
Abstract
Background
Cryptosporidium baileyi is an economically important zoonotic pathogen that causes serious respiratory symptoms in chickens for which no effective control measures are currently available. An accumulating body of evidence indicates the potential and usefulness of metabolomics to further our understanding of the interaction between pathogens and hosts, and to search for new diagnostic or pharmacological biomarkers of complex microorganisms. The aim of this study was to identify the impact of C. baileyi infection on the serum metabolism of chickens and to assess several metabolites as potential diagnostic biomarkers for C. baileyi infection.
Methods
Ultraperformance liquid chromatography-mass spectrometry (UPLC-MS) and subsequent multivariate statistical analysis were applied to investigate metabolomics profiles in the serum samples of chickens infected with C. baileyi, and to identify potential metabolites that can be used to distinguish chickens infected with C. baileyi from non-infected birds.
Results
Multivariate statistical analysis identified 138 differential serum metabolites between mock- and C. baileyi-infected chickens at 5 days post-infection (dpi), including 115 upregulated and 23 downregulated compounds. These metabolites were significantly enriched into six pathways, of which two pathways associated with energy and lipid metabolism, namely glycerophospholipid metabolism and sphingolipid metabolism, respectively, were the most enriched. Interestingly, some important immune-related pathways were also significantly enriched, including the intestinal immune network for IgA production, autophagy and cellular senescence. Nine potential C. baileyi-responsive metabolites were identified, including choline, sirolimus, all-trans retinoic acid, PC(14:0/22:1(13Z)), PC(15:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)), PE(16:1(9Z)/24:1(15Z)), phosphocholine, SM(d18:0/16:1(9Z)(OH)) and sphinganine.
Conclusions
This is the first report on serum metabolic profiling of chickens with early-stage C. baileyi infection. The results provide novel insights into the pathophysiological mechanisms of C. baileyi in chickens.
Graphic abstract

Similar content being viewed by others
Background
Cryptosporidium, an important zoonotic protozoan parasite of humans and animals, has been reported in approximately 30 avian species worldwide, with the prevalence ranging from 0.8 to 44.4% [1]. Among the Cryptosporidium species reported in birds, C. baileyi is the dominant species across all continents, with the exception of Antarctica, especially in chickens [2, 3]. C. baileyi infection can cause serious respiratory symptoms (e.g. coughing, sneezing and dyspnea), decrease in weight gain, higher morbidity and mortality in chickens [4, 5]. Notably, co-infections of C. baileyi with other respiratory and gastrointestinal pathogens (e.g. Escherichia coli and infectious bronchitis virus) have also been reported in chickens [6, 7]. Further, an immunosuppressive effect of C. baileyi infection was also shown following vaccination against reoviruses, infectious bronchitis virus, Newcastle disease virus and avian influenza virus, resulting in significant economic losses to the poultry industry [7,8,9]. More significantly, C. baileyi has been detected in immunodeficient patients [10, 11], suggesting the potential zoonotic risk of this species. However, no effective preventive or treatment agents against C. baileyi infection have been developed to date [12].
The identification of C. baileyi infection is currently mainly based on the presence of oocysts in host feces or of other developmental stages (e.g. schizonts/merozoites, gametocytes/gametes) during histopathological observation [13, 14]. However, the performance of oocyst-based procedures to determine C. baileyi infection is often delayed due to their destructive effects on parasitized tissues (e.g. trachea, bursa of Fabricius, and cloaca), thus relying on the numbers and depth of schizonts prior to oocyst formation [4, 13]. Invasive or injuring histopathological observation is an unattractive option for antemortem diagnosis [14]. In recent decades, metabolomics, an increasingly recognized approach for identifying and quantifying multiple small chemical metabolites in complex biological samples, has been proved to be useful in the search for new diagnostic or pharmacological biomarkers of complex microorganisms (e.g. Mycobacterium tuberculosis, Toxoplasma gondii) [15, 16] as well as for improving our understanding of dormant and intricate interactions between hosts and pathogens [17,18,19,20,21,22]. Many compartments have been used to profile chemical metabolites in humans and animals, such as feces, saliva, urine and blood [23]. Although urine would be the better choice of biofluid in terms of developing a non-invasive diagnostic marker [24], serum samples are more suitable for tracking metabolites with the aim to develop non-invasive diagnostic markers for birds (e.g. chicken) since it’s difficult to purify urine from fecal mixtures due to the specific structural characteristics of the digestive system of birds [25, 26]. Numerous analytical platforms are also applied in metabolomic studies, including nuclear magnetic resonance (NMR) and gas or liquid chromatography coupled to mass spectrometry (GC–MS/LC–MS) [27]. Compared with GC–MS and NMR, LC–MS currently represents the major instrumental and analytical technology used in the global profiling of metabolites [28], and it enables rapid quantification of multiple metabolites ranging from nanograms per liter to grams per liter in body fluids [29]. To further improve the resolution and sensitivity of LC–MS, ultraperformance liquid chromatography-MS (UPLC-MS), one of the more versatile techniques, has been established and used in metabolomics studies [30,31,32]. Previous studies have reported metabolic alterations associated with intestinal permeability in mouse feces following infection with C. parvum using GC–MS [33] and diarrheal aspects of the disease in the mouse gut and COLO-680 N cells during infections of C. parvum or C. hominis [34]. In the present study, we explored the serum metabolomic profiles of chickens in the early stages of C. baileyi infection.
Methods
Chemicals and reagents
All chemicals and solvents were analytical grade. Methanol, acetonitrile and formic acid were purchased from CNW Technologies GmbH (Düsseldorf, Germany). 2-Chloro-L-phenylalanine was obtained from Shanghai Hengchuang Bio-technology Co., Ltd. (Shanghai, China). 1-Heptadecanoyl-2-hydroxy-sn-glycero-3-phosphocholine [LysoPC(17:0)] was purchased from Avanti Polar Lipids (Alabaster, AL, USA).
Experimental design
A total of 18 newly hatched white cockerels (Hy-line variety) were purchased from the Giant Long Company (Shaanxi, China) and randomly divided into experimental (Case 1, E1–E9: C. baileyi-infected chickens) and mock (Con 1, N1–N9) groups with nine chickens per group. Animal care was according to the recommendations of the National Research Council (NRC) as published in the Guide for the Care and Use of Laboratory Animals [35]. All chickens had free access to clean feed and sterile water throughout the whole experimental period. The base diet was also per the recommendation of the NRC, and the components and nutrient levels of the diets are listed in Additional file 1: Table S1. Chickens in the experimental group were orally infected with 1 × 106 C. baileyi oocysts at 3 days after birth according to our previous study [36], while mock birds were orally inoculated with the same volume of phosphate buffer saline.
Sample collection
All chickens were in good nutritional status during the entire experimental period, and no birds died. The blood sample was collected from the heart of each chicken simultaneously into a separate Eppendorf tube to isolate the serum sample on the afternoon of day 5 post-infection (dpi). These samples were used for analyzing the profiles of early serum metabolites since C. baileyi oocyst shedding in chicken feces began at 5 dpi. The obtained serum sample was transferred into a new Eppendorf tube and stored immediately at − 80 °C for further analysis. All chickens in both groups were sacrificed by cervical dislocation, sterilized and stored in specific bags for biological wastes and then later recovered for further processing by the Laboratory Animal Center of Northwest A&F University.
Confirmation of infection
Fecal samples of all chickens in both groups were examined daily using Sheather's sucrose flotation technique [37] and 18S rRNA gene-based nested PCR as reported in [38] to confirm C. baileyi infection. Oocysts per gram feces (OPGs) were counted using the hemocytometer.
Sample preparation for UPLC-MS analysis
For each blood collection, a 100-μl sample of serum was mixed with 10 μl of 2-chloro-L-phenylalanine (0.3 mg/mL) (internal standard) and vortexed for 10 s, followed by the addition of 300 μl of an ice-cold mixture of methanol and acetonitrile (2:1, v/v). The mixed solution was vortexed for 1 min, ultrasonicated in an ice-water bath for 10 min, kept at − 20 °C for 30 min and then centrifuged at 13,000 rpm for 10 min at 4 °C. The supernatant from each tube was then collected and filtered through a 22-μm organic phase pinhole microfilter. Finally, the filtrate was transferred into a UPLC vial and stored at − 80 °C for subsequent UPLC-MS analysis. All extraction reagents were precooled at − 20 °C in advance. Quality control (QC) samples were pooled by mixing all samples at an equal volume.
UPLC–MS analysis for untargeted metabolite profiling
The UPLC-MS analysis was performed on an ACQUITY UPLC I-Class system (Waters Corp., Milford, MA, USA) coupled with a Xevo G2-XS QTof mass spectrometer (Waters Corp.). A 2-μl aliquot of the filtrate described in the previous section was injected into an ACQUITY UPLC BEH C18 column (1.7 μm, 2.1 mm × 100 mm; Waters Corp.) at a flow rate of 0.4 ml/min and a column oven of 45 °C. Mobile phases A and B were water/formic acid (99.9:0.1, v/v) and acetonitrile/formic acid (99.9:0.1, v/v), respectively. Metabolite elution was conducted at the following linear gradient: 0 min, 1% (B); 1 min, 5% (B); 2 min, 30% (B); 3.5 min, 60% (B); 7.5 min, 90% (B); 9.5–12.5 min, 100% (B); 12.7–16 min, 1% (B). All samples were kept at 4 °C during the analysis.
The MS data were acquired in a centroid mean square error (MSE) mode with an electrospray ionization (ESI) source operating in either positive or negative ion mode. The capillary voltage was set to 3 and 2 kV for positive and negative ions, respectively; the sampling cone voltage was set to 40 V. The time-of-flight (TOF) mass range was set from 50 to 1000 m/z, and the scan time was 0.1 s. The QCs were injected at regular intervals (every 9 samples) throughout the analytical run to assess repeatability of the data. All procedures were carried out in the laboratory of Shanghai Luming Biotechnology Co., Ltd. (Shanghai, China).
Data processing and statistical analysis
Raw data were collected from the UPLC-MS analysis platform and preprocessed using the software progenesis QI v2.3 (Nonlinear Dynamics, Newcastle, UK), including baseline filtering, peak picking, integration, retention time (RT) alignment, peak alignment and normalization. The main parameters were set as follows: precursor tolerance, 5 ppm; product tolerance, 10 ppm; product ion threshold, 5%. The metabolites were qualitatively identified by alignment with the Human Metabolome Database (HMDB), Lipidmaps (v2.3) and METLIN Database. Three-dimensional data sets were integrated into an Excel file (Microsoft Corp., Redmond, WA, USA), including the mass-to-charge ratio ( m/z), peak RT and peak intensities, and each ion was identified by using RT–m/z pairs. Any peak with missing values (ion intensity = 0) in > 50% of samples was removed to generate the final matrix. The internal standard was used for QC data (reproducibility). Data for both positive and negative ions were combined into a matrix table and imported into SIMCA software package (v14.0; Umetrics, Umeå, Sweden) for multivariate statistical analysis. Unsupervised principal component analysis (PCA) was utilized to visualize systemic variations and general clustering among all groups, and supervised partial least squares-discriminant analysis (PLS-DA) and orthogonal PLS-DA (OPLS-DA) were used to identify differential metabolites between the mock and experimental groups. The Variable Importance in Projection (VIP) scores in the OPLS-DA model were calculated to select potential differential metabolites, and the variables with a VIP value > 1 were considered relevant for group discrimination. Model overfitting was monitored by using the default seven-fold cross validation and 200-times response permutation testing (RPT). The differential metabolites between two groups were further screened by using the Benjamini–Hochberg adjusted Student’s t-test and fold-change analysis. The log2 fold change (log2FC) represented the ratio of the abundance of the average ion intensities in sera of infected chickens compared to that of mock birds. The P value adjusted by the Benjamini–Hochberg method, also known as the false discovery rate (FDR), was used to identify differentially expressed metabolites and metabolic pathways affected by C. baileyi infection [39]. Metabolites with a VIP value > 1 and FDR (q-value) < 0.05 were identified as significantly differential metabolites. Additionally, the Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) was used to identify important pathways related to metabolic phenotypes following C. baileyi infection.
Biomarker characterization and identification
To identify potential biomarkers for the early diagnosis of C. baileyi infection in chickens, we performed univariate receiver operating characteristic (ROC) curve analyses to quantify the predictive performance of the differential metabolites of interest, and the accuracy (including sensitivity and specificity) was evaluated for each selected metabolite using the area under the ROC curves (AUC), with AUC > 0.7 as the threshold.
Results
Confirmation of C. baileyi infection in chickens
Morphological observation of chicken feces under a microscope (ECLIPSE 80i; Nikon Corp., Tokyo, Japan) found Cryptosporidium oocysts in the feces of the experimental group by using Sheather's sucrose flotation technique without dye, but no oocysts were detected in the mock group (Additional file 2: Figure S1a). The 18S rRNA gene of Cryptosporidium was amplified and sequenced in fecal samples of chickens in the experimental group (Additional file 2: Figure S1b), and the sequence identity was 100% to a C. baileyi isolate from a farmed chicken in Hubei Province, China (MG969393). Oocyst shedding in infected chickens was found from 5 to 28 dpi, with two OPG peaks at 10 and 16 dpi, respectively (Additional file 2: Figure S1c).
Metabolite profiling of chicken serum samples
A total of 4235 metabolites were identified by UPLC-MS (1945 and 2290 for negative and positive ion modes, respectively) in serum samples from the mock and infected chicken groups at 5 dpi, of which 2807, 1147 and 281 were matched against HMDB, Lipidmaps (v2.3) and METLIN Database, respectively (Additional file 3: Table S2). Of these 4235 metabolites, 3865 were clustered into 24 classified super classes, including lipids and lipid-like molecules (2164), organoheterocyclic compounds (437), organic acids and derivatives (396), benzenoids (215), organic oxygen compounds (189), phenylpropanoids and polyketides (189), organic nitrogen compounds (58), alkaloids and derivatives (44), organosulfur compounds (40), organooxygen compounds (37), nucleosides, nucleotides and analogues (27), hydrocarbons (19), homogeneous non-metal compounds (14), organohalogen compounds (13), organometallic compounds (4), lignans, neolignans and related compounds (3), mixed metal/non-metal compounds (3), organic compounds (3), organonitrogen compounds (3), hydrocarbon derivatives (2), organic 1,3-dipolar compounds (2), inorganic compound (1), miscellaneous inorganic compound (1) and organophosphorus compound (1). The good stability and repeatability of the analysis were revealed by the sufficiently close positioning of the QC samples in a PCA score plot (Fig. 1a). Clear separations between the mock and C. baileyi-infected chicken sera were also shown on score plots of PCA, PLS-DA and OPLS-DA (Fig. 1b, c), indicating that the two experimental groups had distinct metabolic profiles. Seven-fold cross validation R2Y (0.95) and Q2 (0.86) showed good fitness and predictability, and the negative Q2 in the 200-times response permutation testing revealed no overfitting in OPLA-DA (Table 1; Fig. 1d).

Score plots of multivariate statistical analysis. a Principal component analysis (PCA) score plots for all samples. Case 1 Cryptosporidium baileyi-infected chickens, Con 1 phosphate buffer saline-inoculated chickens, QC quality control. b Partial least squares-discriminant analysis (PLS-DA) score plots for Case 1 and Con 1 samples, c orthogonal partial least squares-discriminant analysis (OPLS-DA) score plots for Case 1 and Con 1 samples, d results of 200-times response permutation testing of OPLS-DA. Q2 and R2 represent the intercepts of the regression curve and y-axis generated by the linear regression between the R2 and Q2 values of "permuted” model and the R2Y and Q2Y values of the "real" OPLS-DA model, respectively
Effect of C. baileyi infection on metabolite profiles in chicken sera
To explore the impact of C. baileyi infection on serum metabolism in chickens, the metabolic profiles in both groups of chickens were analyzed at 5 dpi. Using the criteria of VIP value > 1 in the OPLS-DA analysis and FDR (q-value) < 0.05 in the Benjamini–Hochberg method, we selected a total of 138 significantly differential metabolites (including 115 upregulated and 23 downregulated metabolites) (Fig. 2; Additional file 4: Table S3) following C. baileyi infection, corresponding to 1945 and 2290 for negative and positive ion modes, respectively. The top 20 significantly altered metabolites based on the VIP values are listed in Table 2. Further analysis showed that these metabolites could be grouped into 11 classified super classes (Additional file 4: Table S3), with lipids and lipid-like molecules being the most altered compounds in terms of metabolite numbers. Within this latter super class, 92 metabolites belonging to seven classes were identified, including glycerophospholipids (45), fatty acyls (20), sphingolipids (10), prenol lipids (7), steroids and steroid derivatives (4), sterol lipids (4) and glycerolipids (2).

Expression levels of metabolites between the experimental (Case 1, E1–E9) and mock (Con 1, N1–N9) samples. a Volcano plot for all differential metabolites. Each dot represents one metabolite with detectable expression in both conditions, with the colored dots marking the threshold [false discovery rate (FDR) < 0.05] for defining a metabolite as differentially expressed. Red and blue points represent the significantly upregulated and significantly downregulated metabolites, respectively; gray points indicate non-significant differential metabolites. b Hierarchical cluster analysis of all differential metabolites (FDR < 0.05). Each sample is visualized in a single column and each metabolite is represented by a single row. Red coloration indicates significantly increased metabolite levels, while green coloration indicates low expression (see color scale on figure)
The secondary super classes in metabolite numbers were organoheterocyclic compounds and organooxygen compounds, and 7 compounds were identified in both super classes. The former super class included azaspirodecane derivative (1), azoline (1), benzoxazine (1), diazanaphthalene (1), heteroaromatic compound (1), naphthofuran (1), and pyridines and derivative (1), and the latter super class only contained carbonyl compounds (7). Metabolites in other super classes were less than 7. Notably, 12 of differential metabolites could not be matched to any known super classes or classes (Additional file 4: Table S3).
Metabolic pathway affected by C. baileyi
To understand potential functional significance of biological metabolisms during C. baileyi infection, all 138 differential metabolites identified in the present study were submitted into KEGG database for metabolic pathway enrichment analysis. Of 15 pathways detected, Six were found to be significantly different with FDR (q-value) < 0.05 by using the Benjamini–Hochberg method in infected chickens compared to mock birds (Fig. 3a, Additional file 5: Table S4). A total of 9 up-regulated metabolites were included into these significantly altered pathways (Fig. 3b, Additional file 6: Table S5).

KEGG pathway enrichment analysis of differential serum metabolites following C. baileyi infection. a Significantly enrichments pathways with FDR (q-value) < 0.05. b Relationships between metabolic pathways and differential serum metabolites enriched. Each oval denotes one metabolic pathway. Triangles denote differentially abundant metabolites, with red representing upregulated metabolites
Two most enriched pathways associated with energy and lipid metabolism were predicted following C. baileyi infection, namely glycerophospholipid metabolism (4 metabolites) and sphingolipid metabolism (2 metabolites). In addition, 3 metabolisms were also enriched into pathways involved into host immune response and defense against pathogenic agents, namely intestinal immune network for IgA production, autophagy, and cellular senescence.
Identification of potential biomarkers
To identify the metabolic biomarkers related with C. baileyi early infection in chickens, all 9 metabolites enriched into significantly altered pathways, namely choline, sirolimus, all-trans-Retinoic acid, PC(14:0/22:1(13Z)), PC(15:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)), PE(16:1(9Z)/24:1(15Z)), phosphocholine, SM(d18:0/16:1(9Z)(OH)) and sphinganine, were selected for ROC analysis by using plotROC package [40]. We found all these metabolites with the AUC > 0.7 (Table 3), including 8 in ESI + mode (Fig. 4a) and 1 in ESI-mode (Fig. 4b).

Identification of potential biomarkers response to C. baileyi infection. a Potential biomarker metabolites detected in ESI+ mode based on receiver operating characteristic curve analysis, b potential biomarker metabolites detected in ESI− mode based on ROC analysis. ESI Electrospray ionization
Discussion
Cryptosporidium baileyi is a potential zoonotic Cryptosporidium species with specific parasitic sites (e.g. epithelial cells of the respiratory tract) that is distinct from gastrointestinal C. parvum and C. hominis and which can cause serious respiratory diseases in humans and birds [2, 11, 36]. It has been suggested that this protozoan parasite can be used as a model to characterize cryptosporidia due to its distinct morphological and biological features and large oocyst production in chickens [36, 41]. However, effective methods to prevent and control this parasite are currently lacking. Increasing evidence shows that metabolomics is an emerging “omics” technique that can provide a functional readout of cellular biochemistry for identifying significant metabolic pathways related to disease processes and potential biomarkers [42, 43], including metabolic diseases (e.g. diabetes, adiposity, metabolic syndrome), cancers (e.g. breast cancer, liver cancer) and microbial diseases (e.g. hepatitis B, tuberculosis) [15, 17, 19, 21, 22, 34, 44, 45]. In 2012, Ng et al. developed an untargeted metabolomics method using GC–MS to compare differences in metabolites present in fecal extractions from Cryptosporidium-positive and Cryptosporidium-negative patients [46], ultimately identifying 30 possible compounds that contributed most to the differences between two groups. Subsequently, metabolome changes following infections by zoonotic C. parvum and C. hominis were investigated in in vivo mouse models (pregnant female BALB/c mouse or neonatal Swiss mouse fecal sample extractions; neonatal ICR mice small intestinal luminal flush samples and cecal contents; protein-deficient diet C67BL/6 mouse urines) and in in vitro COLO-680 N cell culture using NMR [34], GC-TOF MS [47] or GC–MS [33], with the results showing that the host metabolic profile patterns response to Cryptosporidium infections were significantly affected by Cryptosporidium species (C. parvum or C. hominis) and isolates (C. parvum Iowa II or Weru), infection models, sample compartments (fece, urine, intestinal contents) and analytical platforms (NMR, GC–MS). In the present study, we used the UPLC-MS technique to explore the impact of C. baileyi infection on the serum metabolism of chickens at an early stage of infection. A total of 138 significantly differential serum metabolites were found following C. baileyi infection, most (115 differential metabolites) of which were upregulated. Of these, two upregulated organic nitrogen compounds, namely phosphocholine and choline, had previously been found to also be increased in the urine of C67BL/6 mice on a protein-deficient diet that were infected with C. parvum Iowa strain oocysts, at 7 dpi [47], and the up-regulation of choline was also detected in COLO-680 N cells infected with zoonotic Cryptosporidium spp. by using NMR [34]. Choline, an essential substrate in phosphatidylcholine and acetylcholine synthesis, is usually obtained from the diet or the disintegration of membrane phospholipids [48, 49]. Previous studies showed that choline played a significant role in signal transduction, neurotransmitter synthesis and regulation of lipid metabolism in human and animals (e.g. rodents, pigs and chickens) and that it was negatively correlated with weight gain in chickens [50,51,52]. Although few studies have reported the potential actions of host serum phosphocholine during pathogenic infection, the significance of microbial phosphocholine molecules has been demonstrated in host antibacterial immune response and diagnosis of bacterial infections [53,54,55,56].
Previous genomic and biochemical findings indicated that Cryptosporidium survival is highly dependent on host-derived biosynthetic pathways due to the unavailability of some key metabolic pathways and incapability to de novo synthesize nucleosides, fatty acids and amino acids [57, 58]. Significant alterations in metabolites of amino acid biosynthesis pathways have been revealed during Cryptosporidium infection [33, 34, 46, 47]. In the present study, we also detected two significantly differential amino acids, peptides or analogues in serum samples of chickens infected with C. baileyi, and N2-fructopyranosylarginine was upregulated. In addition, one fatty acid or conjugate and one carbohydrate or carbohydrate conjugate were found to be significantly decreased (Additional file 4: Table S3).
The KEGG pathway analysis identified three significantly altered metabolites enriched into four important immunity-associated signal pathways, including intestinal immune network for IgA production, autophagy and cellular senescence. Among these, the intestinal immune network for IgA production, which was the enriched metabolic pathway in this study, was also identified previously in the C. baileyi transcriptomic analysis of chicken tracheal tissues [36]. Specific serum IgA production was detected in patients infected with C. parvum, irrespective of human immunodeficiency virus/immune status [59], and specific IgA antibody response to the coproantigens of C. parvum was also found in serum samples in natural and experimentally infected calves [60, 61]. Serum IgA was also demonstrated in hens infected with C. baileyi [62]. In one study, although Cryptosporidium-induced autophagy-associated molecules were not investigated in host sera, autophagy occurred in intestinal epithelial cells following C. parvum infection [63]. Cellular senescence has been reported in enteroids isolated from neonatal mice and immunocompetent adults following ex vivo C. parvum infection, and senescent cells can communicate with immune cells to invoke an immune response against C. parvum by upregulation of the inflammatory genes Mip-2, Nos2, Dkk1, Icam-1 and IL-6 [64]. Notably, the significance of interplay between metabolic processes and immunity has been reported in several biological processes, including infectious diseases [65, 66]. For example, type I interferon signaling disrupted the hepatic urea cycle and altered systemic metabolism to suppress T-cell function in mice infected with chronic lymphocytic choriomeningitis virus (LCMV) [67]. A combination using genetics and metabolic profiling showed that nine of 12 compounds generated by the gut symbiont Clostridium sporogenes accumulated in host sera and that modulation of serum levels of these metabolites in gnotobiotic mice affected intestinal permeability and systemic immunity [68]. Of three significantly enriched metabolites in immunity-associated pathways during C. baileyi infection, PE(16:1(9Z)/24:1(15Z)), in addition to participating in glycerophospholipid metabolism, could initiate autophagy by covalently binding ATG8 during infections of foreign pathogens and homeostasis maintenance [69]. Rapamycin can inhibit the activity of mechanistic target of rapamycin (mTOR), decrease proliferation of T lymphocytes to reduce adipogenesis and enhance lipogenesis and induce tumor immune evasion [70, 71]. These findings indicate the potential roles of these differential metabolites and that the latter are involved in host immunity or immunopathogenesis during C. baileyi infection.
In recent years, metabolomics has been increasingly recognized as a novel promising tool for developing biomarkers for the early diagnosis of disease [72,73,74]. In our study, ROC analysis of significantly differential metabolites between two groups showed nine C. baileyi-responsive metabolites with AUC > 0.7, including choline, sirolimus, all-trans-retinoic acid, PC(14:0/22:1(13Z)), PC(15:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)), PE(16:1(9Z)/24:1(15Z)), phosphocholine, SM(d18:0/16:1(9Z)(OH)) and sphinganine. As an essential nutrient, choline can modulate immune response through one-carbon metabolism [75], and circulating choline together with its metabolites have been reported to be potential cardiometabolic biomarkers [76]. Sirolimus has been reported to be able to enrich several circulating pro-inflammatory factors, such as interleukin (IL)-12, IL-6 and IL-1β [77]. All-trans-retinoic acid has been found to play an important role in the differentiation of T cells and maintenance of homeostasis [78,79,80,81,82,83], and sphinganine has been identified to be required in programmed cell death, which is recognized as an effective strategy by which plants and animals can defend themselves against infections of pathogens [84]. Consequently, these differential compounds would be potential biomarkers for the early detection of C. baileyi in chickens and also could be used to reveal the suggested interactions between C. baileyi and its host (including chickens).
This study has a number of limitations. First, although several interesting metabolites were found to be responsive to infection by C. baileyi, the small number of chickens in the metabolomic analysis in our study was an unavoidable limitation. More experimental animals are needed to be included in future studies to further confirm our findings. Secondly, only one time point was selected in our study; as such, the dynamic metabolic process during the whole progression of infection can not be perfectly reflected, and limited metabolites of interest were obtained. Last but not the least, future studies should be conducted by integrated application of more available omics technologies (e.g. genomics, transcriptomics, proteomics) to analyze novel interesting findings in serum metabolic processes of chickens infected with C. baileyi. Solving these problems will advance our knowledge in the intricate interactions between Cryptosporidium and hosts.
Conclusion
We have analyzed the serum metabolomics in chickens following Cryptosporidium infection at an early stage of infection for the first time and found differences in certain metabolites between infected birds and healthy ones. These differential compounds were mainly significantly enriched into energy and lipid metabolism and important immunity-associated signal pathways. Nine significantly metabolites were identified as compounds potentially responding to infection of C. baileyi, and these may be used to diagnose chicken cryptosporidiosis at an early stage. The findings in this study suggest that metabolomics profiling provides new insights that will deepen our understanding of the interplay between the host and Cryptosporidium and assist in the development of potential biomarkers for the early detection of Cryptosporidium infections in animals as well as humans.
Availability of data and materials
Data supporting the conclusions of this article are included in the article.
Abbreviations
- AUC:
-
Area under the ROC curve
- dpi:
-
Days post-infection
- ESI:
-
Electrospray ionization
- FDR:
-
False discovery rate
- HMDB:
-
Human Metabolome Database
- GC–MS:
-
Gas chromatography coupled to mass spectrometry
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LC–MS:
-
Liquid chromatography coupled to mass spectrometry
- log2FC:
-
Log2 fold change
- MSE:
-
Mean square error
- NMR:
-
Nuclear magnetic resonance
- NRC:
-
National Research Council
- OPLS-DA:
-
Orthogonal partial least squares-discriminant analysis
- OPG:
-
Number of oocysts per gram
- PCA:
-
Principal component analysis
- PLS-DA:
-
Partial least squares-discriminant analysis
- QC:
-
Quality control
- ROC:
-
Receiver operating characteristic
- RPT:
-
Response permutation testing
- RT:
-
Retention time
- UPLC-MS:
-
Ultra-performance liquid chromatography-mass spectrometry
- VIP:
-
Variable Importance in Projection
References
Nakamura AA, Meireles MV. Cryptosporidium infections in birds—a review. Rev Bras Parasitol Vet. 2015;24:253–67.
Ryan U. Cryptosporidium in birds, fish and amphibians. Exp Parasitol. 2010;124:113–20.
Zahedi A, Ryan U. Cryptosporidium—an update with an emphasis on foodborne and waterborne transmission. Res Vet Sci. 2020;132:500–12.
Fayer R, Xiao L. Cryptosporidium and cryptosporidiosis. Boca Raton: CRC Press; 2008.
Taylor MA, Coop RL, Wall RL. Parasites of poultry and gamebirds. London: Blackwell; 2015.
Blagburn BL, Lindsay DS, Hoerr FJ, Davis JF, Giambrone JJ. Pathobiology of cryptosporidiosis (C. baileyi) in broiler chickens. J Protozool. 1991;38:25S-28S.
Rhee JK, Yang HJ, Yook SY, Kim HC. Immunosuppressive effect of Cryptosporidium baileyi infection on vaccination against avian infectious bronchitis in chicks. Korean J Parasitol. 1998;36:203–6.
Hao YX, Yang JM, He C, Liu Q, McAllister TA. Reduced serologic response to avian influenza vaccine in specific-pathogen-free chicks inoculated with Cryptosporidium baileyi. Avian Dis. 2008;52:690–3.
Ewald AH, Hamed HR, Khalil MR. Consequence of cryptosporidiosis on the immune response of vaccinated broiler chickens against Newcastle disease and/or avian influenza. Vet Res Commun. 2014;38:237–47.
Ditrich O, Palkovic L, Stĕrba J, Prokopic J, Loudová J, Giboda M. The first finding of Cryptosporidium baileyi in man. Parasitol Res. 1991;77:44–7.
Kopacz Ż, Kváč M, Piesiak P, Szydłowicz M, Hendrich AB, Sak B, et al. Cryptosporidium baileyi pulmonary infection in immunocompetent woman with benign neoplasm. Emerg Infect Dis. 2020;26:1958–61.
Cabada MM, White AC Jr. Treatment of cryptosporidiosis: do we know what we think we know? Curr Opin Infect Dis. 2010;23:494–9.
Blagburn BL, Lindsay DS, Giambrone JJ, Sundermann CA, Hoerr FJ. Experimental cryptosporidiosis in broiler chickens. Poult Sci. 1987;66:442–9.
Khurana S, Chaudhary P. Laboratory diagnosis of cryptosporidiosis. Trop Parasitol. 2018;8:2–7.
Subramani E, Jothiramajayam M, Dutta M, Chakravorty D, Joshi M, Srivastava S, et al. NMR-based metabonomics for understanding the influence of dormant female genital tuberculosis on metabolism of the human endometrium. Hum Reprod. 2016;31:854–65.
Zhou CX, Zhou DH, Elsheikha HM, Zhao Y, Suo X, Zhu XQ. Metabolomic profiling of mice serum during toxoplasmosis progression using liquid chromatography-mass spectrometry. Sci Rep. 2016;6:19557.
Xu T, Ping J, Yu Y, Yu F, Yu Y, Hao P, et al. Revealing parasite influence in metabolic pathways in Apicomplexa infected patients. BMC Bioinformatics. 2010;11(Suppl 11):S13.
Newsom SN, McCall LI. Metabolomics: eavesdropping on silent conversations between hosts and their unwelcome guests. PLoS Pathog. 2018;14:e1006926.
Yang W, Zhou G, Zou S, Yang W, Liu A, Sun S, et al. Metabonomics of d-glucaro-1,4-lactone in preventing diethylnitrosamine-induced liver cancer in rats. Pharm Biol. 2018;56:643–8.
Surendran Nair M, Yao D, Chen C, Pieters M. Serum metabolite markers of early Mycoplasma hyopneumoniae infection in pigs. Vet Res. 2019;50:98.
Troisi J, Belmonte F, Bisogno A, Pierri L, Colucci A, Scala G, et al. Metabolomic salivary signature of pediatric obesity related liver disease and metabolic syndrome. Nutrients. 2019;11:274.
Zhang J, Xiao X, Dong Y, Zhou X. Fermented barley extracts with Lactobacillus plantarum dy-1 changes serum metabolomic profiles in rats with high-fat diet-induced obesity. Int J Food Sci Nutr. 2019;70:303–10.
Bujak R, Struck-Lewicka W, Markuszewski MJ, Kaliszan R. Metabolomics for laboratory diagnostics. J Pharm Biomed Anal. 2015;113:108–20.
Khamis MM, Adamko DJ, El-Aneed A. Mass spectrometric based approaches in urine metabolomics and biomarker discovery. Mass Spectrom Rev. 2017;36:115–34.
James EL, Parkinson EK. Serum metabolomics in animal models and human disease. Curr Opin Clin Nutr Metab Care. 2015;18:478–83.
Feng XC. Systematic anatomy of livestock and poultry. Beijing: China Forestry Publishing House; 2014.
Lenz EM, Wilson ID. Analytical strategies in metabonomics. J Proteome Res. 2007;6:443–58.
Gika HG, Theodoridis GA, Plumb RS, Wilson ID. Current practice of liquid chromatography-mass spectrometry in metabolomics and metabonomics. J Pharm Biomed Anal. 2014;87:12–25.
Becker S, Kortz L, Helmschrodt C, Thiery J, Ceglarek U. LC-MS-based metabolomics in the clinical laboratory. J Chromatogr B Anal Technol Biomed Life Sci. 2012;883–884:68–75.
Zhao YY, Lin RC. UPLC-MS(E) application in disease biomarker discovery: the discoveries in proteomics to metabolomics. Chem Biol Interact. 2014;215:7–16.
Li YY, Ghanbari R, Pathmasiri W, McRitchie S, Poustchi H, Shayanrad A, et al. Untargeted metabolomics: biochemical perturbations in Golestan Cohort Study opium users inform intervention strategies. Front Nutr. 2020;7:584585.
Utermann C, Echelmeyer VA, Oppong-Danquah E, Blümel M, Tasdemir D. Diversity, bioactivity profiling and untargeted metabolomics of the cultivable gut microbiota of ciona intestinalis. Mar Drugs. 2020;19:6.
Ng Hublin JS, Ryan U, Trengove R, Maker G. Metabolomic profiling of faecal extracts from Cryptosporidium parvum infection in experimental mouse models. PLoS ONE. 2013;8:e77803.
Miller CN, Panagos CG, Mosedale WRT, Kváč M, Howard MJ, Tsaousis AD. NMR metabolomics reveals effects of Cryptosporidium infections on host cell metabolome. Gut Pathog. 2019;11:13.
National Research Council. Guide for the care and use of laboratory animals. 8th ed. Washington, DC: The National Academies Press; 2011.
Ren GJ, Fan XC, Liu TL, Wang SS, Zhao GH. Genome-wide analysis of differentially expressed profiles of mRNAs, lncRNAs and circRNAs during Cryptosporidium baileyi infection. BMC Genomics. 2018;19:356.
McNabb SJ, Hensel DM, Welch DF, Heijbel H, McKee GL, Istre GR. Comparison of sedimentation and flotation techniques for identification of Cryptosporidium sp. oocysts in a large outbreak of human diarrhea. J Clin Microbiol. 1985;22:587–9.
Xiao L, Morgan UM, Limor J, Escalante A, Arrowood M, Shulaw W, et al. Genetic diversity within Cryptosporidium parvum and related Cryptosporidium species. Appl Environ Microbiol. 1999;65:3386–91.
Saravia CH, Flores C, Schwarz LJ, Bravo L, Zavaleta J, Araujo J, et al. Patterns of mutation enrichment in metastatic triple-negative breast cancer. Clin Med Insights Oncol. 2019;13:1179554919868482.
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011;12:77.
Egyed Z, Sréter T, Széll Z, Beszteri B, Dobos-Kovács M, Márialigeti K, et al. Polyphasic typing of Cryptosporidium baileyi: a suggested model for characterization of cryptosporidia. J Parasitol. 2002;88:237–43.
Vinayavekhin N, Homan EA, Saghatelian A. Exploring disease through metabolomics. ACS Chem Biol. 2010;5:91–103.
Cui L, Hou J, Fang J, Lee YH, Costa VV, Wong LH, et al. Serum metabolomics investigation of humanized mouse model of dengue virus infection. J Virol. 2017;91:e00386-e417.
Dai G, Jia W, Hu X, Xu LX. Study of thermal effect on breast tumor metabolism and growth using metabonomics. Annu Int Conf IEEE Eng Med Biol Soc. 2013;2013:1899–902.
Yue D, Zhang Y, Cheng L, Ma J, Xi Y, Yang L, et al. Hepatitis B virus X protein (HBx)-induced abnormalities of nucleic acid metabolism revealed by (1)H-NMR-based metabonomics. Sci Rep. 2016;6:24430.
Ng JS, Ryan U, Trengove RD, Maker GL. Development of an untargeted metabolomics method for the analysis of human faecal samples using Cryptosporidium-infected samples. Mol Biochem Parasitol. 2012;185:145–50.
VanDussen KL, Funkhouser-Jones LJ, Akey ME, Schaefer DA, Ackman K, Riggs MW, et al. Neonatal mouse gut metabolites influence Cryptosporidium parvum infection in intestinal epithelial cells. MBio. 2020;11:e02582-20.
Leung K. [11C]Choline In: Molecular imaging and contrast agent database (MICAD) [Internet]. Bethesda: National Center for Biotechnology Information; 2004. https://www.ncbi.nlm.nih.gov/books/NBK23549/.
Li Z, Vance DE. Phosphatidylcholine and choline homeostasis. J Lipid Res. 2008;49:1187–94.
Dong XF, Zhai QH, Tong JM. Dietary choline supplementation regulated lipid profiles of egg yolk, blood, and liver and improved hepatic redox status in laying hens. Poult Sci. 2019;98:3304–12.
Gao X, Randell E, Tian Y, Zhou H, Sun G. Low serum choline and high serum betaine levels are associated with favorable components of metabolic syndrome in Newfoundland population. J Diabetes Complic. 2019;33:107398.
Penry JT, Manore MM. Choline: an important micronutrient for maximal endurance-exercise performance? Int J Sport Nutr Exerc Metab. 2008;18:191–203.
Marnell L, Mold C, Du Clos TW. C-reactive protein: ligands, receptors and role in inflammation. Clin Immunol. 2005;117:104–11.
Hergott CB, Roche AM, Naidu NA, Mesaros C, Blair IA, Weiser JN. Bacterial exploitation of phosphorylcholine mimicry suppresses inflammation to promote airway infection. J Clin Invest. 2015;125:3878–90.
Maseda Y, Ohori J, Tanaka N, Nagano H, Miyashita K, Kurono Y. Mucosal and systemic immune response to sublingual or intranasal immunization with phosphorylcholine. Auris Nasus Larynx. 2018;45:273–80.
Ngwa DN, Agrawal A. Structure–function relationships of c-reactive protein in bacterial infection. Front Immunol. 2019;10:166.
Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304:441–5.
Rider SD Jr, Zhu G. Cryptosporidium: genomic and biochemical features. Exp Parasitol. 2010;124:2–9.
Kaushik K, Khurana S, Wanchu A, Malla N. Serum immunoglobulin G, M and A response to Cryptosporidium parvum in Cryptosporidium-HIV co-infected patients. BMC Infect Dis. 2009;9:179.
Peeters JE, Villacorta I, Vanopdenbosch E, Vandergheynst D, Naciri M, Ares-Mazás E, et al. Cryptosporidium parvum in calves: kinetics and immunoblot analysis of specific serum and local antibody responses (immunoglobulin A [IgA], IgG, and IgM) after natural and experimental infections. Infect Immun. 1992;60:2309–16.
Toyoguchi A, Omata Y, Koyama T, Kamiyoshi T, Furuoka H, Kobayashi Y, et al. Specific IgA antibody response to coproantigens of Cryptosporidium parvum in serum and saliva of calves after experimental infection. Vet Parasitol. 2001;96:213–20.
Hornok S, Bitay Z, Széll Z, Varga I. Assessment of maternal immunity to Cryptosporidium baileyi in chickens. Vet Parasitol. 1998;79:203–12.
Priyamvada S, Jayawardena D, Bhalala J, Kumar A, Anbazhagan AN, Alrefai WA, et al. Cryptosporidium parvum infection induces autophagy in intestinal epithelial cells. Cell Microbiol. 2021;23(4):e13298. https://doi.org/10.1111/cmi.13298.
Zhang XT, Gong AY, Wang Y, Chen X, Lim SS, Dolata CE, et al. Cryptosporidium parvum infection attenuates the ex vivo propagation of murine intestinal enteroids. Physiol Rep. 2016;4:e13060.
Wishart DS. Metabolomics for investigating physiological and pathophysiological processes. Physiol Rev. 2019;99:1819–75.
Zhang C, Wang K, Yang L, Liu R, Chu Y, Qin X, et al. Lipid metabolism in inflammation-related diseases. Analyst. 2018;143:4526–36.
Lercher A, Bhattacharya A, Popa AM, Caldera M, Schlapansky MF, Baazim H, et al. Type I Interferon signaling disrupts the hepatic urea cycle and alters systemic metabolism to suppress T cell function. Immunity. 2019;51:1074-1087.e9.
Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature. 2017;551:648–52.
Patel D, Witt SN. Ethanolamine and phosphatidylethanolamine: partners in health and disease. Oxid Med Cell Longev. 2017;2017:4829180.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–76.
Weichhart T. mTOR as regulator of lifespan, aging, and cellular senescence: a mini-review. Gerontology. 2018;64:127–34.
Rysz J, Gluba-Brzózka A, Franczyk B, Jabłonowski Z, Ciałkowska-Rysz A. Novel biomarkers in the diagnosis of chronic kidney disease and the prediction of its outcome. Int J Mol Sci. 2017;18:1702.
Marx D, Metzger J, Pejchinovski M, Gil RB, Frantzi M, Latosinska A, et al. Proteomics and metabolomics for AKI diagnosis. Semin Nephrol. 2018;38:63–87.
Maksymowych WP. Biomarkers for diagnosis of axial spondyloarthritis, disease activity, prognosis, and prediction of response to therapy. Front Immunol. 2019;10:305.
Coleman DN, Alharthi AS, Liang Y, Lopes MG, Lopreiato V, Vailati-Riboni M, et al. Multifaceted role of one-carbon metabolism on immunometabolic control and growth during pregnancy, lactation and the neonatal period in dairy cattle. J Anim Sci Biotechnol. 2021;12:27.
Pan XF, Yang JJ, Shu XO, Moore SC, Palmer ND, Guasch-Ferré M, et al. Associations of circulating choline and its related metabolites with cardiometabolic biomarkers: an international pooled analysis. Am J Clin Nutr. 2021. https://doi.org/10.1093/ajcn/nqab152.
Han Y, Wu L, Ling Q, Wu P, Zhang C, Jia L, et al. Intestinal dysbiosis correlates with sirolimus-induced metabolic disorders in mice. Transplantation. 2021;105:1017–29.
Bono MR, Tejon G, Flores-Santibañez F, Fernandez D, Rosemblatt M, Sauma D. Retinoic acid as a modulator of T cell immunity. Nutrients. 2016;8:349.
Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–74.
Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–64.
Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, et al. Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity. 2011;34:435–47.
Dawson HD, Collins G, Pyle R, Key M, Weeraratna A, Deep-Dixit V, et al. Direct and indirect effects of retinoic acid on human Th2 cytokine and chemokine expression by human T lymphocytes. BMC Immunol. 2006;7:27.
Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35:13-22.84.
Saucedo-García M, Guevara-García A, González-Solís A, Cruz-García F, Vázquez-Santana S, Markham JE, et al. MPK6, sphinganine and the LCB2a gene from serine palmitoyltransferase are required in the signaling pathway that mediates cell death induced by long chain bases in Arabidopsis. New Phytol. 2011;191:943–57.
Acknowledgements
Not applicable.
Funding
This work was funded by the grants from the National Key Research and Development Program of China (Grant Number: 2017YFD0501305), National Natural Science Foundation of China (Grant Number: 32072890) and Open Funds of the State Key Laboratory of Veterinary Aetiological Biology, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Grant Number: SKLVEB2020KFKT015).
Author information
Authors and Affiliations
Contributions
GHZ designed the experiments. XMW and XY performed the experiments, with help from XCF. XMW, XY, XC and YXW analyzed the data. XMW, XY and GHZ wrote the manuscript, with active inputs from LXZ and JKS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was carried out strictly in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Ministry of Health, China. Our protocol with all animal experiments was approved by the Research Ethics Committee of Northwest A&F University. All efforts were made to minimize the pain of animals used in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Ingredient composition and nutrient levels of the basal diets.
Additional file 2: Figure S1.
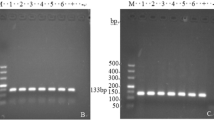
Confirmation of C. baileyi infection in chickens. a Microscopic observation of oocysts in chicken feces of the experimental (left) and mock (right) group. Arrow indicates oocysts of C. baileyi. b Nested-PCR amplification results of 18S rRNA gene of Cryptosporidium. Lanes: M DNA marker DL2000, 1–9 fecal samples of E1–E9 in the experimental group, respectively, 10–18 fecal samples of N1–N9 in the mock group, respectively, 19 negative control. c Oocyst shedding of C. baileyi in chicken feces. The horizontal axis represents the day post infection (dpi) and the vertical axis shows the OPG
Additional file 3: Table S2.
Metabolites in serum samples from mock and C. baileyi-infected chickens.
Additional file 4: Table S3.
Differential metabolites between experimental and mock serum samples responsive to C. bailey infection identified by UPLC-MS in this study.
Additional file 5: Table S4.
KEGG pathway analysis of all differential metabolites following C. baileyi infection in chickens.
Additional file 6: Table S5.
KEGG ID to the differential metabolite name conversion table.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, XM., Yang, X., Fan, XC. et al. Serum metabolomics in chickens infected with Cryptosporidium baileyi. Parasites Vectors 14, 336 (2021). https://doi.org/10.1186/s13071-021-04834-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04834-y