
Abstract
Background
The subfamily Phascolostrongylinae (Superfamily Strongyloidea) comprises nematodes that are parasitic in the gastrointestinal tracts of macropodid (Family Macropodidae) and vombatid (Family Vombatidae) marsupials. Currently, nine genera and 20 species have been attributed to the subfamily Phascolostrongylinae. Previous studies using sequence data sets for the internal transcribed spacers (ITS) of nuclear ribosomal DNA showed conflicting topologies between the Phascolostrongylinae and related subfamilies. Therefore, the aim of this study was to validate the phylogenetic relationships within the Phascolostrongylinae and its relationship with the families Chabertiidae and Strongylidae using mitochondrial amino acid sequences.
Methods
The sequences of all 12 mitochondrial protein-coding genes were obtained by next-generation sequencing of individual adult nematodes (n = 8) representing members of the Phascolostrongylinae. These sequences were conceptually translated and the phylogenetic relationships within the Phascolostrongylinae and its relationship with the families Chabertiidae and Strongylidae were inferred from aligned, concatenated amino acid sequence data sets.
Results
Within the Phascolostrongylinae, the wombat-specific genera grouped separately from the genera occurring in macropods. Two of the phascolostrongyline tribes were monophyletic, including Phascolostrongylinea and Hypodontinea, whereas the tribe Macropostrongyloidinea was paraphyletic. The tribe Phascolostrongylinea occurring in wombats was closely related to Oesophagostomum spp., also from the family Chabertiidae, which formed a sister relationship with the Phascolostrongylinae.
Conclusion
The current phylogenetic relationship within the subfamily Phascolostrongylinae supports findings from a previous study based on ITS sequence data. This study contributes also to the understanding of the phylogenetic position of the subfamily Phascolostrongylinae within the Chabertiidae. Future studies investigating the relationships between the Phascolostrongylinae and Cloacininae from macropodid marsupials may advance our knowledge of the phylogeny of strongyloid nematodes in marsupials.
Graphical Abstract

Similar content being viewed by others


Background
The Phascolostrongylinae is a subfamily of strongyloid nematode (Nematoda: Strongyloidea) belonging to the family Chabertiidae. The subfamily comprising nine genera and 20 species, parasitises macropodid (Family Macropodidae) and vombatid (Family Vombatidae) marsupials. Most of the phascolostrongyline genera occur within the intestines of their hosts. However, a few exceptions are found in the stomachs of their hosts, including Paramacropostrongylus from grey kangaroos (Macropus spp.) and Wallabicola from swamp wallabies (Wallabia bicolor) [1].
Genera of the Phascolostrongylinae are currently subdivided into three tribes (Phascolostrongylinea, Hypodontinea and Macropostrongyloidinea) based primarily on the features of the buccal capsule [2]. The tribe Phascolostrongylinea, characterised by leaf crown elements surrounding the buccal capsule, consists of Phascolostrongylus and Oesophagostomoides and occurs exclusively in wombats. Hypodontinea comprises genera with globular buccal capsules, namely Hypodontus, Macropicola and Corollostrongylus. The genera of the Macropostrongyloidinea, including Macropostrongyloides, Paramacropostrongylus, Torquenema and Wallabicola, possess cylindrical buccal capsules surrounded by teeth or denticles [2]. The morphological classification of the Phascolostrongylinae has been uncertain due to extensive variation in oral morphology that can be challenging to observe [1, 2]. Therefore, molecular markers, including the first and second region of internal transcribed spacers (ITS-1 and ITS-2) of ribosomal DNA (rDNA), have been utilised for specific identification purposes, detection of genetic variation and phylogenetic analyses within the Phascolostrongylinae [3–9]. In a recent study, two species (Macropostrongyloides dissimilis and Paramacropostrongylus toraliformis) were found to be divergent from their congeners using ITS markers [9]. Subsequently, morphological examination of these nematodes led to the description of two new genera, namely Wallabicola dissimilis (formerly M. dissimilis) from the swamp wallaby, Wallabiocola bicolor and Torquenema toraliforme (formerly P. toraliformis) from the eastern grey kangaroo Macropus giganteus [10].
Although ITS sequence data can be useful for inferring phylogenetic relationships at the species and genus level, this has not been the case at higher taxonomic levels. Phylogenetic analyses within the superfamily Strongyloidea determined using the ITS-2 marker showed conflicting topologies and low nodal support for the relationships between the Phascolostrongylinae, Oesophagostominae and Chabertiinae [11]. Amino acid sequences derived from the mitochondrial protein-coding genes have been used to validate phylogenetic relationships within Hypodontus [12] and Macropostrongyloides [13] of the Phascolostrongylinae. These studies validated previous phylogenetic hypotheses based on ITS sequence data with improved nodal support in the phylogenetic analyses.
Therefore, in the present study we assessed the phylogenetic relationships within the subfamily Phascolostrongylinae utilising the amino acid sequence data sets derived from the mitochondrial protein-coding genes. We also assessed the phylogenetic relationship between genera of the Phascolostrongylinae with other subfamilies of the Chabertiidae and with the Strongylidae. Published mitochondrial protein sequences of genera from these families were included in the phylogenetic analyses.
Methods
Sample collection and DNA extraction
Adult male and female nematodes used in the current study (Table 1) were obtained from the frozen parasite collection at the School of Veterinary Science, The University of Melbourne. The nematodes were collected from the gastrointestinal tracts of carcasses of hosts from commercial cullings or vehicle collisions (State-issued permits: Victorian Department of Sustainability and Environment 90-053, 93-016, 10000434, 100003649; Queensland Department of Environment and Heritage Protection WA00006125). The samples had been either frozen at − 80 °C or preserved in 70% ethanol and then frozen at − 80 °C as individuals or pools. If preserved in ethanol, worms were rehydrated in distilled water prior to DNA isolation. For morphological identification, the anterior and posterior extremities of each nematode were removed with a scalpel blade, cleared with lactophenol and identified. They were subsequently deposited in the helminthological collection of the South Australia Museum, Adelaide, as voucher specimens (Registration numbers Australian Helminthological Collection [AHC] 36783, 49028, 49037, 49035, 49052, 49051, 49055, 49108). The mid-sections were used for the DNA isolation.
Genomic DNA was extracted from individual nematodes using the QiaAmp Micro Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol for extracting DNA from tissues. For initial molecular identification, the ITS-1 and ITS-2 sequences were determined for each individual using an established PCR-based sequencing method [9]. Prior to sequencing, the quantity and quality of the DNA were determined using the 2200 TapeStation (Agilent Technologies, Santa Clara, CA, USA).
Sequencing and gene annotation
Illumina TruSeq indexed libraries were prepared using sheared DNA following the manufacturer’s protocol (Illumina Inc., San Diego, CA, USA). Briefly, the steps included: (i) end-repair and A-tailing of the 3′ ends; (ii) ligation of the adaptors; (iii) enrichment of the libraries and purification of the enriched library using Ampure Beads (Beckman Coulter, Brea, CA, USA). The libraries were quantified using the 2200 TapeStation, pooled and sequenced on the Illumina MiSeq platform using the 300 cycle v3 reagent kit (2 × 150 paired-end reads). Illumina library preparation and sequencing were carried out at the Walter and Eliza Hall Institute (WEHI) Genomics Facility, Melbourne, Victoria, Australia.
Raw sequence data in the FASTQ format were filtered for quality in Trimmomatic v.0.38 [14] prior to de novo assembly employing the program Spades v3.13.0 under default parameters. For each assembly, the 12 protein-coding genes of the mitochondrial genome were identified by local sequence alignment (6 reading frames) using the amino acid sequence inferred from corresponding genes of reference mitochondrial genomes, using an established workflow system [15]. The reference mitochondrial genomes used were selected based on sequence similarities using Basic Local Alignment Search Tool (BLAST) for sequence analysis [20]. The mitochondrial genome sequence of Hypodontus macropi (NC023083) was used as a reference for Paramacropostrongylus, Torquenema and Wallabicola, and that of Oesophagostomum dentatum (NC013817) was used as the reference for Oesophagostomoides and Phascolostrongylus. The nucleotide sequences of the 12 protein-coding genes of each species included in this study were deposited in the GenBank database under the accession numbers OK111101–OK111108.
Sequence comparison and phylogenetic analyses
The nucleotide and amino acid sequences of the 12 mitochondrial protein-coding genes were aligned separately using CLUSTAL W [21] and MUSCLE [22] followed by a concatenation of the alignments using MEGA software version X [23]. The alignment included previously published mitochondrial protein-coding gene sequences of species of Phascolostrongylinae, Chabertiinae, Oesophagostominae, Strongylidae and Syngamidae (Table 1). Pairwise comparisons were calculated for the nucleotide and amino acid sequences using MEGA software. The nucleotide diversity across each of the protein-coding genes was determined using sliding window analyses (SWAN) carried out in the DnaSP v.5 program [24] using a sliding window of 100 bp and 25-bp steps.
Phylogenetic analysis was conducted using concatenated and aligned amino acid sequences derived from all 12 mitochondrial protein-coding genes. The phylogenetic analysis was conducted using Bayesian inference (BI) in MrBayes v.3.2.7 [25]. The optimal partitioning schemes and substitution model for the BI analyses were determined using PartitionFinder 2 [26] for amino acids, with a model selection set to the Akaike information criterion and greedy search algorithm. According to PartitionFinder, the amino acid sequence alignment was partitioned into eight subsets consisting of subset 1 (cytochrome c oxidase [cox] subunit 1-2), subset 2 (cox3, nicotinamide adenine dinucleotide hydrogen dehydrogenase [nad] subunit 4), subset 3 (nad5), subset 4 (nad6, nad2), subset 5 (nad4L, nad1), subset 6 (adenosine triphosphate synthase subunit 6 [atp6]), subset 7 (cytochrome b [cob]) subset 8 (cox3). The Mtmam (Mitochondrial Mammalia) model was the evolutionary model for all subsets except subset 6 (general reversible Markov model for amino acid substitution of mitochondrial proteins [Mtrev] model). The BI analysis was conducted with four chains each of 10 million Markov chain Monte Carlo iterations, sampling every 1000th generation for four independent runs. Convergence was determined by the average standard deviation of split frequencies of > 0.01, with the potential scale reduction factor approaching 1. The first 25% of the sampled trees were discarded as burnin and the Bayesian consensus tree was constructed from the remaining trees. The BI trees were visualised in FigTree v.1.4.4 [27]. Syngamus trachea (GQ888718) belonging to the family Syngamidae was used as the outgroup.
Results
Nucleotide and amino acid sequence comparisons
The SWAN revealed that the nucleotide diversity across the alignment of 12 concatenated mitochondrial protein-coding genes ranged from 0.084 to 0.290 (Fig. 1). The 5′ prime end of the nad5 gene exhibited the highest level of nucleotide diversity, whereas cox1 was the most conserved gene.

Nucleotide diversity (Pi) across 12 concatenated mitochondrial protein-coding genes (y-axis) of eight genera of Phascolostrongylinae, two genera of Strongylidae and one genus each of Oesophagostominae, Chabertiinae and Syngamidae. Nucleotide diversity was calculated in DnaSP version 6 software using a window of 100 bp and 25-bp steps. The nucleotide position (base pairs) is indicated on the x-axis next to the boundaries between mitochondrial protein-coding genes. CO1 – 3 = Cytochrome c oxidase subunit 1 – 3; ND1 – 6, 4L = Nicotinamide adenine dinucleotide hydrogen dehydrogenase subunits 1 – 6 and 4L; ATP6 = Adenosine triphosphate synthase subunit 6; COB = Cytochrome B
Pairwise amino acid sequence differences among species of Phascolostrongylinae ranged between 0.60% (between two specimens of Paramacropostrongylus iugalis) and 10.6% (between Phascolostrongylus turleyi and Macropostrongyloides baylisi) (Table 2). The genus Wallabicola was most similar to Paramacropostrongylus typicus, with 5.9% sequence variation, compared to 8.6% variation from Macropostrongyloides in which it was formerly placed. The amino acid sequence of Torquenema was most similar to that of Paramacropostrongylus typicus (6.5% sequence variation), the genus to which it was formerly assigned. Amino acid sequence variation between the wombat-specific genera Phascolostrongylus and Oesophagostomoides spp. was small (4.7–5.0%), with Ph. turleyi and Oesophagostomoides longspicularis sharing the greatest sequence similarities. Interestingly, the amino acid sequences of Phascolostrongylus and Oesophagostomoides spp. shared greater similarities with two species from the subfamily Oesophagostominae, Oe. dentatum and Oesophagostomum quadrispinulatum (6.8–7.3% sequence variation) compared to genera from the same subfamily (9.1–10.6% sequence variation) (Table 2).
Phylogenetic analyses
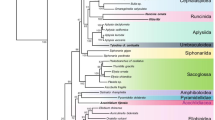
The BI analysis showed strong support for the clustering of the genera of Phascolostrongylinae (Fig. 2). The tree topology showed that the genera of the Phascolostrongylinae distributed between two clades, with the larger clade comprising members of the tribes Macropostrongylinea and Hypodontinea from macropodid marsupials and the smaller clade comprising the tribe Phascolostrongylinea from vombatid marsupials (Fig. 2). The tribe Phascolostrongylinea comprising the wombat-specific genera Phascolostrongylus and Oesophagostomoides formed a strongly supported monophyletic assemblage (posterior probability [pp] = 1), sister to the species occurring in macropodid marsupials and Macropostrongyloides phascolomys from the common wombat. The clade comprising Hypodontus and Macropicola belonging to the tribe Hypodontinea was monophyletic, with strong nodal support (pp = 1). The tribe Macropostrongyloidinea was paraphyletic. Macropostrongyloides was placed as a sister group to the tribe Hypodontinea, and Wallabicola dissimilis from the stomach of the swamp wallaby was placed on a branch external to the clade comprising Hypodontus and Macropostrongyloides. The position of T. toraliforme, sister to Macropostrongyloides lacked nodal support (pp = 0.65). Paramacropostrongylus typicus and P. iugalis from grey kangaroos grouped together with strong nodal support (pp = 1).

Topology of the Bayesian inference phylogenetic analyses inferred from the concatenated alignment of 12 mitochondrial protein-coding genes of the genera of Phascolostrongylinae, Oesophagostominae, Chabertiinae (Chabertiidae) and Strongylidae. Nodal support is indicated as posterior probabilities of the Bayesian inference analysis. Syngamus trachea from the family Syngamidae was used as the outgroup. The host families (Macropodidae or Vombatidae) in which the species of Phascolostrongylinae occur are represented by icons. The scale bar indicates the number of inferred substitutions per amino acid site
Within the tribe Oesophagostominea, Oesophagostomum dentatum and Oesophagostomum quadrispinulatum formed a strongly supported (pp = 1) clade sister to the subfamily Phascolostrongylinae, with the exclusion of Oe. asperum and Oe. columbianum. Oesophagostomum asperum clustered with Chabertia ovina and C. erschowi (tribe Chabertiinea) whilst Oe. columbianum was placed on an external branch. The genera Strongylus vulgaris and Cyclicodontophorus bicoronatus are representative of the family Strongylidae that is sister to the subfamilies Phascolostrongylinae, Oesophagostominae and Chabertiinae, all of which belong to the family Chabertiidae. Overall there were six genera comprising at least two species included in the study, four of these were monophyletic (Macropostrongyloides, Hypodontus, Paramacropostrongylus and Chabertia) and two were paraphyletic (Oesophagostomoides and Oesophagostomum).
Discussion
The current study utilised the amino acid sequences derived from the mitochondrial protein-coding genes to assess the phylogenetic relationships of the subfamily Phascolostrongylinae. The tree topology showed that the nine genera currently placed in the subfamily Phascolostrongylinae clustered together with strong nodal support. Overall, the use of mitochondrial amino acid sequence data sets resulted in well-supported relationships among most but not all taxa included in the phylogenetic analyses. There was good support for the monophyly of the morphologically defined tribes Phascolostrongylinea and Hypodontinea but not Macropostrongyloidinea, consistent with the findings from a previous study using ITS sequence data sets [9].
Contrary to the previous phylogeny based on ITS sequences [9], the BI analysis in the current study showed strong nodal support for the clade containing the genera Macropicola and Hypodontus. This relationship corresponded with the tribe Hypodontinea erected by Beveridge [2]. The classification of Hypodontus and Macropicola has been debated in the past as their morphology shares little resemblance to that of other strongyloid nematodes infecting marsupials. Hypodontus was initially placed in the family Ancylostomatidae as it resembled hookworms, due to its ventrally bent anterior extremity and the presence of two cutting plates within the buccal capsule [28]. Subsequently, Inglis et al. [29] transferred Hypodontus to the subfamily Globocephaloidinae within the Trichostrongyloidea without a clear explanation. However, it was moved back to the Strongyloidea and reassigned as a hookworm in the subfamily Uncinariinae by Durette-Desset et al. [30], having formerly been placed in the hookworm subfamily Bunostomatinae by Skryabin et al. [31]. The genus Macropicola, also in the tribe Hypodontinea, was initially classified as a hookworm in the subfamily Globocephalinae based on its globular buccal capsule and three oesophageal teeth [32]. However, Lichtenfels [33] argued that both Hypodontus and Macropicola belonged to the subfamily Strongylinae in the Strongylidae based on the globular buccal capsule, oesophageal teeth, the Y-shaped female ovejector and a pre-anal vulva. Finally, in the most recent revision, Beveridge [2] assigned Hypodontus and Macropicola to the tribe Hypodontinea, along with Corollostrongylus within the subfamily Phascolostrongylinae, based on their large globular or subglobular buccal capsules that are either straight or bent dorsally or ventrally. This morphological grouping was supported by the mitochondrial protein sequence data in the current study and in a previous study on hookworms which showed the exclusion of Hypodontus and Macropicola from the Strongylinae and Ancylostomatoidea [34]. Although Hypodontus closely resembles hookworms, molecular data suggest that their morphological resemblances could be a result of convergent evolution. Three genotypes of Hypodontus macropi, each from a different host (Osphranter robustus, Thylogale billardierii and Wallabia bicolor) were included in the analyses. The tree topology showed that the specimens of Hypodontus from each host are distinct, with the specimen from the Tasmanian pademelon (T. billardierii) being the most divergent. This finding is consistent with a previous phylogenetic analysis also based on the mitochondrial protein sequence data [12], in addition to ITS-based studies [3, 6]. In the absence of consistent morphological differences between the distinct genotypes, H. macropi remains a cryptic species complex.
Analyses of both the current mitochondrial and previous ITS data sets placed Torquenema external to the clade comprising Macropostrongyloides, but with low branch support. Although Torquenema can be clearly distinguished from Macropostrongyloides by its occurrence in the stomach of its host and its prominent cervical collar, they share some common features, including a Y-shaped ovejector and small peri-oral denticles [10, 35]. Mitochondrial and ITS data support the separation of Torquenema from its previous position within Paramacropostrongylus. However, its current phylogenetic position close to Macropostrongyloides is uncertain due to the lack of nodal support.
The current phylogenetic analyses placed W. dissimilis (formerly Macropostrongyloides dissimilis) external to the clade comprising the genus Macropostrongyloides and the tribe Hypodontinea. This finding differs from that of a previous study using ITS sequence data which showed a strongly supported grouping of W. dissimilis and Paramacropostrongylus spp., both occurring within the stomach of macropodid hosts [9]. However, the current topology based on mitochondrial protein sequence data lacks nodal support (pp = 0.82). Therefore, additional data, including a larger number of representatives of this species and potentially a different genetic marker, are required to validate the phylogenetic position of Wallabicola.
The current and previous study based on ITS sequence data showed strong support for the monophyletic grouping of the genera within tribe Phascolostrongylinea from vombatid marsupials. The topologies of the current and previous phylogenetic analyses based on ITS sequence data showed the grouping of Ph. turleyi and O. longispicularis with O. stirtoni on a sister branch [9]. However, nodal support was higher in the ITS study [9]. Although Oesophagostomoides appears to be paraphyletic due to the position of Phascolostrongylus, there are no morphological features to support this apparent paraphyly. These two genera are considered valid and clearly distinguishable morphologically. The buccal capsules of Phascolostrongylus are shallower with thicker walls compared to Oesophagostomoides and have a greater number of external leaf crown elements [36]. Oesophagostomoides longispicularis and Ph. turleyi often occur at a high prevalence and together within the same host, whereas O. giltneri has been less commonly encountered in the common wombat [36].
The phylogenetic analysis showed strong support for the sister relationship between the genera of the subfamily Phascolostrongylinae with Oe. dentatum and Oe. quadrispinulatum (subfamily Oesophagostominae) which was not evident in a previous study based on ITS-2 sequence data [11]. The genus Oesophagostomum occurs in the caecum of ungulates, rodents and primates [37]. There are several subgenera within Oesophagostomum, some of which corresponded to those included in the current phylogenetic analyses. The separation of Oe. asperum from Oe. columbianum in the phylogenetic tree corresponded with their different subgenera, Hysteracrum and Proteracrum, respectively, whereas the clade closest to the subfamily Phascolostrongylinae, comprising Oe. dentatum and Oe. quadrispinulatum, both belong to the subgenus Oesophagostomum. The genus Chabertia appears to be monophyletic in the current analyses, although C. ovina from a sheep in Australia (GQ888271) was slightly divergent from the one from a goat from China (KF660603), suggesting intraspecific genetic variation within this species. The position of Chabertia, nested among the Oesophagostominae, is consistent with the topology of a previous phylogenetic tree based on the analysis of ITS-2 sequence data [38], suggesting that the current classifications of the Chabertiinae and Oesophagostominae may require further investigation.
The genera Oesophagostomoides and Oesophagostomum were considered synonyms by Popova [39] and Yamaguti [40] based on their morphological similarities. The key differential feature between Oesophagostomoides and Oesophagostomum are the Y-shaped ovejectors in the former genus compared with J-shaped ovejectors in the latter and the shape of their cervical papillae, which are bipartite in the former but conical in the latter [36]. The sister relationship between Oesophagostomum and subfamily Phascolostrongylinae found in the current study raises the question of whether the Oesophagostominae could have been the predecessors of the Strongyloidea in marsupials. However, this proposal was deemed improbable, since the most likely host, rodents, arrived in Australia after the expansive radiation of the marsupials which was believed to coincide with the radiation of their parasitic nematodes [41]. However, in a study using 18S nuclear ribosomal gene sequence data [42], Cyclodontostomum purvursi from Australian rodents grouped closely to Chabertia ovina. Further speculation on the association between the Oesophagostominae and Phascolostrongylinea is beyond the scope of this study and would require additional mitochondrial sequence datasets of other Oesophagostomum spp. and Cyclodontostomum, both of which are currently unavailable.
Although the current phylogenetic analyses suggest the monophyly of the Phascolostrongylinae, not all species were included. These species were Corollostrongylus hypsiprymnodontis (tribe Hypodontinea) from the musky-rat kangaroo Hypsiprymnodon moschatus [43], Macropostrongyloides dendrolagi (tribe Macropostrongyloidinea) from tree kangaroos, Dendrolagus spp. [44], Macropostrongyloides lasiorhini (tribe Macropostrongyloidinea) from the southern hairy-nosed wombat Lasiorhinus latifrons and Oesophagostomoides eppingensis (tribe Phascolostrongylinea) from the northern hairy-nosed wombat Lasiorhinus krefftii [45]. Each of these species occurs in rare or endangered hosts and, therefore, specimens for molecular analyses were not readily available through opportunistic sampling.
Overall, the current study demonstrated that phylogenetic inference based on amino acid sequences of mitochondrial protein-coding genes generated strong nodal support. However, the conflicting evidence between the mitochondrial- and ITS-based analyses in the case of Wallabicola highlights the importance of using multiple genetic markers to independently validate phylogenetic hypotheses. Furthermore, multiple samples from the same host populations should ideally be included in such analyses. However, this can be challenging due to limitations in collecting sufficient samples from wildlife hosts. Sliding window analyses of the mitochondrial protein-coding genes in current and previous studies suggest that there are variable regions flanked by conserved regions in genes such as nad1 and nad5. PCR-coupled sequencing of such potentially phylogenetically informative regions could reduce the cost and amount of DNA required compared to sequencing mitochondrial genomes.
Conclusions
Utilising the amino acid sequences translated from the mitochondrial protein-coding genes, this study provided evidence for the grouping of the currently recognised genera of the Phascolostrongylinae. Findings from the current study contribute to our understanding of the phylogenetic position of the subfamily Phascolostrongylinae within the Chabertiidae. This study suggests that the amino acid sequence data could be used to assess the relationship within the subfamily Cloacininae which has yet to be resolved [46]. The inclusion of genera of the subfamily Cloacininae from macropodid marsupials in future studies could further elucidate the phylogenetic relationships among the strongyloid nematodes from Australian marsupials.
Availability of data and materials
All data generated or analysed during this study are included in this article. The sequence data generated in this study are available in the NCBI database under accession numbers OK111101–OK111108. Molecular vouchers specimens have been deposited in the South Australian Museum under the registration numbers AHC 36783, 49028, 49037, 49035, 49,052, 49051, 49055, 49108.
Abbreviations
- AHC:
-
Australian Helminthological Collection
- atp6:
-
Adenosine triphosphate synthase subunit 6
- BI:
-
Bayesian inference
- BLAST:
-
Basic Local Alignment Search Tool
- cob:
-
Cytochrome b
- cox1-3:
-
Cytochrome c oxidase subunit 1-3
- ITS-1:
-
Internal transcribed spacer region 1
- ITS-2:
-
Internal transcribed spacer region 2
- Mtmam:
-
Mitochondrial Mammalia
- Mtrev:
-
General reversible Markov model for amino acid substitution of mitochondrial proteins
- nad1–6, 4L:
-
Nicotinamide adenine dinucleotide hydrogen dehydrogenase subunits 1–6 and 4L
- MUSCLE:
-
Multiple Sequence Comparison by Log-Expectation program
- pp:
-
Posterior probability
- rDNA:
-
Ribosomal DNA
- SWAN:
-
Sliding window analyses
- WEHI:
-
Walter and Eliza Hall Institute
References
Beveridge I, Mawson PM. A taxonomic revision of the genera Macropostrongyloides Yamaguti and Paramacropostrongylus Johnston & Mawson (Nematoda: Trichonematidae) from Australian marsupials. Aust J Zool. 1978;26:763–87.
Beveridge I. The systematic status of Australian Strongyloidea (Nematoda). Bull Mus Natl Hist Nat Paris. 1987;9:107–26.
Chilton NB, Gasser RB, Beveridge I. Differences in a ribosomal DNA sequence of morphologically indistinguishable species within the Hypodontus macropi complex (Nematoda: Strongyloidea). Int J Parasitol. 1995;25:647–51.
Chilton NB, Huby-Chilton F, Beveridge I, Smales LR, Gasser RB, Andrews RH. Phylogenetic relationships of species within the tribe Labiostrongylinea (Nematoda: Cloacinidae) from Australian marsupials based on ribosomal DNA spacer sequence data. Parasitol Int. 2011;60:381–7.
Chilton NB, Jabbar A, Huby-Chilton F, Jex A, Gasser RB, Beveridge I. Genetic variation within the Hypodontus macropi (Nematoda: Strongyloidea) complex from macropodid marsupial hosts in Australia. Electrophoresis. 2012;33:3544–54.
Gasser RB, Zhu X, Beveridge I, Chilton N. Mutation scanning analysis of sequence heterogeneity in the second internal transcribed spacer (rDNA) within some members of the Hypodontus macropi (Nematoda: Strongyloidea) complex. Electrophoresis. 2001;22:1076–85.
Huby-Chilton F, Beveridge I, Gasser RB, Chilton NB. Single-strand conformation polymorphism analysis of genetic variation in Labiostrongylus longispicularis from kangaroos. Electrophoresis. 2001;22:1925–9.
Sukee T, Beveridge I, Chilton NB, Jabbar A. Genetic variation within the genus Macropostrongyloides (Nematoda: Strongyloidea) from Australian macropodid and vombatid marsupials. Parasitology. 2019;146:1673–82.
Sukee T, Beveridge I, Sabir AJ, Jabbar A. Phylogenetic relationships within the nematode subfamily Phascolostrongylinae (Nematoda: Strongyloidea) from Australian macropodid and vombatid marsupials. Microorganisms. 2021;9:1–13.
Sukee T, Beveridge I, Jabbar A. Torquenema n. g., Wallabicola n. g., and Macropostrongyloides phascolomys n. sp.: new genera and a new species of nematode (Strongylida: Phascolostrongylinae) parasitic in Australian macropodid and vombatid marsupials. Animals. 2021;11:175.
Chilton NB, Gasser RB, Beveridge I. Phylogenetic relationships of Australian strongyloid nematodes inferred from ribosomal DNA sequence data. Int J Parasitol. 1997;27:1481–94.
Jabbar A, Beveridge I, Mohandas N, Chilton NB, Littlewood DTJ, Jex AR, et al. Analyses of mitochondrial amino acid sequence datasets support the proposal that specimens of Hypodontus macropi from three species of macropodid hosts represent distinct species. BMC Evol Biol. 2013;13:259.
Sukee T, Koehler AV, Hall R, Beveridge I, Gasser RB, Jabbar A. Phylogenetic analysis of mitogenomic data sets resolves the relationship of seven Macropostrongyloides species from Australian macropodid and vombatid marsupials. Pathogens. 2020;9:1–11.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Jex AR, Hall RS, Littlewood DTJ, Gasser RB. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010;38:522–33.
Lin RQ, Liu GH, Hu M, Song HQ, Wu XY, Li MW, et al. Oesophagostomum dentatum and Oesophagostomum quadrispinulatum: characterization of the complete mitochondrial genome sequences of the two pig nodule worms. Exp Parasitol. 2012;131:1–7.
Zhao GH, Hu B, Cheng WY, Jia YQ, Li HM, Yu SK, et al. The complete mitochondrial genomes of Oesophagostomum asperum and Oesophagostomum columbianum in small ruminants. Infect Genet Evol. 2013;19:205–11.
Liu GH, Zhao L, Song HQ, Zhao GH, Cai JZ, Zhao Q, et al. Chabertia erschowi (Nematoda) is a distinct species based on nuclear ribosomal DNA sequences and mitochondrial DNA sequences. Parasit Vectors. 2014;7:44.
Gao Y, Wang XX, Ma XX, Zhang ZH, Lan Z, Qiu YY, et al. Characterization of the complete mitochondrial genomes of Coronocyclus labiatus and Cylicodontophorus bicoronatus: Comparison with Strongylidae species and phylogenetic implication. Vet Parasitol. 2021;290:109359.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2017;34:772–3.
Rambaut A. FigTree v1. 4.4. http://tree.bio.ed.ac.uk/software/figtree/2009. Accessed 14 July 2021.
Mönnig HO. Hypodontus macropi n. gen., n. sp., a hookworm of the kangaroo. Union of South Africa 15th Report, Director Vet Services. 1929;1:303–6.
Inglis WG. The geographical and evolutionary relationships of Australian trichostrongyloid parasites and their hosts. Zool J Linn Soc. 1968;47:327–47.
Durette-Desset M-C, Denke MA, Murua R. Présence chez un Rongeur du Chili d’un Nématode Inglamidinae (sub fam nov) appartenant aux Amidostomatidae, famille connue des Mammifères d’Australie. Ann Parasitol Hum Comp. 1976;51:453–60.
Skryabin KI, Schulz RS, Popova TI, Boev SN, Delyyamure SL. Key to parasitic nematodes, volume 3—Strongylata. Leiden: Brill Publishers; 1992.
Mawson PM. Macropicola ocydromi n.g., n.sp. (Nematoda: Strongylidae) from a Western Australian kangaroo. Trans R Soc S Aust. 1978;102:113–5.
Lichtenfels JR. A conventional approach to a new classification of the Strongyloidea, nematode parasites of mammals. Am Zool. 1979;19:1185–94.
Haynes BT, Marcus AD, Higgins DP, Gongora J, Gray R, Šlapeta J. Unexpected absence of genetic separation of a highly diverse population of hookworms from geographically isolated hosts. Infect Genet Evol. 2014;28:192–200.
Sukee T, Beveridge I, Jabbar A. New species of Macropostrongyloides Yamaguti, 1961 (Nematoda: Strongylida) and the redescription of Ma baylisi (Wood, 1930) from Australian macropodid marsupials. Syst Parasitol. 2020;97:267–84.
Beveridge I. A taxonomic revision of the genera Phascolostrongylus Canavan, and Oesophagostomoides Schwartz (Nematoda: Trichonematidae) from wombats. Aust J Zool. 1978;26:585–602.
Lichtenfels JR. CIH keys to the nematode parasites of vertebrates. No. 7. Keys to the genera of the superfamily Strongyloidea. Farnham Royal: Commonwealth Agriculture Bureaux; 1980.
Newton LA, Chilton NB, Beveridge I, Gasser RB. Systematic relationships of some members of the genera Oesophagostomum and Chabertia (Nematoda: Chabertiidae) based on ribosomal DNA sequence data. Int J Parasitol. 1998;28:1781–9.
Popova TI. Principies of nematology, Strongyloidea of animals and man. Trichonematidae. In: Skryabin, editor. Essentials of nematology, vol. 7. Moscow: Izdatelstvo Akademii Nauk SSSR; 1958. p. 424
Yamaguti S. Systema Helminthum. Vol. 111. The nematodes of vertebrates. New York: Interscience Publishers; 1961.
Spratt DM, Beveridge I. Helminth parasites of Australasian monotremes and marsupials. Zootaxa. 2016;4123:1–198.
Chilton NB, Huby-Chilton F, Gasser RB, Beveridge I. The evolutionary origins of nematodes within the order Strongylida are related to predilection sites within hosts. Mol Phylogenet Evol. 2006;40:118–28.
Beveridge I. Corollostrongylus hypsiprymnodontis gen et sp. n. (Nematoda: Strongylidae) from the rat-kangaroo, Hypsiprymnodon moschatus (Marsupialia). J Parasitol. 1978;64:657–60.
Beveridge I. Macropostrongyloides dendrolagi n. sp. and Mbaisonema coronatum n.g., n. sp., two new species of nematodes (Strongyloidea: Cloacinidae) from tree-kangaroos, Dendrolagus spp. (Marsupialia: Macropodidae) from Irian Jaya. Indonesia Syst Parasitol. 1997;38:25–31.
Smales LR. A new species of Oesophagostomoides (Nematoda: Cloacinidae) from the northern hairy-nosed wombat Lasiorhinus krefftii with a key to species of the genus. J Parasitol. 1994;80:638–43.
Beveridge I, Jabbar A, Koehler A, Sukee T. A morphological and molecular phylogenetic analysis of relationships between genera of the nematode sub-family Cloacininae (Stossich) (Strongyloidea: Chabertiidae) parasitic in kangaroos, wallabies and rat-kangaroos (Marsupialia: Macropodoidea). Zootaxa. 2020;4851:271–88.
Acknowledgements
We wish to thank Stephen Wilcox for providing technical assistance with Illumina sequencing.
Funding
This study was financially supported by the Australian Biological Resources Study (Grant Numbers RF217-06 and CBG18-07). TS is a grateful recipient of the Australian Government Research Training Scholarship through the University of Melbourne. Further funding was provided by the University of Melbourne Research Initiatives Fund to AVK and an Australian Research Council Grant to AVK and RBG (LP160101299).
Author information
Authors and Affiliations
Contributions
Conceptualisation: TS and AJ; methodology: TS, AJ, AVK, RBG and RH; software: RH; validation: TS, AJ, AVK, RBG and RH; formal analysis: TS, AVK and RH; investigation: TS; resources: AJ, RBG, TS and AVK; data curation: TS; writing—original draft preparation: TS; writing—review and editing: TS, AJ, IB, AVK, RBG and RH; visualisation: TS; supervision: AJ, IB, AVK, RBG and RH; project administration: AJ, RBG, IB, AVK; funding acquisition: AJ, TS, IB, RBG and AVK. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Specimens were collected under the following state-issued permits: Victorian Department of Sustainability and Environment 90-053, 93-016, 10000434, 100003649; Queensland Department of Environment and Heritage Protection WA 00006125.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sukee, T., Beveridge, I., Koehler, A.V. et al. Phylogenetic relationships of the nematode subfamily Phascolostrongylinae from macropodid and vombatid marsupials inferred using mitochondrial protein sequence data. Parasites Vectors 14, 523 (2021). https://doi.org/10.1186/s13071-021-05028-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-05028-2