thomson’s gazelles, duikers, and lions (Fig. 1). Human genes were primarily found in the tsetse flies of all three regions: 39 (95.12%) in Maswa, 19 (65.52%) in Serengeti, and 27 (90.00%) in Tarangire (Table 1). Genes of humans, common warthogs, African buffaloes, mice, and giraffes were detected in all three regions. In particular, fewer human genes were detected in Serengeti (P = 0.0024), while the African buffalo was identified as a blood-meal source here (P = 0.0010). In the Maswa Game Reserve, genes of one African elephant, one waterbuck, and one duiker were detected. Genes of one Thomson’s gazelle and one lion were detected in Serengeti National Park. Genes of four African elephants and one domestic pig were detected in Tarangire National Park. There were many samples in which the genes of several mammals were simultaneously detected, particularly human genes (Fig. 1; Table 1). In addition, differences in animal blood-meal sources between male and female tsetse flies were not observed (Additional file 1).

Composition of tsetse fly blood-meal sources collected in the A Maswa Game Reserve (N = 41), B Serengeti National Park (N = 29), and C Tarangire National Park (N = 30). Mammalian-specific 12S rRNA gene deep sequencing was performed for each fly sample. D The average relative abundance of tsetse fly blood-meal sources in the Maswa Game Reserve, Serengeti National Park, and Tarangire National Park. Taxa with < 5% relative abundance are included in ‘Others’
Amplicon sequencing targeting the 18S rRNA V9 region was performed to screen for eukaryotic pathogens, including Trypanosoma spp., and 139 samples were successfully sequenced and analyzed. Of these, six tsetse samples harbored Trypanosoma genes: three from Maswa, two from Serengeti, and one from Tarangire (Table 2). In addition, human, fungal, and plant genes were detected (Table 2). Trypanosoma-specific PCR targeting the ITS1 region, conventional DNA sequencing, and homology analysis were performed for six samples to determine the species. Two samples from Serengeti were identified as Trypanosoma godfreyi and one sample from Maswa was identified as Trypanosoma simiae (Fig. 2; Table 3). The remaining three samples were not analyzed.

Trypanosoma-specific PCR targeting the ITS1 region. The M10, M21, and M60 samples were tsetse flies collected from the Maswa Game Reserve; S17 and S55 were from Serengeti National Park; T73 was from Tarangire National Park
Discussion
Of the 139 tsetse flies collected in Tanzania, 2 were identified as Glossina morsitans, and the remaining samples were identified as Glossina swynnertoni. This is similar to a previous study analyzing 21,107 tsetse flies which reported that the major tsetse fly species was G. swynnertoni (55.9%), while G. morsitans (6.0%) was less prevalent [23].
Amplicon deep sequencing was performed using the mammalian-specific 12S rRNA gene to determine the sources of tsetse blood meals. PCR sequencing has previously been performed to determine the species of tsetse flies [24]; however, analyzing the genes of all the collected samples by sequencing requires considerable time and effort. Therefore, deep amplicon sequencing has more recently been utilized to analyze the 12S rRNA gene region of vertebrates [7, 18, 25,26,27].
Analysis of humans, cattle, dogs, bush pigs, African buffaloes, warthogs, greater kudus, rats, and bats was necessary to confirm the sources of tsetse fly blood meals using 12S rRNA gene deep sequencing [7]. Various mammalian genes including those of humans, common warthogs, African buffaloes, mice, giraffes, African elephants, waterbucks, domestic pigs, Thomson's gazelles, duikers, and lions were detected in our tsetse fly samples. This is consistent with previous studies that identified blood-meal sources by analyzing the mitochondrial cytochrome b gene and indicated that humans, hippopotamuses, African buffaloes, African savannah elephants, and giraffes are reservoirs for trypanosomiasis transmission [28, 29].
In Serengeti, the African buffalo was found to be a more significant blood-meal source than in Maswa and Tarangire (Table 1). The African buffalo gene was found in 34.48% of tsetse flies in Serengeti and 7.32% and 3.33% of tsetse flies in Maswa and Tarangire, respectively. This shows that the African buffalo is a major blood-meal source for tsetse flies in Serengeti. In contrast, humans were found to be a significantly less common bloodmeal source in Serengeti. Tsetse flies likely have a greater chance of contact with wild animals such as African buffaloes and common warthogs in Serengeti than that in the other two regions.
Human genes were found in all three of the Maswa Game Reserve, Serengeti National Park, and Tarangire National Park. Several mammalian genes are commonly simultaneously detected in individual flies [18, 25]. The diet of tsetse flies may change if there are not enough animals from which they can draw blood or if there are houses nearby [7, 9]. This means that tsetse flies will suck human blood if given the opportunity.
The Trypanosoma spp. gene was identified by 18S rRNA gene deep sequencing. PCR targeted ITS1 confirmed that two samples from the Serengeti National Park were identified as Trypanosoma godfreyi, and one sample from the Maswa Game Reserve was identified as Trypanosoma simiae (Fig. 2; Table 3). Trypanosoma simiae usually infects pigs [11], and T. godfreyi usually infects cattle. In our study, T. godfreyi was detected in the tsetse fly containing the African buffalo gene and two T. godfreyi samples were detected in flies from Serengeti, where the major blood-meal source is African buffaloes.
There are no reports of Trypanosoma spp. found in this study that infect humans. Trypanosoma infection in animals causes red blood cell phagocytosis and blood catabolism, leading to the accumulation of iron in tissues, hyperbilirubinemia, liver dysfunction, and multiple organ failure [30]. Trypanosomiasis, induced by tsetse blood-feeding, makes animals ill; cattle that are protected from trypanosomiasis are healthier and have significantly reduced disease levels, increased cell volume, and greater body weight [31].
Six samples were positive for Trypanosoma using 18S rRNA gene deep sequencing, and three samples (M21, S17, and S55) were found to be positive using PCR and subsequent DNA sequencing analysis. One sample (M10) showed a very weak band after PCR amplification, and the DNA sequencing of this sample failed. Two other samples (M60 and T73) were found to be negative using PCR, and the samples showed only one and two Trypanosoma reads in deep sequencing, respectively (Table 2). Samples in which Trypanosoma was detected with fewer than ten read counts using deep sequencing were not well detected using PCR. This was probably because deep amplicon sequencing is more sensitive than PCR [32, 33]. Therefore, deep sequencing of the 18S rRNA gene is useful for screening for eukaryotic pathogens in tsetse flies.
In addition, because the primers we used can detect all species of eukaryotic organisms, this method can be applied to screen eukaryotic pathogens in any arthropod vector. Our method can theoretically detect all potentially pathogenic taxa in samples and simultaneously analyze 96 samples at once. The approximate cost of one run of the iSeq 100 machine is US$ 2000, which takes 18 h to complete [19].
As we collected tsetse flies while moving by vehicle between villages, we believe that a large number of tsetse flies that sucked the blood of humans were collected. Because many previous studies reported human as a major blood-meal source of tsetse flies, the possibility of contamination during collection was low [7, 24, 34]. The possibility of degradation of the nucleic acid over a long storage period cannot be ruled out, which might reduce the diversity of animal blood-meal sources of tsetse flies.
Conclusions
Various mammals were identified as blood-meal sources for tsetse flies through 12S rRNA gene deep sequencing, and two species of Trypanosoma spp. that infect animals were identified in tsetse flies through 18S rRNA gene deep sequencing in the Maswa Game Reserve, Serengeti National Park, and Tarangire National Park in Tanzania. This study provides important information on the patterns of parasitism of tsetse flies, affected wild animals, and Trypanosoma spp. in this region.
Availability of data and materials
Raw sequence data are available in NCBI GenBank under BioProject PRJNA817381.
Abbreviations
- AAT:
-
Trypanosomiasis in animals
- HAT:
-
African human trypanosomiasis
- ITS:
-
Internal transcribed spacer
- OUT:
-
Operational taxonomic unit
- PCR:
-
Polymerase chain reaction
- rRNA:
-
Ribosomal RNA
References
Signaboubo D, Payne VK, Moussa IMA, Hassane HM, Berger P, Kelm S, et al. Diversity of tsetse flies and trypanosome species circulating in the area of Lake Iro in southeastern Chad. Parasit Vector. 2021;14:293.
Malele II. Fifty years of Tsetse control in Tanzania: challenges and prospects for the future. Tanzan J Heal Res. 2011;13:399–406.
Vreysen MJB, Seck MT, Sall B, Bouyer J. Tsetse flies: their biology and control using area-wide integrated pest management approaches. J Invertebr Pathol. 2013;112:S15-25.
Farikou O, Njiokou F, Simo G, Asonganyi T, Cuny G, Geiger A. Tsetse fly blood meal modification and trypanosome identification in two sleeping sickness foci in the forest of southern Cameroon. Acta Trop. 2010;116:81–8.
Ngonyoka A, Gwakisa PS, Estes AB, Nnko HJ, Hudson PJ, Cattadori IM. Variation of tsetse fly abundance in relation to habitat and host presence in the Maasai Steppe, Tanzania. J Vector Ecol. 2017;42:34–43.
Votýpka J, Rádrová J, Skalický T, Jirků M, Jirsová D, Mihalca AD, et al. A tsetse and tabanid fly survey of African great apes habitats reveals the presence of a novel trypanosome lineage but the absence of Trypanosoma brucei. Int J Parasitol. 2015;45:741–8.
Gaithuma A, Yamagishi J, Hayashida K, Kawai N, Namangala B, Sugimoto C. Blood meal sources and bacterial microbiome diversity in wild-caught tsetse flies. Sci Rep. 2020. https://doi.org/10.1038/s41598-020-61817-2.
Makhulu EE, Villinger J, Adunga VO, Jeneby MM, Kimathi EM, Mararo E, et al. Tsetse blood-meal sources, endosymbionts and trypanosome-associations in the Maasai Mara National Reserve, a wildlife-human-livestock interface. PLoS Neglect Trop D. 2021;15:e0008267.
Clausen PH, Adeyemi I, Bauer B, Breloeer M, Salchow F, Staak C. Host preferences of tsetse (Diptera: Glossinidae) based on bloodmeal identifications. Med Vet Entomol. 1998;12:169–80.
Njiru ZK, Constantine CC, Guya S, Crowther J, Kiragu JM, Thompson RCA, et al. The use of ITS1 rDNA PCR in detecting pathogenic African trypanosomes. Parasitol Res. 2005;95:186–92.
Desquesnes M, Dávila AMR. Applications of PCR-based tools for detection and identification of animal trypanosomes: a review and perspectives. Vet Parasitol. 2002;109:213–31.
Aksoy E, Telleria EL, Echodu R, Wu Y, Okedi LM, Weiss BL, et al. Analysis of multiple tsetse fly populations in Uganda reveals limited diversity and species-specific gut microbiota. Appl Environ Microbiol. 2014;80:4301–12. https://doi.org/10.1128/aem.00079-14.
Ebhodaghe FI, Bastos ADS, Kidambasi KO, Kalayou S, Masiga DK, Okal MN. Molecular characterization of Trypanosoma vivax in tsetse flies confirms the presence of the virulent Tvv4 genotype in Kenya: potential implications for the control of trypanosomiasis in Shimba Hills. Infect Genet Evol. 2021;93:104953.
Edslev SM, Andersen PS, Agner T, Saunte DML, Ingham AC, Johannesen TB, et al. Identification of cutaneous fungi and mites in adult atopic dermatitis: analysis by targeted 18S rRNA amplicon sequencing. Bmc Microbiol. 2021;21:72.
Moreno Y, Moreno-Mesonero L, Amorós I, Pérez R, Morillo JA, Alonso JL. Multiple identification of most important waterborne protozoa in surface water used for irrigation purposes by 18S rRNA amplicon-based metagenomics. Int J Hyg Environ Health. 2018;221:102–11.
Mishra P, Tulsani NJ, Jakhesara SJ, Dafale NA, Patil NV, Purohit HJ, et al. Exploring the eukaryotic diversity in rumen of Indian camel (Camelus dromedarius) using 18S rRNA amplicon sequencing. Arch Microbiol. 2020;202:1861–72.
Kounosu A, Murase K, Yoshida A, Maruyama H, Kikuchi T. Improved 18S and 28S rDNA primer sets for NGS-based parasite detection. Sci Rep. 2019;9:15789.
Arias-Giraldo LM, Muñoz M, Hernández C, Herrera G, Velásquez-Ortiz N, Cantillo-Barraza O, et al. Identification of blood-feeding sources in Panstrongylus, Psammolestes, Rhodnius and Triatoma using amplicon-based next-generation sequencing. Parasit Vectors. 2020;13:434.
Kim JY, Yi M, Mahdi AAS, Yong T-S. iSeq 100 for metagenomic pathogen screening in ticks. Parasit Vectors. 2021;14:346.
Chen X, Li S, Aksoy S. Concordant evolution of a symbiont with its host insect species: molecular phylogeny of genus Glossina and Its bacteriome-associated Endosymbiont, Wigglesworthia glossinidia. J Mol Evol. 1999;48:49–58.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Albakri NN, Bouqellah NA, Shabana II. A metagenomic survey of lamb’s pre- and post-weaning fecal microbiomes. J Taibah Univ Sci. 2020;14:1233–42.
Malele II, Ouma JO, Nyingilili HS, Kitwika WA, Malulu DJ, Magwisha HB, et al. Comparative performance of traps in catching tsetse flies (Diptera: Glossinidae) in Tanzania. Onderstepoort J Vet Res. 2016;83:1057.
Konnai S, Mekata H, Odbileg R, Simuunza M, Chembensof M, Witola WH, et al. Detection of Trypanosoma brucei in field-captured tsetse flies and identification of host species fed on by the infected flies. Vector-borne Zoonot. 2008;8:565–74.
Dumonteil E, Ramirez-Sierra M-J, Pérez-Carrillo S, Teh-Poot C, Herrera C, Gourbière S, et al. Detailed ecological associations of triatomines revealed by metabarcoding and next-generation sequencing: implications for triatomine behavior and Trypanosoma cruzi transmission cycles. Sci Rep. 2018;8:4140.
Ushio M, Fukuda H, Inoue T, Makoto K, Kishida O, Sato K, et al. Environmental DNA enables detection of terrestrial mammals from forest pond water. Mol Ecol Resour. 2017;17:e63–75.
Moloo SK. The distribution of Glossina species in Africa and their natural hosts. Insect Sci Appl. 1993;14:511–27.
Auty H, Cleaveland S, Malele I, Masoy J, Lembo T, Bessell P, et al. Quantifying heterogeneity in host-vector contact: Tsetse (Glossina swynnertoni and G. pallidipes) host choice in Serengeti National Park, Tanzania. PLoS ONE. 2016;11:e0161291.
Mwandiringana E. Polymerase chain reaction (PCR) detection of mixed trypanosome infection and blood meal origin in field-captured tsetse flies from Zambia. Afr J Biotechnol. 2012;11:14490–7.
Kasozi KI, Zirintunda G, Ssempijja F, Buyinza B, Alzahrani KJ, Matama K, et al. Epidemiology of trypanosomiasis in wildlife—implications for humans at the wildlife interface in Africa. Front Vet Sci. 2021;8:621699.
Saini RK, Orindi BO, Mbahin N, Andoke JA, Muasa PN, Mbuvi DM, et al. Protecting cows in small holder farms in East Africa from tsetse flies by mimicking the odor profile of a non-host bovid. Plos Neglect Trop D. 2017;11:e0005977.
da Fonseca AJ, Galvão RS, Miranda AE, de Ferreira LCL, Chen Z. Comparison of three human papillomavirus DNA detection methods: next generation sequencing, multiplex-PCR and nested-PCR followed by Sanger based sequencing. J Med Virol. 2016;88:888–94.
Early AM, Daniels RF, Farrell TM, Grimsby J, Volkman SK, Wirth DF, et al. Detection of low-density Plasmodium falciparum infections using amplicon deep sequencing. Malaria J. 2019;18:219.
Rodrigues CMF, Garcia HA, Sheferaw D, Rodrigues AC, Pereira CL, Camargo EP, et al. Genetic diversity of trypanosomes pathogenic to livestock in tsetse flies from the Nech Sar National Park in Ethiopia: a concern for tsetse suppressed area in Southern Rift Valley? Infect Genetics Evol. 2019;69:38–47.
Acknowledgements
Not applicable.
Funding
This study was supported by a National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MEST; nos. NRF‑2020R1I1A2074562).
Author information
Authors and Affiliations
Contributions
JYK and TSY designed this study. JHC conducted laboratory experiments. SHN and RF performed sampling. JYK, JHC, and TSY analyzed and interpreted the data to create the manuscript. All authors have read and accepted the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The number of tsetse flies harboring 12S ribosomal RNA genes from several animals.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kim, J.Y., Choi, J.H., Nam, SH. et al. Parasites and blood-meal hosts of the tsetse fly in Tanzania: a metagenomics study. Parasites Vectors 15, 224 (2022). https://doi.org/10.1186/s13071-022-05344-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05344-1