
Abstract
Background
Diplozoidae are monogenean (Monogenea: Polyopisthocotylea) fish parasites characterised by a unique life history: two larvae permanently fuse into an X-shaped “Siamese” organism. Taxonomy and phylogeny of Diplozoidae and Polyopisthocotylea remain unresolved due to the unavailability of molecular markers with sufficiently high resolution. Mitogenomes may be a suitable candidate, but there are currently only 12 available for the Polyopisthocotylea (three for Diplozoidae). The only available study of diplozoid mitogenomes found unique base composition patterns and elevated evolution rates in comparison with other Monogenean mitogenomes.
Methods
To further explore their evolution and generate molecular data for evolutionary studies, we sequenced the complete mitogenomes of two Diplozoidae species, Paradiplozoon homoion and Paradiplozoon yarkandense, and conducted a number of comparative mitogenomic analyses with other polyopisthocotyleans.
Results
We found further evidence that mitogenomes of Diplozoidae evolve at a unique, elevated rate, which was reflected in their exceptionally long branches, large sizes, unique base composition, skews, and very low gene sequence similarity levels between the two newly sequenced species. They also exhibited remarkably large overlaps between some genes. Phylogenetic analysis of Polyopisthocotylea resolved all major taxa as monophyletic, and Mazocraeidea was split into two major clades: (Diplozoidae) + (all four remaining families: Diclidophoridae, Chauhaneidae, Mazocraeidae and Microcotylidae). It also provided further confirmation that the genus Paradiplozoon is paraphyletic and requires a taxonomic revision, so the two species may have to be renamed Indodiplozoon homoion and Diplozoon yarkandense comb. nov.
Conclusions
Although our findings indicate that mitogenomes may be a promising tool for resolving the phylogeny of Polyopisthocotylea, elevated evolutionary rates of Diplozoidae may cause phylogenetic artefacts, so future studies should pay caution to this problem. Furthermore, as the reason for their elevated evolution remains unknown, Diplozoidae are a remarkably interesting lineage for other types of evolutionary mitogenomic studies.
Graphical Abstract

Similar content being viewed by others


Background
Flatworms are a phylum (Platyhelminthes) of largely parasitic animals of high importance for medicine, as they cause diseases in a variety of host animal groups, including domestic animals and humans. They also exhibit a broad range of parasitic strategies, including some lineages exhibiting remarkably high host-specificity [1,2,3]. As Platyhelminthes also comprises free-living lineages, these factors make Platyhelminthes a good phylogenetic system for studying the evolution of parasitism. The predominantly parasitic three classes of flatworms are classified together as the superclass Neodermata: Monogenea (primarily ectoparasitic), Trematoda (endoparasitic flukes), and Cestoda (endoparasitic tapeworms). Whereas the monophyly of Neodermata is relatively robust, the relationships among the above three main clades, as well their monophyly, remain debated [2,3,4,5,6]. Notably, there is evidence that Monogenea, which comprises a major part of the obligate parasitic flatworm diversity, might be paraphyletic, and split into two independent radiations (subclasses): Polyopisthocotylea and Monopisthocotylea [6, 7]. Mitogenomic nucleotide sequences and gene order rearrangements indicate that Polyopisthocotylea may even be a sister group to all other Neodermata [6, 8]. Another intriguing feature of this subclass is that it also exhibits mitogenomes with rearranged gene orders; this is uncommon within the Neodermata, which mostly exhibit a highly conserved architecture [6].
Along with the family Octomacridae, Diplozoidae are the only polyopisthocotyleans primarily parasitizing freshwater teleosts [9]. They are predominantly ectoparasites, commonly found on the gills of cyprinid and characid fishes, and they have a direct life-cycle (no intermediate host), which is typical for monogeneans. However, Diplozoidae are also characterised by a unique life history: two larvae (diporpae) permanently fuse into a pair, morphing into an X-shaped “Siamese” organism [9,10,11]. They are also important for aquaculture because they cause notable damage to the gill tissue of their hosts, which may cause secondary infections and mortality [11].
Taxonomy and phylogeny of diplozoids are mainly based on morphology (central hooks, clamps and spermatozoid ultrastructure) and host fishes, but reliance on these parameters often causes taxonomic and phylogenetic artefacts [11,12,13]. Therefore, the availability of molecular markers is a prerequisite for the reliable identification and phylogenetic and taxonomic studies of diplozoid parasites. Previous phylogenetic studies mostly relied on the 28S and ITS-2 rDNA (second internal transcribed spacer of ribosomal DNA) sequences, which often produce incongruent results between different datasets and methods [11, 13,14,15,16]. This lack of reliable phylogenetic marker results in multiple unresolved phylogenetic and taxonomic questions within the Diplozoidae and Monogenea [6, 13]. Mitochondrial genomes (mitogenomes) can provide a phylogenetic resolution superior to the traditionally used single-gene markers, so they are becoming an increasingly popular tool in evolutionary, population genetic, taxonomic, phylogenetic and diagnostic studies of Platyhelminthes [7, 11, 17, 18]. The only previous study that applied mitochondrial phylogenomics to Diplozoidae produced some novel family-level relationships, which indicates that the usefulness of mitogenomes for inferring the phylogeny of Polyopisthocotylea should be further explored [11, 13].
In most animal lineages, mitogenomes are circular molecules, ranging 13 ~ 16 kb in size, that encode 37 genes—13 protein-coding genes (PCGs), two ribosomal RNA genes, and 22 transfer RNA (tRNA) genes—but comparative mitogenomic architecture analyses (e.g., gene arrangement, base composition skews, etc.) often reveal lineages that exhibit intriguing patterns of mitogenomic evolution [19,20,21,22]. The only published study of Diplozoidae mitogenomes found unique base composition patterns and elevated evolution rates in comparison with other Monogenean mitogenomes [11]. As there are currently (Dec. 2021) only 12 sequenced and annotated complete mitogenomes for the Polyopisthocotylea, this scarcity of data hampers progress in the understanding of mitogenomic architecture evolution and phylogeny of Polyopisthocotylea and Monogenea. To address this dearth of data and further explore the unique base composition of diplozoid mitogenomes, we sequenced complete mitogenomes of two Diplozoidae species: Paradiplozoon homoion (Bychowsky & Nagibina, 1959) and a recently described [23] new species Paradiplozoon yarkandense (Arken et al. [23]).
Methods
Paradiplozoon yarkandense was collected from the host Diptychus maculatus in the Taxkorgan river (a tributary of the Yarkand River) (37° 41′ 14″ N; 75° 18′ 9″ E), Xinjiang, China. Paradiplozoon homoion was collected from the host Leuciscus baicalensis in the Kelan River (a tributary of the Irtysh River) (47° 42′ 40″ N, 88° 13′ 25″ E). Xinjiang, China. Both species were morphologically identified according to our previous publications [23, 24] (Additional file 1: Text S1 and Figs. S1 to S5). DNA extraction, mitogenome amplification and sequencing, and sequence annotation and analyses were conducted exactly as described before [11]. The methodology only differed in selecting different reference mitogenomes: Polylabris halichoeres [25] and the three available Diplozoidae species [11] and different primers (Additional file 1: Tables S1 and S2). tRNAs were first identified using ARWEN [26] and MITOS [27] programs, and then R2DT [28] was further used to predict and visualise the secondary structure of selected tRNAs. ORF-Finder [29] was further used to search for genes resembling atp8 in large non-coding regions of both mitogenomes. Phylogenetic analysis was conducted on a dataset comprising all available Polyopisthocotylea mitogenomes (Additional file 2: Dataset S1). Following the evidence that using multiple outgroups produces better results than using a single outgroup [30], we used two different lineages as outgroups, two mitogenomes from the relatively closely related neodermatan clade, Monopisthocotylea: Gyrodactylus salaris (Gyrodactyloidea) [31] and Dactylogyrus lamellatus (Dactylogyridea) [32]; and two mitogenomes from Rhabditophora, belonging to the basal radiation of largely free-living flatworms [5]: Bipalium kewense [33] and Obrimoposthia wandeli [34]. PhyloSuite [35] was used to standardise annotation, extract data, and conduct phylogenetic analysis in the Flowchart mode using nucleotide sequences of 12 concatenated (but partitioned) PCGs with the help of several plug-in programs. Genes were aligned using the codon mode and the accurate G-INS-i strategy in MAFFT [36], concatenated using PhyloSuite, best-suited evolutionary models for partitions inferred using ModelFinder [37], and phylogeny was reconstructed using IQ-TREE [38] with 10,000 ultrafast bootstraps [39]. iTOL [40] was used to visualise the phylogeny and architecture using files generated by PhyloSuite. OGDRAW was used to create to-scale circular maps of the mitogenomic architecture [41].
Results
Phylogeny and identity
The ITS-2 sequence of the putative P. yarkandense exhibited 100% identity to a number of orthologous P. yarkandense sequences published in the original description study [23], whereas that of P. homoion exhibited a 100% identity to a P. homoion ITS-2 sequence sequenced by Benovics et al. [42] (Additional file 1: Figs. S5 and S6). The phylogenetic analysis resolved all major taxa, including the Diplozoidae, as monophyletic (Fig. 1). Statistical support for clades was very high (mostly over 90%). Mazocraeidea was split into two major clades; one comprising only Diplozoidae and the other containing all four remaining families included in the dataset (Diclidophoridae, Chauhaneidae, Mazocraeidae, and Microcotylidae). Within the Diplozoidae, however, the genus Paradiplozoon was paraphyletic: P. yarkandense formed a sister lineage with Paradiplozoon opsariichthydis, whereas P. homoion formed a sister lineage with Sindiplozoon sp. Eudiplozoon sp. was the sister group to the remaining four species.

The mitogenomic phylogeny of Polyopisthocotylea. Species names are followed by the GenBank accession number, statistical support is shown next to branches, and the tree scale is included in the figure. Yellow bars correspond to the A+T composition of entire mitogenomes, red bars correspond to the GC skew, and blue bars to the mitogenome length. Taxonomic identity is shown to the right: family and order (apart from Rhabditophora)
Mitochondrial architecture and size
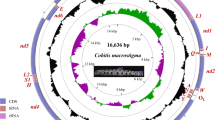
All genes are encoded on the same strand and the atp8 gene is missing from both newly sequenced mitogenomes (Fig. 2; Table 1). Both exhibit a gene order identical to that of Eudiplozoon sp. and Sindiplozoon sp. The only difference they exhibited was in the distribution of non-coding regions larger than 100 base pairs (bp) (Figs. 2 and 3), which is indicative of the fast evolution of non-coding sections. Both mitogenomes were the largest among the sequenced polyopisthocotylean mitogenomes: P. homoion was the largest with 17,321 bp and P. yarkandense was the second largest, with 16,816 bp (Fig. 1, Additional file 2: Dataset S1). Moreover, all other available mitogenomes were much smaller: 12,290 to 15,527 bp (some of the smallest mitogenomes were incomplete, so the lower end of the range is almost certainly a sequencing or assembly artefact). Aside from the outlier of P. halichoeres (Microcotylidae; 15,527 bp), the top five largest mitogenomes belonged to Diplozoidae. Aside from Eudiplozoon sp. (14,334 bp), all other Diplozoidae mitogenomes were larger than 15 kbp.

To-scale circular architectural maps of mitogenomes of P. homoion and P. yarkandense. The transcription direction is clockwise

Gene orders in Polyopisthocotylea. Species names are followed by GenBank accession numbers. The two newly sequenced species are shaded yellow. Family-level taxonomic identity is shown to the right. Partial mitogenomes are labelled with the letter P
Base composition and skews
The A+T-base content was high in the polyopisthocotylid mitogenomes: 65.6–72.7% (Fig. 1, Fig. 4, Additional file 2: Dataset S1). However, Diplozoidae exhibited the three lowest values in the dataset. This was particularly strongly pronounced in the P. homoion mitogenome, which shared the lowest AT content of 65.6% with Paradiplozoon hemiculteri. The elevated AT content in the dataset was largely driven by the high T-base content (39.2–50.7%). Somewhat surprisingly, the six Diplozoidae were among the top seven taxa with the highest T-content (45.2–50.7%), along with the Polystomatidae species, Diplorchis hangzhouensis (49.7%). Therefore, the reduced AT content in Diplozoidae was driven solely by the strongly reduced A-content (18.6–20.4%) in all Diplozoidae aside from Eudiplozoon sp. (23.6%) in comparison with other polyopisthocotylids (22.5–29.9%). The C-content was strongly reduced in all species (7.3–12.1%), but Diplozoidae were not consistently different from the rest of the dataset; the only minor exception was the newly sequenced P. homoion with the highest C-content in the dataset (12.1%). In terms of the G-content, Diplozoidae on average exhibited comparatively reduced values (five out of seven lowest values).

The base composition of mitogenomes of Polyopisthocotylea
The unusual base composition was also reflected in base composition skews (Fig. 1, Fig. 5, Additional file 2: Dataset S1). Diplozoidae had much higher AT skews (−0.34 to −0.45) than the rest of the dataset (−0.15 to −0.22). The only other species that had skews comparable to Diplozoidae was D. hangzhouensis (−0.38). As regards GC skews, a majority of Diplozoidae grouped in the upper end of the range (0.358 to 0.454), but the highest value was exhibited by D. hangzhouensis (0.479), and P. homoion exhibited a low value of 0.294 (the overall range: 0.25 to 0.48).

AT and GC skews in mitogenomes of Polyopisthocotylea
Gene comparison
For nominally congeneric species, the two newly sequenced Paradiplozoon species exhibited remarkably low identity values between genes (Fig. 6). Only three tRNA genes exhibited similarity values larger than 75%: trnF (79), trnI (77), and trnC (75). The two species also exhibited surprisingly divergent start and stop codons (Table 1). Otherwise, start/stop codons were standard for polyopisthocotyleans (Additional file 2: Dataset S2).

Gene identity (%) values between the genes of P. yarkandense and P. homoion
This rapid sequence evolution was reflected in a high number of insertions, deletions, and in general poorly aligned gene segments within the Polyopisthocotylea dataset. For example, nad6 exhibited a 3′ elongation in P. homoion, which caused a 7-bp overlap with the downstream trnY (Table 1, Additional file 1: Fig. S8). Nad5 and nad3 were highly divergent among the available species, with few universally conserved sites, and 5′ and 3′ ends exhibiting many insertions and deletions. Nad1, nad2 and nad4 were relatively conserved, aside from their highly divergent 5′ ends. Maybe it should be noted that nad2 in D. hangzhouensis had a 3′ extension unmatched in the dataset, so we hypothesise that this is most probably an artefact. Somewhat surprisingly, nad4L was relatively conserved within the dataset. As regards cytb, 5′ and 3′ ends were highly divergent in the dataset, but the rest was conserved. cox3 was relatively poorly conserved, with insertions and deletions throughout the sequences, but also with some universally conserved sites. Start codons for this gene were perfectly conserved among all species, but the two newly sequenced species exhibited a 3′ elongation of 6 to 10 bases. Cox2 was more conserved, with fewer insertions and deletions. In P. homoion, cox2 exhibited a 3′ end elongation of 12 bases. This occurred due to a loss of the T base, which changed the stop codon TAG into –AG and caused a frameshift mutation. As a consequence, there may exist a 15 bp overlap with the downstream trM (discussed above). We sequenced this segment twice to confirm this, and found no indications of an annotation artefact. The central part of cox1 was highly conserved, higher than any other gene, but both 5′ and 3′ ends were remarkably divergent, at least for this usually highly conserved gene [43]. Paradiplozoon homoion exhibited an elongation of 15 bp at the 5′ end as compared with P. yarkandense and several other species, but these were not the only species that exhibited a 5′ elongation. Paradiplozoon homoion exhibited a slightly truncated 3′ end in comparison with most other species (but the 3′ end was not conserved among species so there is no reliable benchmark to compare it with), whereas P. yarkandense exhibited a slightly elongated 3′ end. As 3′ was highly divergent in the dataset, we have no reason to suspect sequencing or annotation artefacts. Atp6 was generally very poorly conserved, with only a few globally conserved sites and highly divergent 3′ end. Py exhibited a large insertion, which resulted in a slightly increased size in comparison with available orthologues (636 bp; compared with 556 in P. homoion).
Gene overlaps
Locations and sizes of intergenic regions and gene overlaps were also remarkably divergent for nominal congenerics (Table 1). Furthermore, several overlaps were very large. Accordingly, in the two studied species, most gene overlaps involved a tRNA gene, but we also identified some overlaps between two PCGs. A relatively large putative overlap between cytb and nad4L was conserved in both species: P. yarkandense = 10 bp, P. homoion = 16 bp (Table 1). There was also a putative remarkably large overlap of 49 bases between nad4 and nad4L genes in P. homoion. Equally surprisingly, these two genes had 59 bases of intergenic space in P. yarkandense. Although nad4L exhibited only a handful of universally conserved amino acids in the alignment of polyopisthocotylean orthologues (Additional file 1: Fig. S9), the P. yarkandense and P. homoion orthologues exhibited the identity of 60% (Fig. 6), which was an average similarity value for PCGs.
Other overlaps involved tRNA genes. In P. yarkandense, trnI exhibited a large overlap of 19 bases with the downstream nad3. The alignment of trnI genes of Polyopisthocotylea indicated that only the 3′ was not highly conserved, and the P. yarkandense orthologue was remarkably similar to other genes (Additional file 1: Fig. S10), as additionally indicated by some of the highest identity levels between the two newly sequenced species of almost 78% (Fig. 6). Notably, this gene was annotated by MITOS, whereas ARWEN and R2DT failed to fold it into a cloverleaf structure. The trnI of P. homoion was successfully folded into a standard structure (Additional file 1: Fig. S11). It is unclear whether this is an indication of an algorithm artefact, posttranslational editing [44], or non-functionality. trnG of P. yarkandense overlapped by 18 bases with the upstream cox1. This gene was less conserved than trnI (Additional file 1: Fig. S12), but it was successfully recognised as a tRNA by all three algorithms employed. It could be folded into a cloverleaf structure, but its T-arm appeared rather crippled (Fig. 7a) as compared with the P. homoion orthologue (Fig. 7b) and other polyopisthocotylean orthologues [11]. In P. homoion, trnM overlapped by 15 bases with the upstream cox2. Its 3′ end was rather poorly conserved among the orthologues (Additional file 1: Fig. S13). Structurally, it was successfully folded into a perfectly standard cloverleaf structure, apart from its 3′ end (acceptor stem), which was truncated (Fig. 7c and d).

Secondary structures of tRNA genes. a P. yarkandense trnG. b P. homoion trnG. c P. homoion trnM. (d) P. yarkandense trnM
Discussion
Phylogenetic analysis of Polyopisthocotylea resolved all major taxa as monophyletic, and Mazocraeidea was split into two major clades: (Diplozoidae) + (all four remaining families: Diclidophoridae, Chauhaneidae, Mazocraeidae and Microcotylidae). It also provided further confirmation that the genus Paradiplozoon Akhmerov, 1974 is paraphyletic and requires a taxonomic revision. The paraphyly of Paradiplozoon is a recognised scientific fact [13, 16, 23, 42, 45]. In the study that described P. yarkandense, the ITS-2 sequence grouped it with Paradiplozoon bingolense and Paradiplozoon krugerense [23]. This clade was recognised as “Genus 3” in a recent review of diplozoid phylogeny [13]. No other species nominally included in the “Genus 3” were available for our analysis, so P. yarkandense clustered with the only available “Genus 2” species—P. opsariichthydis. This is in agreement with the proposed sister group relationship of “Genus 2” (Paradiplozoon) and “Genus 3”, which together form the Clade2 [13]. Clade 1 comprised a number of different genera: Eudiplozoon, Sindiplozoon, Inustiatus and Genus 1. The Genus 1 clade comprised Diplozoon and some Paradiplozoon species, including the P. homoion complex [13]. Sindiplozoon sp. and P. homoion forming a separate clade in our analysis supports the above topology, as they correspond to the above described “Clade 1”. The authors proposed that the “Genus 1” clade should revert to the type genus Diplozoon von Nordmann, 1832 [13]. Following this proposal, which is also supported by our study, the species currently denominated P. homoion may have to be renamed Diplozoon homoion. Remarkably, “Genus 3” contains species from Africa, Turkey and an unidentified sequence from China [13]. Our findings provide an additional indirect indication that this may be a unique genus, sufficiently evolutionarily old to have such a wide distribution (alternatively the distribution may be of a recent origin due to global trade, but in that case, this should show as a poorly resolved topology). The authors also proposed that the appropriate designation for Genus 3 may be Indodiplozoon, given that all specimens were collected from river systems that eventually enter the Indian Ocean, which may indicate that members of this proposed genus have an Indo-Pacific ancestor [13]. Accordingly, P. yarkandense may have to be renamed Indodiplozoon yarkandense. This taxonomic revision is further supported by remarkably low identity values between genes of nominally congeneric P. yarkandense and P. homoion. Additionally, the geographic distribution of the genus Indodiplozoon indicates that it may be evolutionarily rather old. In this light, it should be noted that the species inhabits the endorheic Tarim Basin in Xinjiang, China. However, a number of rivers from Xinjiang and geographically close Tibet and Qinghai empty into the Indian Ocean, so this finding may not directly contradict the above hypothesis. We should note that the accuracy of taxonomic revision is affected by a limited number of morphological parameters useful for the identification and classification of these species, as well as insufficiently detailed previous morphological descriptions and unavailability of sufficiently detailed and complete morphological maps [12, 13]. As a result, currently we do not have morphological comparative data that perfectly support the molecular results, or we even have conflicting signals from molecular and morphological data. For example, molecular data indicate that Paradiplozoon homoion should be assigned to the genus Diplozoon, but morphologically this species better resembles the classical Paradiplozoon features: the anterior part of the body between the opisthaptor and the area of the reproductive organs does not have a prominent disc dilation [46, 47] (more details in Additional file 1: Text S1 and Figs. S1 to S5). Similarly, the three Diplozoon species that putatively belong to the Genus 3 (Indodiplozoon) also share the basic characteristics of the genus Diplozoon (a prominent disc dilation in the anterior part of the body) [47]. Therefore, in cases where morphological data do not directly support the molecular data, we should seek agreement between both mitochondrial DNA (mtDNA) and nuclear DNA (nucDNA) data before going forth with taxonomic revision.
Unfortunately, in some cases, mtDNA and nucDNA data sometimes also produce contradictory results. For example, Eudiplozoon forming a sister group to all other available Diplozoidae is in disagreement with the topology produced by the ITS-2 sequences, which indicates that more studies with new molecular data are needed to resolve the phylogeny of Diplozoidae. It may also be important to note that a study based on nuclear 18S and 28S genes found that Mazocraeidae was the only polyopisthocotylean family that exhibited an elevated evolutionary rate [9], but mitogenomic sequences do not follow this pattern: Diplozoidae exhibited the longest branches in the dataset. Although our results provide further indications that mitogenomes may be a useful tool in the phylogenetics of Diplozoidae and Polyopisthocotylea, there are currently too few mitogenomic sequences available to conclude this with confidence.
Our study provides further evidence that mitogenomes of Diplozoidae are evolving at elevated rates within the polyopisthocotylean dataset. This was reflected in their exceptionally long branches, large sizes, unique base composition, large base composition skews, and very low gene sequence similarity levels between the two newly sequenced species. As an example, even the normally highly conserved cox1 gene [43] exhibited an identity value of only 71.67%, which is exceptionally low for metazoans on average, but not uncommon in Platyhelminthes [48]. Similar values were also observed in the previous Diplozoidae analysis [11]. Unique base composition and increased skews in Diplozoidae were also observed in the previous study [11]. Increased skews might be reflective of reduced purifying selection pressures [49], which would also explain long branches. The A+T-base content is commonly high in monogenean mitogenomes [2, 31, 32]. Remarkably, the elevated AT content in the Polyopisthocotylea is largely driven by the high T-base content, whereas the reduced AT content in Diplozoidae was driven solely by the strongly reduced A-content in comparison with other polyopisthocotylids. As their reduced A+T-base composition bias is reflective of their faster, and not slower, evolution, this indicates that the AT content of Diplozoidae might be evolving in the opposite direction of most other Monogenean mitogenomes, towards lower values. In some other aspects, the two newly sequenced mitogenomes possessed typical general characteristics of most flatworm and all neodermatan mitogenomes; e.g. all genes are transcribed from the same strand and the atp8 gene could not be identified [8, 11, 50, 51]. It should be noted that we identified a number of putative ORFs in both large NCRs, but comparison with previously annotated flatworm atp8 genes did not reveal any similarity. Without gene and protein expression data, it remains impossible to assess whether these putative ORFs are expressed and functional. The gene order exhibited by Eudiplozoon sp. and Sindiplozoon sp. was previously recognised as the ancestral architecture for the Diplozoidae, and putatively even for Polyopisthocotylea [11]. Our study offers further support for this hypothesis, as both newly sequenced mitogenomes exhibit the putative ancestral gene order.
We identified remarkably large overlaps between genes. Usually, overlaps in metazoan mitogenomes involve tRNA genes, which is believed to be a consequence of lesser evolutionary constraints on tRNA sequences [44]. The overlaps between atp6/atp8 and nad4/nad4L are common in a broad range of metazoan lineages, perhaps due to their evolutionary conserved translation from a bicistronic mRNA [19, 52,53,54,55,56,57] and the fact that atp8 and nad4L are small genes that appear to evolve under relaxed evolutionary constraints, as evidenced by the absence of atp8 from several major metazoan lineages [19]. Overlaps between PCGs of less than 10 bp have been experimentally confirmed in a nematode species [58], but the exceptionally large overlap between cox1 and nad4L observed here appears to be very rare. Much larger overlaps involving tRNA genes have been observed and experimentally confirmed in Armadillidium vulgare (Arthropoda: Isopoda) [44]. However, this was explained by the apparent existence of strong evolutionary pressure for a reduced mitogenomic size [44], which does not appear to be the case in the newly sequenced Diplozoidae species, as they possess larger than average mitogenomes. While relaxed purifying selection pressures may also be a putative explanation for the existence of unusually large overlaps, their existence remains an evolutionary puzzle. Finally, although we did our best to confirm these overlaps by conducting detailed comparative analyses and considering alternative start and stop codons, without the transcriptomic data, we cannot be 100% confident that these are not annotation artefacts.
Conclusions
Due to their comparative advantages, mitogenomic sequences are a popular marker for phylogenetic studies, but their applicability is still somewhat curbed by the fact that many taxonomic categories remain poorly or not at all represented. Furthermore, multiple studies have indicated that in some cases, mitogenomes may also produce artefactual relationships [59,60,61]. Although our initial findings are promising, future studies should be very cautious in this aspect, and seek agreement between the topology produced by mitogenomic, nuclear, and ideally morphological data, to assess whether mitogenomic data can be used with confidence to conduct a thorough revision of the taxonomy and phylogeny of Diplozoidae. Furthermore, as the reasons for their elevated evolutionary rates remain unknown, Diplozoidae are a remarkably interesting lineage for other types of evolutionary mitogenomic studies.
Availability of data and materials
The two newly sequenced mitogenomes are available from GenBank under the accession numbers: OM525853 (Paradiplozoon yarkandense), and OM525852 (Paradiplozoon homoion). The ITS-2 sequences are available under OM432149 (Paradiplozoon yarkandense), OM422697 (Paradiplozoon homoion). Other datasets supporting the conclusions of this article are included within the article and its additional files.
Change history
18 May 2022
Following the original publication of this article, the authors corrected figures 3, 4 and 5, and corrected information concerning an enzyme in the ‘Gene overlaps’ part of the Results section; please see the linked correction article for further details.
16 May 2022
A Correction to this paper has been published: https://doi.org/10.1186/s13071-022-05308-5
Abbreviations
- PCG:
-
Protein-coding gene
- bp:
-
Base pair
References
Huyse T, Volckaert FAM. Comparing host and parasite phylogenies: Gyrodactylus flatworms jumping from goby to goby. Syst Biol. 2005;54:710–8.
Park J-K, Kim K-H, Kang S, Kim W, Eom KS, Littlewood D. A common origin of complex life cycles in parasitic flatworms: evidence from the complete mitochondrial genome of Microcotyle sebastis (Monogenea: Platyhelminthes). BMC Evol Biol. 2007;7:11.
Hahn C, Fromm B, Bachmann L. Comparative genomics of flatworms (Platyhelminthes) reveals shared genomic features of ecto- and endoparasitic Neodermata. Genome Biol Evol. 2014;6:1105–17.
Laumer CE, Hejnol A, Giribet G. Nuclear genomic signals of the ‘microturbellarian’ roots of platyhelminth evolutionary innovation. Elife. 2015;4:e05503.
Egger B, Lapraz F, Tomiczek B, Müller S, Dessimoz C, Girstmair J, et al. A transcriptomic-phylogenomic analysis of the evolutionary relationships of flatworms. Curr Biol. 2015;25:1347–53.
Zhang D, Li WX, Zou H, Wu SG, Li M, Jakovlić I, et al. Homoplasy or plesiomorphy? Reconstruction of the evolutionary history of mitochondrial gene order rearrangements in the subphylum Neodermata. Int J Parasitol. 2019;49:819–29.
Perkins EM, Donnellan SC, Bertozzi T, Whittington ID. Closing the mitochondrial circle on paraphyly of the Monogenea (Platyhelminthes) infers evolution in the diet of parasitic flatworms. Int J Parasitol. 2010;40:1237–45.
Egger B, Bachmann L, Fromm B. Atp8 is in the ground pattern of flatworm mitochondrial genomes. BMC Genomics. 2017;18:414.
Sicard M, Desmarais E, Lambert C. Is the Octomacridae the sister family of the Diplozoidae? Parasite. 2002;9:85–7.
Hodová I, Sonnek R, Gelnar M, Valigurová A. Architecture of Paradiplozoon homoion: a diplozoid monogenean exhibiting highly-developed equipment for ectoparasitism. PLoS ONE. 2018;13:e0192285.
Zhang D, Zou H, Wu SG, Li M, Jakovlić I, Zhang J, et al. Three new Diplozoidae mitogenomes expose unusual compositional biases within the Monogenea class: implications for phylogenetic studies. BMC Evol Biol. 2018;18:133.
Matejusová I, Koubková B, Gelnar M, Cunningham CO. Paradiplozoon homoion Bychowsky & Nagibina, 1959 versus P. gracile Reichenbach-Klinke, (Monogenea): two species or phenotypic plasticity? Syst Parasitol. 2002;53:39–47.
Dos Santos QM, Avenant-Oldewage A. Review on the molecular study of the Diplozoidae: analyses of currently available genetic data, what it tells us, and where to go from here. Parasit Vectors. 2020;13:539.
Gao Q, Chen MX, Yao WJ, Gao Y, Song Y, Wang GT, et al. Phylogeny of diplozoids in five genera of the subfamily Diplozoinae Palombi, 1949 as inferred from ITS-2 rDNA sequences. Parasitology. 2006;134:695–703.
Civáňová K, Koyun M, Koubková B. The molecular and morphometrical description of a new diplozoid species from the gills of the Garra rufa (Heckel, 1843) (Cyprinidae) from Turkey—including a commentary on taxonomic division of Diplozoidae. Parasitol Res. 2013;112:3053–62.
Avenant-Oldewage A, le Roux LE, Mashego SN, van Vuuren BJ. Paradiplozoon ichthyoxanthon n. sp. (Monogenea: Diplozoidae) from Labeobarbus aeneus (Cyprinidae) in the Vaal River, South Africa. J Helminthol. 2014;88:166–72.
Huyse T, Buchmann K, Littlewood DTJ. The mitochondrial genome of Gyrodactylus derjavinoides (Platyhelminthes: Monogenea)—a mitogenomic approach for Gyrodactylus species and strain identification. Gene. 2008;417:27–34.
Solà E, Álvarez-Presas M, Frías-López C, Littlewood DTJ, Rozas J, Riutort M. Evolutionary analysis of mitogenomes from parasitic and free-living flatworms. PLoS ONE. 2015;10:e0120081.
Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–80.
San Mauro D, Gower DJ, Zardoya R, Wilkinson M. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol Biol Evol. 2006;23:227–34.
Nguyen DT, Wu B, Xiao S, Hao W. Evolution of a record-setting AT-rich genome: indel mutation, recombination, and substitution bias. Genome Biol Evol. 2020;12:2344–54.
Jakovlić I, Zou H, Zhao X-M, Zhang J, Wang G-T, Zhang D. Evolutionary history of inversions in directional mutational pressures in crustacean mitochondrial genomes: implications for evolutionary studies. Mol Phylogenet Evol. 2021;164:107288.
Arken K, Hao C-L, Guo A-M, Zhang W-R, Rong M-J, Kamal W, et al. A new species of Paradiplozoon (Monogenea: Diplozoidae), a gill parasite of the Schizothorax fish (Cyprinidae: Schizothoracinae) from the Yarkand River, Xinjiang, China. Acta Parasitol. 2021. https://doi.org/10.1007/s11686-021-00466-5.
Xin W, Li J, Jia S, Wang N, Hao C, Zhu M, et al. A new record of Diplozoidae in China. Arid Zone Res. 2014;31:1121–4.
Zhang J, Wu X, Xie M, Xu X, Li A. The mitochondrial genome of Polylabris halichoeres (Monogenea: Microcotylidae). Mitochondrial DNA. 2011;22:3–5.
Laslett D, Canbäck B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–5.
Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–9.
Sweeney BA, Hoksza D, Nawrocki EP, Ribas CE, Madeira F, Cannone JJ, et al. R2DT is a framework for predicting and visualising RNA secondary structure using templates. Nat Commun. 2021;12:3494.
Rombel IT, Sykes KF, Rayner S, Johnston SA. ORF-FINDER: a vector for high-throughput gene identification. Gene. 2002;282:33–41.
Zou H, Jakovlić I, Zhang D, Hua C-J, Chen R, Li W-X, et al. Architectural instability, inverted skews and mitochondrial phylogenomics of Isopoda: outgroup choice affects the long-branch attraction artefacts. R Soc Open Sci. 2020;7:191887.
Huyse T, Plaisance L, Webster BL, Mo TA, Bakke TA, Bachmann L, et al. The mitochondrial genome of Gyrodactylus salaris (Platyhelminthes: Monogenea), a pathogen of Atlantic salmon (Salmo salar). Parasitology. 2006;134:739–47.
Zhang D, Zou H, Wu SG, Li M, Jakovlić I, Zhang J, et al. Sequencing, characterization and phylogenomics of the complete mitochondrial genome of Dactylogyrus lamellatus (Monogenea: Dactylogyridae). J Helminthol. 2018;92:455–66.
Gastineau R, Justine J-L, Lemieux C, Turmel M, Witkowski A. Complete mitogenome of the giant invasive hammerhead flatworm Bipalium kewense. Mitochondrial DNA Part B. 2019;4:1343–4.
Yang H-M, Ji S-J, Min G-S. The complete mitochondrial genome of the Antarctic marine triclad, Obrimoposthia wandeli (Platyhelminthes, Tricladida, Maricola). Mitochondrial DNA Part B. 2019;4:2515–6.
Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, et al. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 2020;20:348–55.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44:W232–5.
Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30:1188–95.
Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–8.
Greiner S, Lehwark P, Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019;47:W59-64.
Benovics M, Koubková B, Civáňová K, Rahmouni I, Čermáková K, Šimková A. Diversity and phylogeny of Paradiplozoon species (Monogenea: Diplozoidae) parasitising endemic cyprinoids in the peri-Mediterranean area, with a description of three new Paradiplozoon species. Parasitol Res. 2021;120:481–96.
Castellana S, Vicario S, Saccone C. Evolutionary patterns of the mitochondrial genome in Metazoa: exploring the role of mutation and selection in mitochondrial protein-coding genes. Genome Biol Evol. 2011;3:1067–79.
Doublet V, Ubrig E, Alioua A, Bouchon D, Marcade I, Marechal-Drouard L. Large gene overlaps and tRNA processing in the compact mitochondrial genome of the crustacean Armadillidium vulgare. RNA Biol. 2015;12:1159–68.
Jirsová D, Ding X, Civáňová K, Jirounková E, Ilgová J, Koubková B, et al. Redescription of Paradiplozoon hemiculteri (Monogenea, Diplozoidae) from the type host Hemiculter leucisculus, with neotype designation. Parasite. 2018;25:4.
Galli P, Pugachev O, Kritsky D. Guide to Monogenoidea of freshwater fish of Palaearctic and Amur regions. Ledizioni; 2010.
Dos Santos QM, Avenant-Oldewage A. The description of a new diplozoid species, Paradiplozoon krugerense n. sp., from Labeo rosae Steindachner, 1894 and Labeo congoro Peters, 1852 in the Kruger National Park, South Africa with notes on the effect of water quality on its infection variables. Hydrobiologia. 2016;777:225–41.
Hebert PDN, Ratnasingham S, de Waard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond B Biol Sci. 2003;270:S96–9.
Jakovlić I, Zou H, Chen J-H, Lei H-P, Wang G-T, Liu J, et al. Slow crabs—fast genomes: locomotory capacity predicts skew magnitude in crustacean mitogenomes. Mol Ecol. 2021;30:5488–502.
Wey-Fabrizius AR, Podsiadlowski L, Herlyn H, Hankeln T. Platyzoan mitochondrial genomes. Mol Phylogenet Evol. 2013;69:365–75.
Nam U-H, Whang I, Kim J-H. The complete mitochondrial genome sequence of Microcotyle caudata (Platyhelminthes: Monogenea) from dark-banded rockfish (Sebastes inermis) in Korea. Mitochondrial DNA Part B. 2020;5:1817–9.
Boore JL. Complete mitochondrial genome sequence of the Polychaete Annelid Platynereis dumerilii. Mol Biol Evol. 2001;18:1413–6.
Gissi C, Pesole G. Transcript mapping and genome annotation of Ascidian mtDNA using EST data. Genome Res. 2003;13:2203–12.
Wang J-G, Zhang D, Jakovlić I, Wang W-M, Gui J, Hwang D-F. Sequencing of the complete mitochondrial genomes of eight freshwater snail species exposes pervasive paraphyly within the Viviparidae family (Caenogastropoda). PLoS ONE. 2017;12:e0181699.
Zhang D, Li WX, Zou H, Wu SG, Li M, Jakovlić I, et al. Mitochondrial genomes of two diplectanids (Platyhelminthes: Monogenea) expose paraphyly of the order Dactylogyridea and extensive tRNA gene rearrangements. Parasit Vectors. 2018;11:601.
Zou H, Jakovlić I, Chen R, Zhang D, Zhang J, Li W-X, et al. The complete mitochondrial genome of parasitic nematode Camallanus cotti: extreme discontinuity in the rate of mitogenomic architecture evolution within the Chromadorea class. BMC Genomics. 2017;18:840.
Zhang D, Zou H, Wu SG, Li M, Jakovlić I, Zhang J, et al. Sequencing of the complete mitochondrial genome of a fish-parasitic flatworm Paratetraonchoides inermis (Platyhelminthes: Monogenea): tRNA gene arrangement reshuffling and implications for phylogeny. Parasit Vectors. 2017;10:462.
He Y, Jones J, Armstrong M, Lamberti F, Moens M. The mitochondrial genome of Xiphinema americanumsensu stricto (Nematoda: Enoplea): considerable economization in the length and structural features of encoded genes. J Mol Evol. 2005;61:819–33.
Rubinoff D, Holland BS, Savolainen V. Between two extremes: mitochondrial DNA is neither the Panacea nor the Nemesis of phylogenetic and taxonomic inference. Syst Biol. 2005;54:952–61.
Zhang D, Zou H, Hua C-J, Li W-X, Mahboob S, Al-Ghanim KA, et al. Mitochondrial architecture rearrangements produce asymmetrical nonadaptive mutational pressures that subvert the phylogenetic reconstruction in Isopoda. Genome Biol Evol. 2019;11:1797–812.
Borges R, Boussau B, Szöllősi GJ, Kosiol C. Nucleotide usage biases distort inferences of the species tree. Genome Biol Evol. 2022;14:evab290.
Acknowledgements
The authors would like to thank all laboratory members for their help in conducting the experiments. We thank Prof. Lin-Gang Cai, Prof. Jian-Gong Niu, and Mr. Hong Liu from the Xinjiang Fishery Research Institute for their help during the field studies and fish sampling.
Funding
The study was supported by the National Natural Science Foundation of China (Grants Nos. 31960737 and 31860738), Natural Science Foundation of Xinjiang Uygur Autonomous Region (2021D01B56), and Xinjiang key laboratory of new drug study and creation for herbivorous animals (XJ-KLNDSCHA).
Author information
Authors and Affiliations
Contributions
CLH participated in the study conception and design, sample collection, molecular lab work, mitogenome assembly and annotation, data analysis, data visualisation, and manuscript preparation. KA participated in the sample collection, molecular lab work and data analysis. MK participated in the sample collection, molecular lab work and data analysis. WRZ and MJR participated in the sample collection. NWW and YJL participated in the data analysis. CY participated in the study conception and design and supervised the study. All authors have approved the submitted version and agree to be personally accountable for their own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures contributing to this work comply with the ethical standards of the relevant national and institutional guides on the care and use of laboratory animals, and the study was approved by the Animal Care and Use Committee of Xinjiang Agricultural University (Permit No. 2019021).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Text S1.
Morphology of Diplozoidae. Figure S1. A drawing of a P. homoion specimen. Figure S2. P. homoion—anchor. Figure S3. P. homoion—clamp. Figure S4. P. yarkandense—holotype. Figure S5. Opisthaptor of P. yarkandense. Figure S6. BLAST results for the ITS-2 of P. yarkandense. Figure S7. BLAST results for the ITS-2 of P. homoion. Figure S8. Nad6 3′ elongation in P. homoion. Figure S9. Alignment of translated nad4L gene products of Polyopisthocotylea. Figure S10. Alignment of trnI genes of Polyopisthocotylea. Figure S11. Secondary structure of the trnI gene of P. homoion. Figure S12. Alignment of trnG genes. Figure S13. Alignment of trnM genes. Table S1. Primers used for sequencing and amplification of the complete mitogenome of P. yarkandense. Table S2. Primers used for sequencing and amplification of the complete mitogenome of P. homoion.
Additional file 2: Dataset S1.
Taxonomic details and comparative mitogenomic data for the polyopisthocotylean mitogenomic dataset used. Dataset S2. Comparative gene data for the polyopisthocotylean mitogenomic dataset used.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hao, CL., Arken, K., Kadir, M. et al. The complete mitochondrial genomes of Paradiplozoon yarkandense and Paradiplozoon homoion confirm that Diplozoidae evolve at an elevated rate. Parasites Vectors 15, 149 (2022). https://doi.org/10.1186/s13071-022-05275-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05275-x