
Abstract
Background
In Brazil, malaria is concentrated in the Amazon Basin, where more than 99% of the annual cases are reported. The main goal of this study was to investigate the population structure and genetic association of the biting behavior of Nyssorhynchus (also known as Anopheles) darlingi, the major malaria vector in the Amazon region of Brazil, using low-coverage genomic sequencing data.
Methods
Samples were collected in the municipality of Mâncio Lima, Acre state, Brazil between 2016 and 2017. Different approaches using genotype imputation and no gene imputation for data treatment and low-coverage sequencing genotyping were performed. After the samples were genotyped, population stratification analysis was performed.
Results
Weak but statistically significant stratification signatures were identified between subpopulations separated by distances of approximately 2–3 km. Genome-wide association studies (GWAS) were performed to compare indoor/outdoor biting behavior and blood-seeking at dusk/dawn. A statistically significant association was observed between biting behavior and single nucleotide polymorphism (SNP) markers adjacent to the gene associated with cytochrome P450 (CYP) 4H14, which is associated with insecticide resistance. A statistically significant association between blood-seeking periodicity and SNP markers adjacent to genes associated with the circadian cycle was also observed.
Conclusion
The data presented here suggest that low-coverage whole-genome sequencing with adequate processing is a powerful tool to genetically characterize vector populations at a microgeographic scale in malaria transmission areas, as well as for use in GWAS. Female mosquitoes entering houses to take a blood meal may be related to a specific CYP4H14 allele, and female timing of blood-seeking is related to circadian rhythm genes.
Graphical Abstract

Similar content being viewed by others

Background
Malaria is the most impactful arthropod-borne disease in developing countries. According to the WHO world malaria report 2019 [1], there were 229 million malaria cases and an estimated 409,000 deaths related to malaria in 2018, 94% of which were concentrated in Africa. In addition to Africa, this disease affects other poor populations in tropical and subtropical areas because environmental conditions are favorable for the development of the vectors and dissemination of the causative agent [2].
Brazil presents a high incidence of malaria, with the majority of the 194,000 cases registered in 2018 concentrated in the Brazilian Amazon rainforest [3]. Nyssorhynchus darlingi, the main malaria vector in Brazil, is highly susceptible to human Plasmodium and capable of transmitting the parasite inside and outside houses, even when at low density [4]. Preferred breeding sites for this species are collections of clear, shallow water that are shaded, with vegetation and a low salt concentration [5,6,7]. Nyssorhynchus darlingi is both anthropophilic and opportunistic [8, 9] and, as the natural environment becomes more modified, or deforested, local populations tend to cohabit with humans, invading their homes, thereby increasing the importance of this species as a vector [10]. In the Amazon rainforest, it is the anopheline vector that most quickly and efficiently benefits from the changes humans produce to the natural environment [10, 11].
Recent studies support the hypothesis of a Ny. darlingi species complex, and the mosquitoes present in the Amazon correspond to one of three lineages within this complex [12]. Moreover, microgeographic scale studies with markers across the Ny. darlingi genome have demonstrated genetic differentiation that could represent phenotypic differences related to malaria transmission dynamics [13, 14].
The rapid development of technologies involved in whole-genome sequencing (WGS) has resulted in dramatic reductions in the per base sequencing cost. However, studies that require the sequencing of large numbers of samples remain costly, possibly prohibitively so in some laboratories. One low-cost strategy is genotyping-by-sequencing for low-coverage WGS (L-WGS), which is associated with imputation that provides sufficient genomic information to select markers accurately [15]. The accuracy of variant detection is low in genomes with low coverage depth and tends to have a high false positive rate, but this is attenuated when information between samples is combined, providing good common variant identification power [15, 16]. The inference of genotypes by imputation for both panel-based genotyping and sequencing genotyping has been shown to be accurate, allowing for the potential use of extreme low-coverage WGS (EXL-WGS) to discover variants at a dramatic reduction in cost when compared to standard WGS [17, 18].
Li and collaborators [19] demonstrated that rare variants in L-WGS samples are more challenging to detect because of the difficulty in distinguishing genuine rare alleles from sequencing errors. The number of variants identified is higher when the proportion of polymorphisms among the sequenced individuals in the segregated population is higher. Since different approaches can be conducted in EXL-WGS analysis, the sensitivity of each method must be carefully adjusted as the reduction in coverage inevitably amplifies the possibility of false positive detection.
In the present study, we used low-coverage sequencing markers to investigate the population of Ny. darlingi collected in Mâncio Lima, Acre state, Brazil. Genetic data were correlated with information such as specimen collection and location (larva or adult), adult capture time, house-to-house capture location (intra- or peridomestic) and distance of larvae capture sites from forest areas.
Methods
Sample collection
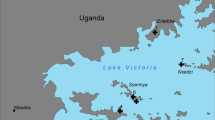
Larval and adult samples were captured from three different collection points in the municipality of Mâncio Lima, Acre state (Fig. 1) during December 2016 and February, May and September 2017. Adult anophelines were collected by human landing catch, by authors DPA, SMK, PRM and PEMR during 12-h collections, from 18:00 to 06:00 hours, for 2 days at each collection point. There were two volunteers indoors and two outdoors, who rotated locations to mitigate collector-specific bias. The three collection points were located around three houses, as depicted in Fig. 1. The three samples are: (i) sampling site A, houses relatively distant from the city center and main streets as well as from forested areas; (ii) sampling site B, houses located in close proximity to the city center, alongside paved streets and distant from forested areas; and (iii) sampling site C, houses relatively distant from city center and in close proximity to forested areas. Biting behavior was recorded and classified as indoor or peridomestic (outdoor). The approximate linear distances between the collection points were: 1.96 km from A to B, 3.39 km from A to C and 2.51 km from B to C. Larvae were also collected in the community of Salvador, Loreto, Peru (sampling site D on Fig. 1), for long-range comparisons (site D is approximately 432 km distant from site A).

Nyssorhynchus darlingi collection sites in Mâncio, Lima city, Acre State, Brazil and in the Salvador community on the Napo River, Iquitos city, Loreto, Peru. A–D represent the collection sites: A 7°37′12.9″S, 72°53′06.7″W; B 7°38′02.1"S, 72°52′26.5"W; C 7°39′05.3″S, 72°53′20.9″W; D 3°44′17.1″S 73°14′19.9″W. The schematic representation of respective sites shows the houses where adults were captured (red dots), all breeding sites analyzed within approximately 1 km of each house (blue), forest areas (green) and the main roads (black lines)
Sample preparation and sequencing
For DNA extraction, heads and thoraces of mosquitoes were separated from the rest of the body with a sterile scalpel. Each adult (head and thorax) and larva (whole body) were extracted individually using the Glass Fiber Plate DNA Extraction Kit (Canadian Center for DNA Barcoding, Guelph, ON, Canada) following the Center’s recommendations. DNA quantification was performed by fluorometric quantitation using QuBit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's recommendations.
DNA libraries were prepared using one fifth of the total volume recommended for the Nextera XT Library prep kit (Illumina Inc., San Diego, CA, USA) following the manufacturer's recommendations. DNA samples were multiplexed to a total of 60 samples per run and sequenced in the NextSeq500 (Illumina) platform in a 151-cycle single-read run. Sequence quality analysis was performed using the FastQC [20] program, and reads were used if results from all analysis modules were approved without errors.
Species identification
Sequencing data was aligned with the Ny. darlingi cytochrome oxidase subunit I (COI) reference sequence (available at https://www.ncbi.nlm.nih.gov/nuccore/KP193458.1/) using Burrows-Wheeler Aligner (BWA) software [21]. After alignment, the individual COI consensus sequences were generated using the SamTools [22] software package. The BLASTn tool [23] was used for multi-species identification using the individual generated COI consensus sequence. Only the highest matching result from BLAST was used. Specimens were discarded if e-value > 1e-100, identities < 200, and identity < 90%, and if the matching sequence was not identified as Ny. darlingi.
Variant calling and genotype imputation
The Ny. darlingi reference genome available in the VectorBase database, version AdarC3 [24], was used. Alignments were performed with BWA software and the variant calling was performed with the SamTools software package. A genotype panel was generated in VCF 4.2 format. Single nucleotide polymorphisms (SNPs) were removed from the pre-imputation panel by minimum allele frequency (MAF) < 0.1, as were missing data (MD) > 0.5 using the LCVCFtools program [25]. Genotypes with sequencing depth (DP) < 5 or genotype quality of phred quality score (GQ) < 20 were imputed with BEAGLE 4.1 software [26] using genotype normalized probability values (PL). After imputation, genotypes were removed from the panel if the probability of the imputed genotype (GP) < 0.95. Finally, SNPs were filtered by MAF > 0.1, MD < 0.3 and Hardy–Weinberg Equilibrium (HWE) < 0.001. HWE was calculated within locations (collection points) to test for existing Wahlund effect between groups.
The unimputed genotypic data used in secondary analyses were generated by the same variant call workflow described above, except for the imputation and post-imputation steps. VCF quality control was applied with LCVCFtools. Genotypes were removed if DP < 5 and GQ < 20 and SNPs were filtered for MAF > 0.1 and MD < 0.8 (at least 15 non-missing genotypes from each strata). HWE analysis control was also applied within strata (HWE < 0.001), including samples from Peru.
Marker selection
The linkage disequilibrium decay was estimated from the r2 pairwise linkage disequilibrium for all markers, calculated by the PLINK 1.9 program [27]. Linkage disequilibrium averages (r2) were calculated by 500-bp windows for 40 adjacent windows, for a total of 20 kb. The prediction of the linkage disequilibrium decay function \(\hat{Y}\) was calculated according to the nonlinear model \(\hat{Y} = \beta_{0} + \beta_{1} \frac{1}{Log\left( x \right)} + e_{res}\) where \(\beta_{0}\), is the intercept value, \(\beta_{1}\) is the coefficient for variable one over the logarithm of the distance of the markers in base pairs and eres is the value of residual error. SNPs were selected by pruning, considering as window size that the average r2 value at that distance is approximately between 0.1 and 0.05.
Population stratification analysis
Stratification signals were estimated by FST according to the mathematical model of Weir and Cockerham [28]. The FST value was calculated using the PLINK 1.9 program according to the model \(F_{ST} = \frac{GS - GT}{{1 - GT}}\)., where GT and GS are the probabilities that two randomly selected alleles in the population and between house-to-house groups, respectively, will be identical by state. Pairwise FST values were calculated using all pruned markers, and a permutation test (10,000 permutations) was performed to verify the statistical significance of the genome-wide average FST and per SNP FST values. The genome-wide average FST and per SNP FST estimates were considered significant when P ≤ 0.05 after false discovery rate (FDR; Benjamini–Hochberg procedure) correction. Principal component analysis (PCA) was performed using the PLINK 1.9 program [27] and k-means clustering was performed using R (R Foundation for Statistical Computing, Vienna, Austria). Both analyses were performed using only the statistically significant SNP for stratification.
Genome-wide association study
A genome-wide association study (GWAS) was performed using the Cochran-Mantel–Haenszel Test statistical model [29]. The test assumes a case control 2 × 2 × K for k strata under the null hypothesis \(H_{0}\) that MH ~ χ2 (Chi-square) with 1 degree of freedom. The MH value can be calculated as \(\chi_{MH}^{2} = \frac{{\left( {\left| {\mathop \sum \nolimits_{i = 1}^{k} \left[ {a_{i} - \frac{{\left( {a_{i} + b_{i} } \right)\left( {a_{i} + c_{i} } \right)}}{{n_{i} }}} \right]} \right| - \frac{1}{2} } \right)^{2} }}{{\mathop \sum \nolimits_{i = 1}^{k} \frac{{(a_{i} + b_{i} )\left( {a_{i} + c_{i} } \right)\left( {b_{i} + d_{i} } \right)\left( {c_{i} + d_{i} } \right)}}{{\left( {n_{i}^{3} - n_{i}^{2} } \right)}}}}\), given that in any biallelic site (alleles A and B) for the kth stratum, a and c are equal to the total number of alleles A for the case and control, respectively. In the same way, b and d are equal to the total number of alleles B for the case and control, respectively. n is the total number of observed alleles for the kth stratum, where n = a + b + c + d.
Case and control categories were considered indoor and outdoor for biting behavior, and samples were collected between 06:00 and 22:00 hours (dusk); samples collected between 02:00 and 06:00 hours (dawn) were considered as blood seeking at dusk or dawn. Stratum groups were determined by collection location (A, B and C). Table 1 shows the number of samples collected within the studied categories.
The FDR multiple test correction method was applied to control for false positives, assuming statistical significance when corrected P-value < 0.05. Manhattan Plot images were generated by R scripting in RStudio [30, 31]. Adjacent genes up to 10 kb from FDR-significant SNPs were investigated using AdarC3 [30, 31] from the annotated Ny. darlingi genome available in the gff3 format in VectorBase.
Results
A total of 436 samples were captured and sequenced, of which 394 and 42 samples were from Mâncio Lima and Salvador, respectively. Following species identification and genome alignment, 73 samples from Mâncio Lima and three from Salvador were discarded due to minimum sequencing coverage threshold, low confidence BLAST result or non-darlingi BLAST result for species identification. The samples used in the population analysis are described in Table 1 and Additional file 1: Table A.
The imputed genotypes panel from 321 Brazilian samples (Table 1) resulted in 1,070,802 markers, about 8.16 SNPs/kbp and a genotyping rate of 83.6%. The non-imputed genotype panel from 360 Brazilian and Peruvian samples resulted in 330,885 markers (29.6% of the imputed panel), around 2.41 SNPs/Kbp and a genotyping rate of 14.2%. The linkage disequilibrium decay was estimated and the observed nonlinear function coefficients were approximately − 0.40 (P < 0.001) and 4.76 (P < 0.001) for \({\beta }_{0}\) and \({\beta }_{1}\) , respectively, with R2 approximately = 0.97. At approximately 12.57 kbp away, the estimated average linkage disequilibrium was 0.1 for the lower confidence interval curve. For practical purposes, 14 kb was adopted as the window size for the pruning process. Figure 2 shows the observed average linkage disequilibrium values as a function of distance. Marker selection was performed by pruning, resulting in 123,620 markers (about 0.91 SNPs/kbp) and a genotyping rate of 86.06%.

Estimated LD decay. Black dashed vertical line represents the estimated distance (in kbp) for r2 ≤ 0.1. Black dashed horizontal line represents mean r2 = 0.1. Black error bars represent the mean standard error. Green dashed line describes the estimated LD decay (nonlinear regression). Abbreviations: LD linkage disequilibrium
Mean FST values obtained by pairwise comparisons of geographically and behaviorally distinct groups are described in Table 2, considering imputed after pruning (IMPUT) and non-imputed (RAW) data. All geographically distinct populations presented significant FST values, and no behaviorally distinct ones had significant values. For comparison, 39 larvae of Ny. darlingi collected from Salvador (Peru), an estimated 465 km from Mâncio Lima (Brazil), were compared with all Brazilian mosquitoes, resulting in significant FST (0.0420; P-value < 0.0001) that was 15-fold greater than the highest FST obtained when groups within Mâncio Lima, about 2–3 km apart, were compared. The per SNP permutation analysis resulted in a subset of 34 microgeographic informative SNPs. The results from the PCA and clustering analysis shown in Fig. 3 indicate an optimal k value of k = 3. Clusters 1, 2 and 3 contain 7, 18 and 70 samples from location A, 34, 11 and 15 samples from location B and 35, 83 and 48 samples from location C. The relationship between clusters and locations was found to be not independent (χ2 = 95.257, df = 4, P-value < 2.2e-16).

The left image shows the principal component analysis and k-means clustering analysis. The right images show the optimal k for k-means analysis (top right) and SNP FST histogram for the statistically significant microgeographic informative SNPs (bottom right). For the optimal k plot, the blue line with dots represents the TWSS for the k-th value (left axis) and the red bars represent the difference (in percentage) between the TWSS for the k-th value and (k - 1)-th value (right axis). Abbreviations: SNP, Single nucleotide; TWSS, total within sum of squares polymorphism
Cochran-Mantel–Haenszel model genome-wide association tests were performed between mosquitoes collected outdoors and indoors (Fg. 4-I; Table 3) and mosquitoes collected during dusk (06:00 to 10:00 PM) and dawn (02:00 to 06:00 AM) (Fig. 4-II; Table 3). For the indoor and outdoor analysis, three different scaffolds had significantly associated SNPs that present genes < 10 kb apart from the significant SNPs (cyp4H14, period and prp4). For the blood-seeking period, three scaffolds had significantly associated SNPs, of which two present genes < 10 kb apart from the significant SNPs (timeless-2 and rdgC) (Fig. 4).

Results for the genome-wide association study on biting behavior (outdoor vs indoor) and blood-seeking (dusk vs dawn). Dashed lines represent false discovery rate-corrected P-value threshold of 0.05. Highlighted colors represent the scaffolds containing significantly associated SNPs. Labels represent genes located < 10 kb apart from the significant SNPs
Discussion
Population stratification and diversity
Significant FST values were observed between groups from different collection sites in both the analyses (imputed and non-imputed data). Considering pairwise comparisons of the groups from Brazil and the comparison between groups from Brazil and Peru, groups collected in Acre showed signs of stratification that were approximately 15-fold lower when compared to the FST values between the groups from Brazil and Loreto, Peru. Although the Mâncio Lima groups showed significance in the permutation tests, the FST values showed a relatively weak signal of stratification. Gélin and collaborators [32] evaluated stratification between populations of Anopheles gambiae in Muheza, Tanzania using microsatellite markers from 172 mosquito samples (43, 27 and 102 from Mamboleo, Songa Kibaoni and Zeneth villages, respectively). The linear distances between the studied locations ranged from 5 to 10 km. FST values of 0.001, 0.003 and 0.009 were observed at distances of 6.5, 9.2 and 3.5 km, respectively, but none of the results were significant in the permutation test. Our study presents similar observed values regarding the magnitude of the stratification signal for short distances and, interestingly, the stratification signal of our data is significant. Stratification analysis detected convergent results between the imputed and non-imputed panels, indicating that imputation does not generate significant bias in stratified populations. The groups collected for biting behavior and blood-seeking period did not show significant FST values. The PCA and clustering analysis indicated an optimal k-value of k = 3 because this value had the optimal TWSS difference when compared to k < 3; in addition the TWSS difference reached a plateau when k > 3. The three main clusters indicate a high association with the microgeographic structure, with samples from location A mostly in the third cluster (73.7%), samples from location B mostly in the first cluster (56.7%) and samples from location C mostly in the second cluster (50%). Interestingly, the mosquito population structure presented here seems to be similar with Plasmodium vivax population structure reported by Salla and collaborators [33], where the city contains clusters of genetically correlated parasites. We suggest that mosquito population structure could contribute to parasite population structure.
Genome-wide association study
Our investigation of the gene regions adjacent to the four significant SNPs in the GWAS for biting behavior revealed some genes that should be highlighted, including prp4 and CYP4H14 (CYP450 superfamily). Two SNPs (ADMH02000641:85,279 and ADMH02000641:86,788) were < 10 kbp apart from CYP4H14 (FDR-corrected P-value < 0.05). CYP450 is a well-known superfamily containing members that are important in determining insecticide resistance in insects [34], including in anophelines [35,36,37]. The relationship between the use of pyrethroid insecticides indoors at locations where the samples were collected and the presence of markers associated with CYP450 genes was evident, since individuals with higher degrees of insecticide resistance were found to have a higher chance of survival in an environment where the insecticide was applied. Gao and collaborators [38] studied the response to five different types of insecticides on Plutella xylostella based on transcriptome analysis to identify genes that responded to these treatments. The tested insecticides were chlorantraniliprole, cypermethrin, dinotefuran, indoxacarb and spinosad. Differential expression of prp4 genes was detected, indicating the functional importance of this gene in insecticide resistance. The role of prp4 is not yet clear, but the results suggest the importance of further studies to disclose the relationship between prp4 and insecticide resistance.
The results of our GWAS on blood-seeking at dusk or dawn and on adjacent gene regions related to the three significantly associated SNPs highlight two genes and their biological roles. The SNP ADMH02000929:14,323 (FDR-corrected P-value < 0.05) is located < 1.5 kb downstream from the retinal degeneration C protein (rdgC) gene locus, and the SNP ADMH02001945:12,409 (FDR-corrected P-value < 0.05) is located approximately 1 kb downstream from the timeout/timeless-2 (tim2 or timeout) gene locus. Rhodopsin phosphatase (rdgC) plays an important role in the dephosphorylation of rhodopsin Rh1, the most abundant photosensory protein in Drosophila melanogaster. Rh1 is required for molecular synchronization with light and circadian rhythm behavior [39]. The loss of the rdgC gene function is associated with Rh1 hyperphosphorylation, leading to photoreceptor degeneration in the presence of light in D. melanogaster adults [40]. Adewoye[41] described several candidate genes associated with the circadian cycle in D. melanogaster, including rdgC. Timeout is a timeless gene (tim1) paralog, and both have been described as components of the circadian cycle in D. melanogaster. Timeout is mainly involved in the perception of luminosity and circadian photoreception in adults [42]. Considering that all of the analyzed samples in the GWAS analysis were adult Ny. darlingi (from Brazil), the functional association of timeout with the light stimulus at the time that anophelines were collected is rather remarkable. Honnen and collaborators [43] studied differentially expressed genes in response to overnight artificial light treatment in Culex pipiens and timeout was observed in males.
Conclusion
The genetic association related to the behavior of females entering houses seems to be selection mediated by the use of indoor insecticides. On the other hand, genetic control of the blood-seeking period could be an ecological adaptation to host availability. Taken together, the data presented here suggest that L-WGS with adequate processing represents a powerful tool to genetically characterize vector populations at a microgeographic scale in malaria transmission areas, as well as for GWAS to disclose behavioral processes, such as the findings that females entering the houses to take a blood meal might be related to a specific CYP4H14 allele and that female time of blood-seeking is related to circadian rhythm genes.
Availability of data and materials
Data are available at NCBI with the following BIOSAMPLE numbers: SAMN17015725 to SAMN17016048 and SAMN21386746 to SAMN21386784 as described in the Additional file 1: Table A.
References
World Health Organization. World malaria report 2018. World Health Organization; 2019. https://www.who.int/publications/i/item/9789241565653.
Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–7.
Pan-American Health Organization/WHO. Interactive malaria statistics. Pan American Health Organization/World Health Organization. http://www.paho.org/hq/index.php?option=com_content&view=article&id=2632:2010-interactive-malaria-statistics&Itemid=2130&lang=en. Accessed 15 Nov 2019
Hiwat H, Bretas G. Ecology of Anopheles darlingi root with respect to vector importance: a review. Parasit Vectors. 2011;4:177.
Forattini OP. Culicidologia médica: identificaçäo, biologia e epidemiologia: v 2. Sao Paulo: Editora da Universidade de Sao Paulo. 2002.
dos Reis IC, Codeço CT, Degener CM, Keppeler EC, Muniz MM, de Oliveira FGS, et al. Contribution of fish farming ponds to the production of immature Anopheles spp. in a malaria-endemic Amazonian town. Malar J. 2015;14:452.
Reis IC, Codeço CT, Câmara DCP, Carvajal JJ, Pereira GR, Keppeler EC, et al. Diversity of Anopheles spp (Diptera: Culicidae) in an Amazonian Urban Area. Neotrop Entomol. 2018;47(3):412–17. https://doi.org/10.1007/s13744-018-0595-6.
Moreno M, Saavedra MP, Bickersmith SA, Prussing C, Michalski A, Tong Rios C, et al. Intensive trapping of blood-fed Anopheles darlingi in Amazonian Peru reveals unexpectedly high proportions of avian blood-meals. PLoS Negl Trop Dis. 2017;11:e0005337.
Saavedra MP, Conn JE, Alava F, Carrasco-Escobar G, Prussing C, Bickersmith SA, et al. Higher risk of malaria transmission outdoors than indoors by Nyssorhynchus darlingi in riverine communities in the Peruvian Amazon. Parasit Vectors. 2019;12:374.
Rozendaal JA. Observations on the distribution of anophelines in Suriname with particular reference to the malaria vector Anopheles darlingi. Mem Inst Oswaldo Cruz. 1990;85:221–34.
Consoli R, Lourenço-de-Oliveira R. Principais mosquitos de importância sanitária no Brasil. Rio de Janeiro: Fiocruz Google Scholar; 1994.
Emerson KJ, Conn JE, Bergo ES, Randel MA, Sallum MAM. Brazilian Anopheles darlingi root (Diptera: Culicidae) clusters by major biogeographical region. PLoS ONE. 2015;10:e0130773.
Campos M, Conn JE, Alonso DP, Vinetz JM, Emerson KJ, Ribolla PEM. Microgeographical structure in the major Neotropical malaria vector Anopheles darlingi using microsatellites and SNP markers. Parasit Vectors. 2017;10:76.
Campos M, Alonso DP, Conn JE, Vinetz JM, Emerson KJ, Ribolla PEM. Genetic diversity of Nyssorhynchus (Anopheles) darlingi related to biting behavior in western Amazon. Parasit Vectors. 2019;12:242.
Gorjanc G, Dumasy J-F, Gonen S, Gaynor RC, Antolin R, Hickey JM. Potential of low-coverage genotyping-by-sequencing and imputation for cost-effective genomic selection in biparental segregating populations. Crop Sci. 2017;57:1404–20.
Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15:121–32.
Pasaniuc B, Rohland N, McLaren PJ, Garimella K, Zaitlen N, Li H, et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat Genet. 2012;44:631–5.
Rustagi N, Zhou A, Watkins WS, Gedvilaite E, Wang S, Ramesh N, et al. Extremely low-coverage whole genome sequencing in South Asians captures population genomics information. BMC Genomics. 2017;18:396.
Li Y, Sidore C, Kang HM, Boehnke M, Abecasis GR. Low-coverage sequencing: implications for design of complex trait association studies. Genome Res. 2011;21:940–51.
Cathcart R, Roberts A. Evaluating Google Scholar as a tool for information literacy. Internet Ref Serv Q. 2005. https://doi.org/10.1300/j136v10n03_15.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST: architecture and applications. BMC Bioinform. 2009;10:421. https://doi.org/10.1186/1471-2105-10-421.
Giraldo-Calderón GI, Emrich SJ, MacCallum RM, Maslen G, Dialynas E, Topalis P, et al. VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucl Acids Res. 2015;43:D707-13. https://doi.org/10.1093/nar/gku1117.
Alvarez MVN. LCVCFtools v1.0.0-alpha. 2020. https://zenodo.org/record/4243800. Accessed 26 Nov 2020.
Browning BL, Browning SR. Genotype imputation with millions of reference samples. Am J Hum Genet. 2016;98:116–26.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70.
Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22:719–48.
R Development Core Team. The R reference manual: base package. Network Theory; 2003.
TEAM, RStudio et al. RStudio: integrated development for R. RStudio, Inc., Boston, MA. http://www.rstudio.com, 2015;42:14.
Gélin P, Magalon H, Drakeley C, Maxwell C, Magesa S, Takken W, et al. The fine-scale genetic structure of the malaria vectors Anopheles funestus and Anopheles gambiae (Diptera: Culicidae) in the north-eastern part of Tanzania. Int J Trop Insect Sci. 2016;36(4):161-170. https://doi.org/10.1017/s1742758416000175.
Salla LC, Rodrigues PT, Corder RM, Johansen IC, Ladeia-Andrade S, Ferreira MU. Molecular evidence of sustained urban malaria transmission in Amazonian Brazil, 2014–2015. Epidemiol Infect. 2020;148:e47.
Scott JG. Cytochromes P450 and insecticide resistance. Insect Biochem Mol Biol. 1999;29:757–77.
Balabanidou V, Kampouraki A, MacLean M, Blomquist GJ, Tittiger C, Juárez MP, et al. Cytochrome P450 associated with insecticide resistance catalyzes cuticular hydrocarbon production in Anopheles gambiae. Proc Natl Acad Sci USA. 2016;113:9268–73.
Ibrahim SS, Ndula M, Riveron JM, Irving H, Wondji CS. The P450 CYP6Z1 confers carbamate/pyrethroid cross-resistance in a major African malaria vector beside a novel carbamate-insensitive N485I acetylcholinesterase-1 mutation. Mol Ecol. 2016;25:3436–52.
Donnelly MJ, Isaacs AT, Weetman D. Identification, validation, and application of molecular diagnostics for insecticide resistance in malaria vectors. Trends Parasitol. 2016;32:197–206.
Mo W, Jian-Xia T, Ju-Lin L, Mei-Hua Z, Jing C, Sui X, et al. Study on expression characteristics of cytochrome P450 genes in Anopheles sinensis. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi. 2018;30:149–54.
Gao Y, Kim K, Kwon DH, Jeong IH, Clark JM, Lee SH. Transcriptome-based identification and characterization of genes commonly responding to five different insecticides in the diamondback moth Plutella xylostella. Pestic Biochem Physiol. 2018;144:1–9.
Ogueta M, Hardie RC, Stanewsky R. Non-canonical phototransduction mediates synchronization of the drosophila melanogaster circadian clock and retinal light responses. Curr Biol. 2018;28:1725-35.e3.
Xiong B, Bellen HJ. Rhodopsin homeostasis and retinal degeneration: lessons from the fly. Trends Neurosci. 2013;36:652–60.
Adewoye AB, Nuzhdin SV, Tauber E. Mapping quantitative trait loci underlying circadian light sensitivity in Drosophila. J Biol Rhythm. 2017;32(5):394–405. https://doi.org/10.1101/135129.
Benna C, Bonaccorsi S, Wülbeck C, Helfrich-Förster C, Gatti M, Kyriacou CP, et al. Drosophila timeless2 is required for chromosome stability and circadian photoreception. Curr Biol. 2010;20:346–52.
Acknowledgements
Not applicable.
Funding
This research was funded by TDR/WHO (201460655) to DG. GC-E was supported by NIH/Fogarty International Center Global Infectious Diseases Training Program (D43 TW007120). MVNA was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) 2018/07406-6. JMV received funding from the National Institutes of Health, USA, International Centers for Excellence in Malaria Research (U19AI089681).
Author information
Authors and Affiliations
Contributions
PEMR and DPA designed the field and laboratory work. DPA, SMK, PR-M, GC-E, MM, DG, JEC and PEMR participated in field collections. DPA, SMK, IAFB, ACFM, ACS performed the wet laboratory research. MVNA and PEMR worked on bioinformatics. MVNA, DPA and PEMR analyzed the data. All authors actively contributed to the interpretation of the findings. PEMR, MVNA and DPA wrote, JEC and DG revised the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical review and approval were waived for this study because only the professionally trained authors conducted the human landing catches and the use of this method to collect anopheline mosquitoes was considered to be a risk management issue, not a human subjects issue. All normal safety precautions were taken. The authors (DPA, SMK, PRM and PEMR) involved in the human landing catches were fully informed about the details of the procedures, potential risks and mitigation plans, and were subject to checks by medical doctors for 2 weeks after the collection.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table A
. List of statistically significant taxonomy identifications using BLAST and COI gene sequence as target. Individual sample data can be accessed using the BioSample accession code on NCBI.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Alvarez, M.V.N., Alonso, D.P., Kadri, S.M. et al. Nyssorhynchus darlingi genome-wide studies related to microgeographic dispersion and blood-seeking behavior. Parasites Vectors 15, 106 (2022). https://doi.org/10.1186/s13071-022-05219-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05219-5