
Abstract
Background
Giardia duodenalis causes giardiasis, with diarrhea as the primary symptom. The trophozoite proliferation of this zoonotic parasite is mainly affected by telomerase, although the mechanism of telomerase regulation has not been thoroughly analyzed.
Methods
This study was performed to identify the telomerase RNA-binding domain (TRBD)-interacting protein in G. duodenalis and its regulation of telomerase. Interaction between TRBD and interacting proteins was verified via pulldown assays and co-immunoprecipitation (co-IP) techniques, and the subcellular localization of the protein interactions was determined in vivo via split SNAP-tag labeling. The hammerhead ribozyme was designed to deplete the mRNA of TRBD-interacting proteins.
Results
Using TRBD as bait, we identified zinc-finger domain (ZFD)-containing proteins and verified it via pulldown and co-IP experiments. Protein-protein interaction occurred in the nuclei of 293T cells and both nuclei of G. duodenalis. The hammerhead ribozyme depleted ZFD mRNA levels, which reduced the reproduction rate of G. duodenalis, telomerase activity and telomere length.
Conclusions
Our findings suggest that ZFD may regulate telomere function in G. duodenalis nuclei.
Similar content being viewed by others


Background
Telomeres are specialized nucleoprotein complexes at the end of a linear chromosome in eukaryotic cells [1,2,3] and are maintained by telomerase, an enzyme that is responsible for telomere extension and adds repeated sequences to telomeric ends in cells [4]. Telomerase complexes generally contain three common subunits: telomerase reverse transcriptase (TERT), telomerase RNA (TER), and proteins that bind RNA or are involved in the stabilization and maturation of telomerase complexes. TERT is the catalytic component of telomerase that monitors cellular changes, restores telomerase activity and maintains telomeres [5,6,7]. TERT can be divided into four main consecutive structural domains: the telomerase essential N-terminus (TEN) domain, the telomerase RNA-binding domain (TRBD), the reverse transcriptase (RT) domain and the C-terminal extension (CTE) domain. The TRBD is responsible for telomerase ribonucleoprotein assembly and is the key component of TERT [8].
Based on in-depth analyses of telomerases, various telomerase-associated proteins have been reported to play important roles in regulating telomerase activity. Studies have shown that hTERT interacts with TPP1 to protect chromosome ends in tumor cells [9]. Additionally, an hTERT fragment (567–594 aa) participates in regulating telomerase activity by interacting with DDRGK1. Moreover, two telomerase proteins from Tetrahymena thermophila, p80 and p95, were identified on the basis of their association with telomerase activity and TER [10]. Although the role of telomere-associated proteins in regulating telomerase has been demonstrated, related mechanisms are less well understood.
Giardia spp. are zoonotic parasites that greatly impact the health of humans and other mammals [11,12,13]. Giardia duodenalis is a parasite of the duodenum and jejunum that causes giardiasis, with diarrhea being the major symptom. Because G. duodenalis is a kind of model organism with a highly reduced genome, it is important to understand complex biological processes in eukaryotic cells. The endomembrane system of G. duodenalis is simple and contains only the endoplasmic reticulum (ER) and peripheral vesicles (PVs) [14]. Because G. duodenalis can be cultured in vitro, it is more amenable for studying such organisms and related diseases than other parasites. At present, the control of giardiasis mainly depends on anti-Giardia drugs, such as metronidazole, tinidazole and albendazole. However, since G. duodenalis proliferation appears “immortalized” and because a detailed understanding of the molecular biological characteristics of Giardia and its regulatory mechanisms is lacking, there is no commercialized vaccine can be used clinically to control giardiasis [15,16,17,18,19]. Thus, the molecular biological characteristics of G. duodenalis must be examined, and the nucleus is a key topic in such experiments.
The complete TERT sequence (GenBank: AF195121.1) and repeat sequence (TAGGG)n in G. duodenalis have been identified [20,21,22], and the TRBD and RT of TERT (cl13446 and cd01648) have been found using tools from NCBI. Nonetheless, only a few studies thus far have analyzed TERT localization, telomere length and telomere activity in G. duodenalis [23]. Therefore, identifying TERT-associated proteins will complement the available data on the regulatory network of G. duodenalis and may contribute to explaining the mechanisms underlying G. duodenalis immortalization.
In G. duodenalis, daughter cells inherit copies of parental nuclei, but there is little evidence defining the position of the TRBD or telomerase-interacting proteins in G. duodenalis nuclei or for whether protein-protein interactions (PPIs) occur in both nuclei or in only one nucleus [18]. Therefore, after identifying TRBD-associated proteins by yeast double-hybrid screening and pulldown and co-immunoprecipitation (co-IP) assays, we explored the location of the TRBD and its associated proteins. Moreover, to ensure the function of TRBD-associated proteins in regulating telomerase, the viral vector-mediated hammerhead ribozyme was used to evaluate telomere length and telomerase activity.
Methods
Parasites, yeast strains, and cell culture
The G. duodenalis wildtype strain isolated from a dog was genotyped as assemblage A in Changchun, China. Yeast strains Y187 and AH109 were grown at 30 °C in yeast extract peptone dextrose agar (YPDA) medium (LABest, Beijing, China). The 293T cell line was cultured in 1640 medium supplemented with 10% FBS in 5% CO2 at 37 °C. To construct a G. duodenalis cDNA library, total RNA was extracted from G. duodenalis (1 × 108/ml) using TRIzol reagent (Roche, Basel, Switzerland), and first and double-strand cDNAs were synthesized according to the SMARTTM cDNA Library Construction kit protocol (Clontech, Palo Alto, USA). The first-strand cDNA sample was amplified using long-distance PCR (LD-PCR), and double-stranded cDNA was purified with CHROMA SPIN+TE 400 columns (Clontech, Saint-Germain-en-Laye, France). Giardivirus vector pC631 was a gift from Wang CC (Department of Pharmaceutical Chemistry, University of California, San Francisco, CA, USA).
Construction of the G. duodenalis cDNA library and yeast two-hybrid screening
A full-length, normalized G. duodenalis cDNA library was introduced into the pGADT7-Rec vector and used as prey, as previously described [24]. The prey, which contains a Gal4 activation domain, was transformed into AH109 cells. The TRBD of G. duodenalis TERT (808–1464 bp) was amplified by PCR (forward, 5′-GAA TTC ATT ACA AGT ACT AGA GTA GTA AAT T-3′; reverse, 5′-GGA TCC CGA TAG ACA AAC GAT AGC CTA C-3′) to construct an EcoRI-BamHI fragment of G. duodenalis cDNA and ligated into pGBKT7 as the bait. The bait, which contains a Gal4 DNA-binding domain, was transformed into Y187. An α-galactosidase assay was performed to assess autoactivation.
The yeast two-hybrid screen was carried out in accordance with Matchmaker Library Construction and Screening Kit (Clontech). The clones were spread on SD/-His/-Leu/-Trp/3-AT and SD/-Ade/-His/-Leu/-Trp/X-α-gal selection media, and the appearance of blue colonies indicated a positive result. Positive (co-transformed with pGBKT7-53 and pGADT7-T) and negative (co-transformed with pGBKT7-Lam and pGADT7-T) controls were also included. The positive transformants were sequenced by Sangon Biotech (Shanghai, China).
Expression, purification, and identification of the TRBD and ZFD
ZFD was cloned into pGEX-4T-1 vector to generate a GST tag. TRBD was ligated with 6X His-tag vector pET-32a (+), generating His-tagged proteins. For protein expression, Escherichia coli BL21(DE3) was grown in 200 mL LB-Amp media at 37 °C until an OD600 of 0.6–0.8 was reached. After induction with 0.25 mmol/l IPTG at 16 °C for another 20 h, the cells were harvested and lysed by sonication in chilled lysis buffer. Cell lysates contained His-TRBD or GST-ZFD were clarified three times by centrifugation at 30,000×g for 30 min. The supernatants were subjected to Ni-NTA affinity chromatography using a column pre-equilibrated with buffer (Qiagen, Chatsworth, CA) or glutathione-triethyleneglycocyl-sepharose 6B (CoWin Biotech, Beijing, China) according to instructions by manufacturers, respectively. Unbound proteins were removed with wash buffer, and target proteins were eluted with elution buffer. The final protein concentration was determined using a BCA kit (Thermo Fisher Scientific, Waltham, MA, USA).
Pulldown assays
Pulldown experiments were performed as described previously [24]. His-TRBD was incubated with GST-ZFD in lysis buffer (10 mmol Tris-HCl (pH 7.5), 150 mmol NaCl, 1% NP-40, 1 mM EDTA, and complete protease inhibitor cocktail from Roche) at 4 °C overnight. The beads were washed with lysis buffer five times, and the bound proteins were analyzed by western blotting. The GST protein alone incubated with His-TRBD was used as a negative control. Moreover, the groups of the whole TERT and the C-terminal part (360 aa) of TERT in pulldown are also used as control groups. The intensity of each band was measured using ImageJ software.
Co-immunoprecipitation and western blotting analyses
293T cells were transfected with the recombinant plasmids pcDNA3.1-Myc-TRBD/pcDNA3.1-HA-ZFD, pcDNA3.1-Myc-TRBD, and pcDNA3.1-HA-ZFD using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA). The proteins were immunoprecipitated with anti-HA (Sigma-Aldrich, St Louis, MO, USA) or anti-Myc (Sigma-Aldrich) antibodies and protein A beads. The beads were washed three times with 1 ml of IP buffer, SDS sample buffer was added, the samples were boiled to elute the bound proteins, and the eluates were analyzed by WB.
SNAP-tag protein complementation assay in 293T cells
To verify interaction between TRBD and ZFD, TRBD and ZFD were amplified by PCR and inserted into cSNAP and nSNAP, respectively, to generate pTRBD-cSNAP and pZFD-nSNAP plasmids in accordance with previously described methods. The PCR primers for ZFD were forward (5′-CTA GCT AGC TAG ATG AGC ATA GTG ATG CGC AAT A-3′) and reverse (5′-TCG CGA TCG CGA TTA CTT GGC TGT GGC TGC TCC G-3′) and for TRBD were forward (5′-CTA GCT AGC TAG GAC AAG AAA CCC CAA TCT CT-3′) and reverse (5′-CCG CTC GAG CGG CGA TAG ACA AAC GAT AGC CTA C-3′). Combinations of pTRBD-cSNAP and pZFD-nSNAP plasmids were co-transformed into 293T cells in accordance with previously described methods. GFP fluorescence was visualized and imaged by laser scanning confocal microscopy (FluoView FV1000, Olympus, Japan).
Virus-mediated SNAP-tag protein complementation assay in G. duodenalis
To verify the PPI location in G. duodenalis, the TRBD-cSNAP and ZFD-nSNAP fragments were inserted into the pC631 viral vector. GFP fluorescence was visualized and imaged by laser scanning confocal microscopy (FluoView).
Structure of the hammerhead ribozyme
Using RNA structure software, the full-length Giardia lamblia ATCC 50803 ZFD (GL50803_20802) was analyzed, and the secondary structure of the mRNA was predicted. According to the position of the GUC sequence and the nucleotide sequences flanking it, a triplet GUC located at position 257 of ZFD mRNA was chosen as the target site for the ribozyme. The designed hammerhead ribozyme consisted of 52 nucleotides, including a 23 nt catalytic core and 29 nt of flanking antisense sequences. The sequence was 5’-GTG TTT GGA AGC GCT GAT GAG TTC CGT GAG GAC GAA ACA AGC TCT TTA TGC T-3’ (the underlined nucleotides are the catalytic core). The primers used for the ribozyme were as follows: forward (5′-CGC GGA TCC GCG GTG TTT GGA AGC GCT GAT GAG TTC CGT GAG GAC GAA ACA AGC TCT TTA TGC TTC CCC GCG GGG A-3′ (BamHI); reverse, 5′-TCC CCG CGG GGA AGC ATA AAG AGC TTG TTT CGT CCT CAC GGA ACT CAT CAG CGC TTC CAA ACA C-3′ (SacII). After amplification by PCR, the double-stranded DNA was inserted into the pC631-SNAP vector to construct pC631-SNAP-Ham-ZFD. DH5α was transformed with the plasmid and plated on LB solid medium containing ampicillin. Positive colonies were sequenced by Sangon Biotech (Sangon Biotech, Shanghai, China).
In vitro transcription
To construct pC631-SNAP-ZFD, ZFD was amplified by PCR: forward (5′-CGC GGA TCC GCG ATG AGC ATA GTG ATG CGC AAT A-3′) (BamHI); reverse (5′-TCC CCG CGG GGA TTA CTT GGC TGT GGC TGC TCC G-3′) (SacII), and the product was inserted into the pc631-SNAP vector. Both pc631-SNAP-ZFD and pC631-SNAP-Ham-ZFD were linearized by NruI at the 3’-end. The two transcripts were each synthesized in vitro with RNA Production System (Promega, Madison, WI, USA).
In vitro ribozyme cleavage of ZFD mRNA
For the in vitro ribozyme cleavage assay, pC631-SNAP-ZFD and pC631-SNAP-Ham-ZFD were mixed at a 1:5 molar ratio in Tris-HCl (50 mM, pH 8.0). The mixture was vortexed and maintained at 95 °C for 3 min, centrifuged briefly at room temperature and incubated at 37 °C for another 5 min. Finally, MgCl2 was added to start the cleavage reaction (final concentration, 10 mM), which was catalyzed at 37 °C for 3 h. The reaction product was then analyzed by RT-PCR.
Electrotransformation
For the in vivo ribozyme cleavage assay, trophozoites of virus-free G. duodenalis mixed with pC631-SNAP-Ham-ZFD were electroporated at 2.5 kV, 25 mF, 400 fQ, and a 0.2 time constant (three times) using a 0.1-cm electrode gap cuvette. The cells were incubated for an additional 15 min on ice after electroporation. Control samples were treated similarly but without electroporation. Negative control samples were electroporated with pC631-SNAP vectors.
Telomerase assay
The TRAP assay was performed as follows. First, cell extracts were added to the telomerase extension reactions and incubated for 20 min at 37 °C. PCR was performed using the following primers: forward (5′-AAT CCG TCG AGC AGA GTT-3′); reverse (5′-CAA CAT CTC CAC TAC CTT CCT ACC CTA C-3′). As an internal telomerase assay standard, internal primers were added to the PCR mixture as described previously. The telomerase products were resolved by 12% nondenaturing polyacrylamide gel electrophoresis. Bands were detected for densitometry analysis using BandScan software.
qPCR test for telomere length measurement
qPCR was employed to measure TL using the method described by Cawthon. Briefly, TL primers (forward, 5′-GCT TTA GCC CAG CCC AGC CCA-3′; reverse, 5′-CTT TCG AGG AGG GGA GGG GA-3′) and a single gene primer (GenBank: L29032.1) (forward, 5′-TTA CCA CGA GCG CAG AGT TT-3′; reverse, 5′-TAG GCG AGA CCC CAC TTG TA-3′) were used. All qPCR assays were conducted using a Rotor-Gene Q (Qiagen) real-time PCR system with the following cycling parameters: 5 min at 95 °C, followed by 30 cycles of 7 s at 98 °C and 10 s at 58 °C. The TL for each sample was determined using the telomere to single-copy gene ratio (T/S ratio) by calculating the ΔCt [Ct(telomere)/Ct(single gene)]. The T/S ratio for each sample (x) was normalized to the mean T/S ratio of the reference sample [2−(ΔCtx−ΔCtr) = 2−ΔΔCt], which was also applied for the standard curve, both as a reference sample and as a validation sample.
Experimental design and statistical analysis
The G. duodenalis wildtype strain, yeast strains Y187, AH109 and 293T cell line are stored at Jilin University in China. At least three independent biological replicates of each treatment were carried out for statistical analyses. P-values were determined by one-way and two-tailed ANOVAs. The results are expressed as the mean ± standard error, SE. Asterisks indicate statistically significant correlations at the 95 % confidence level.
Results
ZFD is a novel TRBD-interacting protein
To identify proteins involved in regulating the TRBD, we screened a G. duodenalis cDNA library using the TRBD of G. duodenalis TERT (808–1464 bp) as bait in a yeast two-hybrid system. Three interacting clones were identified from 1.2 × 107 transfectants. NCBI BLAST software showed that the clone sequence was identical to that of Giardia lamblia ATCC 50803 ZFD (GL50803_20802) (Additional file 1: Figure S1a, n = 3). Direct two-hybrid binding assay results confirmed the specificity of the interaction. In addition, an α-galactosidase assay indicated that TRBD did not activate the reporter gene, suggesting a specific interaction between TRBD and ZFD (Additional file 1: Figure S1b). pGBKT7-TRBD plasmid toxicity was also tested (Additional file 1: Figure S1c), and the results showed no significant difference between the empty vector and pGBKT7-TRBD.
To demonstrate interaction between the TRBD and ZFD in vitro, TRBD, TERT, C-terminal part of TERT and ZFD were amplified and cloned into pET-32a and pGEX-4T-1 to construct pET-32a-TRBD, pET-32a-TERT, pET-32a-C-terminal part of TERT and pGEX-4T-1-ZFD expression plasmids, respectively. His-TRBD, His-TERT, His-C-terminal part of TERT and GST-ZFD proteins were purified from E. coli (Additional file 2: Figure S2), pulled down and analyzed by western blot (WB) assays with anti-His or anti-GST antibodies. As shown in Fig. 1a–c, the WB analysis indicated that His-TRBD bound directly to GST-ZFD in vitro but not to GST alone, which was consistent with the results of the two-hybrid analysis. To further confirm the interaction, co-IP was performed in 293T cells transfected with Myc-TRBD and/or HA-ZFD. When the cell lysates were immunoprecipitated with the anti-HA or anti-Myc antibody, co-precipitation was detected only with the Myc-TRBD/HA-ZFD group and not in the single plasmid or control groups. Moreover, the TRBD-ZFD expression level increased from 10 h to 40 h (Fig. 1d, e). These results suggest that the TRBD directly interacts with ZFD, which is consistent with the results of the pulldown assays. These findings indicate that ZFD is a novel binding protein of TRBD.

A pulldown assay and co-immunoprecipitation validate the interaction between TRBD and ZFD. a Binding of His-TRBD to GST-ZFD immobilized on glutathione beads was determined by WB analysis. Total protein was probed with an anti-His or anti-GST antibody as indicated. b Binding of His- C-terminal part of TERT to GST-ZFD immobilized on glutathione beads was determined by WB analysis. Total protein was probed with an anti-His or anti-GST antibody as indicated. c Binding of His-TERT to GST-ZFD immobilized on glutathione beads was determined by WB analysis. Total protein was probed with an anti-His or anti-GST antibody as indicated. d Interaction between TRBD and ZFD in 293T cells. Immunoprecipitation was detected by WB with anti-HA and anti-Myc antibodies. e TRBD-ZFD protein was detected at 10 h, 20 h, and 40 h. *P < 0.05
Interaction between the TRBD and ZFD in living cells was also investigated using the SNAP-tag protein complementation assay, which enables visualization of protein complex formation and the location of protein interactions in living cells. The TRBD was fused to cSNAP (pTRBD-cSNAP), and ZFD was fused to nSNAP (pZFD-nSNAP). Co-expression of pTRBD-cSNAP and pZFD-nSNAP bimolecular fluorescence complementation (BiFC) vectors in 293T cells resulted in fluorescence complementation, which was observed as green dots predominantly in the nucleus; in contrast, no fluorescence was detected in reactions containing pTRBD-cSNAP or pZFD-nSNAP alone. Fluorescence complementation between NLS-FRB and FKBP12 fused to cSNAP (pcDNA-NLS-FRB-cSNAP) and nSNAP (pcDNA-nSNAP-FKBP12), respectively, was used as a positive control (Fig. 2a, b). Subsequently, TRBD-cSNAP and ZFD-nSNAP fragments were inserted into the Giardia viral vector pC631 to form pC631-TRBD-cSNAP and pC631-ZFD-nSNAP, respectively. Fluorescence complementation between Jun and Fos fused to cSNAP (pC631-Jun-cSNAP) and nSNAP (pC631-nSNAP-Fos), respectively, was used as the positive control. The results showed that PPIs occurred in both nuclei of G. duodenalis (Fig. 2c–e).

Interactions between the TRBD and ZFD in cells. Interactions between the TRBD and ZFD in 293T cells and G. duodenalis. a Co-infection of 293T cells with pcDNA-NLS-FRB-cSNAP and pcDNA-nSNAP-FKBP12. b Co-infection of 293T cells with pTRBD-cSNAP and pZFD-nSNAP. c Co-infection of G. duodenalis with pTRBD-cSNAP and pZFD-nSNAP. d Co-infection of G. duodenalis with pC631-Jun-cSNAP and pC631-Fos-nSNAP. e Negative control. Scale-bars: 5 µm
Reduction in ZFD inhibits the proliferation of G. duodenalis
A hammerhead ribozyme targeting ZFD was designed to knock down ZFD expression in cells. Using a computer-predicted RNA secondary structure, we selected the triplex GUC of the TERT mRNA as the cleavage site and then constructed the hammerhead ribozyme (Ham-ZFD), which included a ZFD antisense sequence and catalytic core. Ham-ZFD was inserted into the viral expression vector pC631-SNAP, which contains the 631 nt 5′-end and the 2022 nt 3′-end of the Giardia viral genome cDNA and T7 promoter. After transcription in vitro, the RNA concentrations of pC631-SNAP-Ham-ZFD, pC631-SNAP-ZFD, pC631-SNAP-Ham-GDH and pC631-SNAP-ZFD-Sub were 11.2 μg/µl, 11.5 μg/µl, 10.8 μg/µl and 10.7 μg/µl, respectively.
pC631-SNAP-ZFD was mixed with pC631-SNAP-Ham-ZFD, pC631-SNAP-Ham-GDH and pC631-SNAP-ZFD-Sub, and the remainder of the pC631-SNAP-ZFD RNA was reverse transcribed into cDNA and analyzed by real-time PCR. The results confirmed 84% knockdown of ZFD, whereas less than 3% of ZFD was cleaved in the negative control (Ham-GDH) and Sub-ZFD (only ZFD antisense sequence) groups (Fig. 3a, Additional file 3: Figure S3).

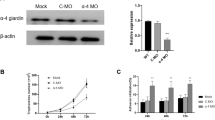
The effect of Ham-ZFD in G. duodenalis cell lines. a Cleavage activity of the hammerhead ribozyme in vitro. *P < 0.05. b Trophozoite count. Control: trophozoites without electroporation; pC631-SNAP: trophozoites electroporated with a plasmid without the ribozyme; pC631-SNAP-Ham-GDH: trophozoites electroporated with a plasmid harboring the Ham-GDH ribozyme; pC631-SNAP-Ham-ZFD: trophozoites electroporated with a plasmid harboring the Ham-ZFD ribozyme. *P < 0.05. c Relative Ham-ZFD mRNA levels in Ham-ZFD-transfected G. duodenalis cell lines. d Relative ZFD mRNA levels in Ham-ZFD-transfected G. duodenalis cell lines. *P < 0.05
To confirm the results in vivo, we knocked down ZFD expression in G. duodenalis by transfecting ZFD-targeting ribozymes. Because pC631-SNAP-Ham-ZFD contained Giardia viral genome cDNA, green fluorescence was visible in the whole trophozoite. The Ham-ZFD proliferation rate was similar to that of wildtype cells in the first two days, but the cell density on the 5th day was only approximately 41% of that of wildtype cells (Fig. 3b). Moreover, the presence of Ham-ZFD and decrease in ZFD were monitored by RT-PCR (Fig. 3c, d). These data suggest that electroporation with Ham-ZFD in G. duodenalis may reduce cell growth.
Reduction in ZFD decreases telomere length in G. duodenalis
qPCR analysis of G. duodenalis was employed to measure telomere length (TL) as previously described with the formula \( \left[ {{{ 2^{{{\text{C}}({\text{telomeres}})}}_{\text{t}} } \mathord{\left/ {\vphantom {{ 2^{{{\text{C}}({\text{telomeres}})}}_{\text{t}} } { 2^{{C({\text{single}})}}_{\text{t}} }}} \right. \kern-0pt} { 2^{{C({\text{single}})}}_{\text{t}} }}} \right]^{ - 1} = 2^{{{-}\Delta C}}_{\text{t}} \). The actin gene (GenBank: L29032.1) was used as a single-copy gene control. The relative telomere to single-copy gene ratio (T/S) ratio (T/S of one sample relative to the T/S of another sample) was calculated using the formula \( 2^{{{-}(\Delta C 2}}_{\text{t}} -^{{{-}\Delta C 1)}}_{\text{t}} = 2^{{{-}\Delta \Delta C}}_{\text{t}} \). To verify the effect of the hammerhead ribozyme, the TLs in the Ham-ZDF, ZFD-Sub and Ham-GDH were analyzed. The TL in the Ham-ZFD group was 0.91 ± 0.07 relative to that in the control group TL after 20 h. The TLs of the Sub-ZFD and Ham-GDH groups were 0.97 ± 0.05 and 0.96 ± 0.02 relative to the control group TL, respectively (Fig. 4a). These results suggest that the hammerhead ribozyme does not affect the TL and ZFD may be partly involved in TL control.

TLs and Telomerase activity in ribozyme Ham-ZFD-transfected G. duodenalis cell lines. a TLs in ribozyme Ham-ZFD-transfected G. duodenalis cell lines. Control: non-transfected G. duodenalis cell lines; ZFD-Sub: G. duodenalis with the pC631-SNAP-ZFD-Sub plasmid. Ham-GDH: G. duodenalis with the pC631-SNAP-Ham-GDH plasmid; Ham-ZFD: G. duodenalis with the pC631-SNAP-Ham-ZFD plasmid; *P < 0.05. b Telomerase activity in G. duodenalis cell lines. M: DL500 DNA marker; 1: TRAP products from HeLa cells; 2: TRAP products from untreated G. duodenalis cells; 3: TRAP products from G. duodenalis cells with pC631-SNAP; 4: TRAP products from G. duodenalis cells with pC631-SNAP-Ham-GDH; 5: TRAP products from G. duodenalis cells with pC631-SNAP-Ham-ZFD. c Telomerase activity in G. duodenalis cell lines. *P < 0.05. Control: G. duodenalis without pC631-SNAP plasmid; SNAP: G. duodenalis with pC631-SNAP plasmid; Ham-GDH: G. duodenalis with pC631-SNAP-Ham-GDH plasmid; Ham-ZFD: G. duodenalis with pC631-SNAP-Ham-ZFD plasmid; *P < 0.05
Reduction in ZFD decreases telomerase activity in G. duodenalis
A TRAP assay was performed to reveal whether ZFD affects telomerase activity in G. duodenalis. A ladder of typical TRAP products was observed in all of the groups, with the Ham-GDH and Sub-ZFD groups not differing from the control group. However, loss of telomerase activity was observed in the Ham-ZFD group (Fig. 4b, c), indicating that the decrease in ZFD reduced telomerase activity in G. duodenalis. In addition, telomerase activity in the Ham-ZFD group was only approximately 62% of that in the control group.
Discussion
Since Muller [25] discovered special sequences at the end of chromosomes and named them telomeres, telomeres, telomerase, and telomerase-related proteins have become hot research topics [26]. Telomere assembly, activation and attrition are complex processes driven by telomerase that show similarities and differences in various organisms [27]. Studying telomerase is helpful for understanding the mechanisms involved and providing research directions for cell life-cycle control. Telomerase complexes generally contain TERT, TER, and proteins that bind to RNA or are involved in the stabilization and maturation of telomerase complexes.
The TERT component was first identified in Euplotes aediculatus and later in numerous eukaryotes, including humans, mice, Saccharomyces cerevisiae, Oxytricha trifallax, Leishmania amazonensis and Trichinella spiralis [28,29,30,31]. Regarding parasites, the gene encoding TERT (2385 nt) was detected from Plasmodium falciparum in 2005, and remarkable sequence diversity has been observed in different Plasmodium species [18, 32]. The TERT gene sequence of Leishmania major was used to clone the same gene from other parasite species identified by Miriam [33] (L. amazonensis, L. braziliensis and L. donovani). Miriam [33] also showed that the TERT catalytic subunit is a universally conserved feature of telomerases in these species. In 2004, Xu et al. [34] reported the TERT gene in C. parvum (4407 nt) but did not conduct relevant mechanistic research.
TERT regulation at the transcriptional level is considered the main mechanism affecting telomerase activity, but some studies have noted that PPIs and protein-level regulation also control telomerase [35, 36]. PPIs are important for various life processes, such as signal transduction, gene expression regulation, energy and material metabolism, and cell cycle regulation [37]. Studying PPIs in biological systems is key for understanding protein mechanisms, responses to biological signals and energy metabolism under special physiological conditions such as disease, and functional links between proteins [38].
Because telomerase characteristics differ among species, the function of telomeric DNA-binding proteins in telomerase and telomere regulation also differ. In ciliates, yeasts and vertebrates, telomerase-related proteins have been shown to play important roles in regulating telomerase assembly, posttranslational modification, localization, and enzyme function [39, 40]. The protein p80 in mammalian cells is related to telomerase activity in vivo [41]. MKRN1 is a well-known telomerase modifier in the ubiquitin-proteasome pathway, and it promotes hTERT degradation, leading to a reduction in telomerase activity and TL [42]. The hTERT protein level reportedly increases significantly after gene silencing of PCDH10, indicating that PCDH10 likely affects hTERT protein stability via PPIs and thus regulates telomerase activity [43]. In yeast, Ku binds specifically to the TER neck-ring and promotes telomerase aggregation at telomere ends [44]. In parasite research, extraction of telomeres from T. thermophila has revealed p80 and p95 to be as telomerase-related proteins, and the data suggest that p80 and p95 have good affinity for telomerase [45]. Moreover, Zhao et al. [24] found that TERT-related proteins in Eimeria tenella can interact with 14-3-3 proteins and negatively regulate telomerase activity. These results suggest that TERT may play an important role in regulating telomerase activation via PPIs. However, research into the relevant mechanism is lacking.
Giardia duodenalis is a model organism that presents immortal proliferation mainly due to telomerase complex-induced regulation. Because the molecular biological characteristics of Giardia are poorly understood, few ideal vaccines can be used in the clinic [15,16,17,18,19]. Although GlTERT (2883 nt), a repeat sequence (TAGGG)n and a retrotransposable element in the G. duodenalis genome were identified in 2000, only one study on G. duodenalis telomerase has been conducted to date [46]. To investigate TERT/telomerase-related proteins in G. duodenalis, we first constructed a cDNA library for G. duodenalis and screened it using the yeast double-hybrid method to identify a related zinc-finger protein, ZFD (GL50803-20802), interacting with the TRBD. The screening results were verified using co-IP and pulldown experiments.
Zinc fingers proteins were first identified in a study of transcription in the African clawed frog Xenopus laevis [37], and they are now known to be widely distributed in animals, plants, and microorganisms [38, 47, 48]. Indeed, nearly 1% of the human genome may encode proteins containing zinc-finger structures [49]. Many zinc-finger proteins are transcription factors that play an important role in gene regulation. ZFD-containing proteins are involved in regulating cell replication, repair, transcription, translation, metabolism, signal conduction, cell proliferation and apoptosis through specific binding to target molecular DNA, RNA, and proteins [50, 51]. Thus far, the study of the zinc finger proteins of G. duodenalis has primarily focused on variant-specific surface proteins (VSPs), and an oral vaccine against G. duodenalis was designed to target VSPs [52]. However, the protein sequence obtained in this study does not belong to the VSP family because no CRGKA motif exists at the C-terminus. Notably, the protein sequences obtained in G. duodenalis are less than 6% homologous to those of other species, suggesting that ZFD may have a special role in G. duodenalis.
The most obvious feature of G. duodenalis is the two morphologically identical nuclei, though whether these two nuclei are indeed identical is unknown, as are the locations of the TRBD and telomerase-interacting proteins. Protein-fragment complementation assays (PCAs) were applied to determine the site of interaction between ZFD and the TRBD in both mammalian cells and G. duodenalis, and BiFC PCAs have been widely used to observe PPIs in living cells [53,54,55]. This assay was first described by Regan, who used two proteins of interest fused to the N-terminal or C-terminal non-fluorescent fragment of a fluorescent protein; the proteins were then co-expressed in cells [56]. Using fluorescence microscopy, an interaction occurring between the two proteins of interest is visualized by a fluorescent signal. To date, YFP and GFP have been used in BiFC assays in yeast, plant and mammalian cells and in drug discovery related to a specific PPI [57,58,59]. However, few reports have used the BiFC assay in G. duodenalis because GFP must be oxygenated for maturation and to become fluorescent. To solve this problem, we used the SNAP-tag protein complementation assay to visualize PPIs in living cells, as previously reported [58, 60].
We first utilized the split SNAP-tag labeling method and fused ZFD and TRBD to cSNAP and nSNAP, respectively and then inserted them into a mammalian plasmid. The results suggested that the interaction between ZFD and TRBD occurs in the nucleus of 293T cells. ZFD and TRBD were also fused to cSNAP and nSNAP, respectively, and inserted into the Giardia viral vector pC631, and the results confirmed that PPIs consistently occurred in both nuclei. Construction of a new virus-mediated split SNAP-tag labeling plasmid offers a new tool for imaging PPI events in G. duodenalis and may provide a new method of discovering PPIs in other anaerobic organisms via virus-mediated plasmids.
TERT variants influence TL and telomerase activity, though the correlation between TL and telomerase activity is unclear. Some studies have shown a lack of correlation between TL and telomerase activity and expression in leukemic cells [56]. Others have shown that the mean telomere length (TRF) in peripheral blood leukocytes correlates negatively with telomerase activity in aplastic anemia patients [60]. Moreover, modified TERT transiently enhances telomerase activity and rapidly extends telomeres [61]. To determine the relationship between ZFD and TL and telomerase activity in G. duodenalis, we constructed a ZFD ribozyme and found that a reduction in ZFD mRNA led to a decreased reproduction rate and a lower number of G. duodenalis. Telomerase activity and TL measurements revealed that reducing the ZFD mRNA level can decrease telomerase activity and shorten the TL. Because the mechanism by which ZFD regulates TL and telomerase activity remains not well understood, we will conduct further systematic studies of ZFD with regard to cell activity and other aspects of telomeres in future research.
Conclusions
In this study, we used the yeast double-hybrid assay to identify a new type of ZFD that interacts with the TRBD and examined the interaction using co-IP and pulldown assays. We then used split SNAP-tag labeling to determine the site of the PPI in 293T cells. To ensure that split SNAP-tag labeling can be used in G. duodenalis, a new type of virus-mediated split SNAP-tag labeling plasmid was constructed. Our results showed that PPIs consistently occurred in both G. duodenalis nuclei. Moreover, we constructed a ZFD ribozyme and found that decreases in ZFD mRNA reduced the reproduction rate, the number of G. duodenalis and telomerase activity and shortened the TL.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- CTE:
-
c-terminal extension domain
- ER:
-
endoplasmic reticulum
- PPI:
-
protein–protein interaction
- PVs:
-
peripheral vesicles
- RT:
-
reverse transcriptase domain
- TER:
-
telomerase RNA
- TERT:
-
telomerase reverse transcriptase
- TRBD:
-
telomerase RNA-binding domain
- ZFD:
-
zinc finger domain
References
Lee SHC, Jefferies R, Watt P, Hopkins R, Sotzik F, Reid S, et al. In vitro analysis of the TAT protein transduction domain as a drug delivery vehicle in protozoan parasites. Exp Parasitol. 2008;118:303–7.
Tangtrongsup S, Scorza V. Update on the diagnosis and management of Giardia spp. infections in dogs and cats. Top Companion Anim Med. 2010;25:155–62.
Vidal F, Vidal JC, Gadelha AP, Lopes CS, Coelho MG, Monteiro-Leal LH. Giardia lamblia: the effects of extracts and fractions from Mentha x piperita Lin. (Lamiaceae) on trophozoites. Exp Parasitol. 2007;115:25–31.
Ducray C, Pommier JP, Martins L, Boussin FD, Sabatier L. Telomere dynamics, end-to-end fusions and telomerase activation during the human fibroblast immortalization process. Oncogene. 1999;18:4211–23.
Lan J, Zhu Y, Xu L, Yu H, Yu J, Liu X, et al. The 68-kDa telomeric repeat binding factor 1 (TRF1)-associated protein (TAP68) interacts with and recruits TRF1 to the spindle pole during mitosis. J Biol Chem. 2014;289:14145–56.
Lin KW, McDonald KR, Guise AJ, Chan A, Cristea IM, Zakian VA. Proteomics of yeast telomerase identified Cdc48-Npl4-Ufd1 and Ufd4 as regulators of Est1 and telomere length. Nat Commun. 2015;6:8290.
Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun. 2018;9:579.
Knight JS, Cotter MA, Robertson ES. The latency-associated nuclear antigen of Kaposiʼs sarcoma-associated herpesvirus transactivates the telomerase reverse transcriptase promoter. J Biol Chem. 2001;276:22971–8.
Lee JH, Chung IK. Curcumin inhibits nuclear localization of telomerase by dissociating the Hsp90 co-chaperone p23 from hTERT. Cancer Lett. 2010;290:76–86.
Lee JH, Khadka P, Baek SH, Chung IK. CHIP promotes human telomerase reverse transcriptase degradation and negatively regulates telomerase activity. J Biol Chem. 2010;285:42033–45.
Gilley D, Tanaka H, Hande MP, Kurimasa A, Li GC, Oshimura M, et al. DNA-PKcs is critical for telomere capping. Proc Natl Acad Sci USA. 2001;98:15084–8.
Kim H, Li F, He Q, Deng T, Xu J, Jin F, et al. Systematic analysis of human telomeric dysfunction using inducible telosome/shelterin CRISPR/Cas9 knockout cells. Cell Discov. 2017;3:17034.
Renaud S, Loukinov D, Alberti L, Vostrov A, Kwon YW, Bosman FT, et al. BORIS/CTCFL-mediated transcriptional regulation of the hTERT telomerase gene in testicular and ovarian tumor cells. Nucleic Acids Res. 2011;39:862–73.
Varela E, Munoz-Lorente MA, Tejera AM, Ortega S, Blasco MA. Generation of mice with longer and better preserved telomeres in the absence of genetic manipulations. Nat Commun. 2016;7:11739.
Harkisheimer M, Mason M, Shuvaeva E, Skordalakes E. A motif in the vertebrate telomerase N-terminal linker of TERT contributes to RNA binding and telomerase activity and processivity. Structure. 2013;21:1870–8.
Marchesini M, Matocci R, Tasselli L, Cambiaghi V, Orleth A, Furia L, et al. PML is required for telomere stability in non-neoplastic human cells. Oncogene. 2016;35:1811–21.
Sekulovic S, Gylfadottir V, Vulto I, Gasparetto M, Even Y, Brookes C, et al. Prolonged self-renewal activity unmasks telomerase control of telomere homeostasis and function of mouse hematopoietic stem cells. Blood. 2011;118:1766–73.
Uzlikova M, Fulneckova J, Weisz F, Sykorova E, Nohynkova E, Tumova P. Characterization of telomeres and telomerase from the single-celled eukaryote Giardia intestinalis. Mol Biochem Parasitol. 2017;211:31–8.
Yoo JE, Park YN, Oh BK. PinX1, a telomere repeat-binding factor 1 (TRF1)-interacting protein, maintains telomere integrity by modulating TRF1 homeostasis, the process in which human telomerase reverse transcriptase (hTERT) plays dual roles. J Biol Chem. 2014;289:6886–98.
Feng Y, Xiao L. Zoonotic potential and molecular epidemiology of Giardia species and giardiasis. Clin Microbiol Rev. 2011;24:110–40.
Kraft MR, Klotz C, Bucker R, Schulzke JD, Aebischer T. Giardiaʼs epithelial cell interaction in vitro: mimicking asymptomatic infection? Front Cell Infect Microbiol. 2017;7:421.
Maʼayeh SY, Liu J, Peirasmaki D, Hornaeus K, Lind SB, Grabherr M, et al. Characterization of the Giardia intestinalis secretome during interaction with human intestinal epithelial cells: the impact on host cells. PLoS Negl Trop Dis. 2017;11:e0006120.
Tian HF, Chen B, Wen JF. Giardiasis, drug resistance, and new target discovery. Infect Disord Drug Targets. 2010;10:295–302.
Zhao N, Gong P, Cheng B, Li J, Yang Z, Li H, et al. Eimeria tenella: 14-3-3 protein interacts with telomerase. Parasitol Res. 2014;113:3885–9.
Muller HJ. The remaking of chromosomes. Collect Net Woods Hole. 1938;13:181–98.
Liu C-W, Zhang X-C, Li J-H, Gong P-T, Chen L-F. Clone and sequence analysis of telomerase reverse transcriptase gene from Giardia canis. J Pathog Biol. 2006;6:424–7.
Fernandez MF, Castellari RR, Conte FF, Gozzo FC, Sabino AA, Pinheiro H, et al. Identification of three proteins that associate in vitro with the Leishmania (Leishmania) amazonensis G-rich telomeric strand. Eur J Biochem. 2004;271:3050–63.
Figueiredo LM, Freitas-Junior LH, Bottius E, Olivo-Marin JC, Scherf A. A central role for Plasmodium falciparum subtelomeric regions in spatial positioning and telomere length regulation. EMBO J. 2002;21:815–24.
Hovel-Miner GA, Boothroyd CE, Mugnier M, Dreesen O, Cross GA, Papavasiliou FN. Telomere length affects the frequency and mechanism of antigenic variation in Trypanosoma brucei. PLoS Pathog. 2012;8:e1002900.
Khalfallah O, Ravassard P, Lagache CS, Fligny C, Serre A, Bayard E, et al. Zinc finger protein 191 (ZNF191/Zfp191) is necessary to maintain neural cells as cycling progenitors. Stem Cells. 2009;27:1643–53.
Liu C-W, Zhang X-C, Li J-H, Liu H, Gong P-T, Zhang G-C. Southern blotting analysis of telomeric repetitive DNA sequences in Giardia canis. J Pathog Biol. 2008;35:183–5.
Krebs CJ, Zhang D, Yin L, Robins DM. The KRAB zinc finger protein RSL1 modulates sex-biased gene expression in liver and adipose tissue to maintain metabolic homeostasis. Mol Cell Biol. 2014;34:221–32.
Giardini MA, Lira CBB, Fábio F, Conte FF, et al. The putative telomerase reverse transcriptase component of Leishmania amazonensis: gene cloning and characterization. Parasitol Res. 2006;9:447–54.
Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, et al. The genome of Cryptosporidium hominis. Nature. 2004;431:1107–12.
Sandhu R, Sanford S, Basu S, Park M, Pandya UM, Li B, et al. A trans-spliced telomerase RNA dictates telomere synthesis in Trypanosoma brucei. Cell Res. 2013;23:537–51.
Yang X, Figueiredo LM, Espinal A, Okubo E, Li B. RAP1 is essential for silencing telomeric variant surface glycoprotein genes in Trypanosoma brucei. Cell. 2009;137:99–109.
Qiao A, Zhao Z, Zhang H, Sun Z, Cui X. Gene expression profiling reveals genes and transcription factors associated with dilated and ischemic cardiomyopathies. Pathol Res Pract. 2017;213:548–57.
Cavga AD, Karahan N, Keskin O, Gursoy A. Taming oncogenic signaling at protein interfaces: challenges and opportunities. Curr Top Med Chem. 2015;15:2005–18.
Liu Y, Snow BE, Hande MP, Baerlocher G, Kickhoefer VA, Yeung D, et al. Telomerase-associated protein TEP1 is not essential for telomerase activity or telomere length maintenance in vivo. Mol Cell Biol. 2000;20:8178–84.
Mohanty P, Gupta A, Bhatnagar S. Modeling of Plasmodium falciparum telomerase reverse transcriptase ternary complex: repurposing of nucleoside analog inhibitors. Assay Drug Dev Technol. 2015;13:628–37.
Pandya UM, Sandhu R, Li B. Silencing subtelomeric VSGs by Trypanosoma brucei RAP1 at the insect stage involves chromatin structure changes. Nucleic Acids Res. 2013;41:7673–82.
Kim JH, Park SM, Kang MR, Oh SY, Lee TH, Muller MT, et al. Ubiquitin ligase MKRN1 modulates telomere length homeostasis through a proteolysis of hTERT. Genes Dev. 2005;19:776–81.
Zhou LN, Hua X, Deng WQ, Wu QN, Mei H, Chen B. PCDH10 interacts with hTERT and negatively regulates telomerase activity. Medicine (Baltimore). 2015;94:e2230.
Chan SW, Blackburn EH. New ways not to make ends meet: telomerase, DNA damage proteins and heterochromatin. Oncogene. 2002;21:553–63.
Miller MC, Collins K. The Tetrahymena p80/p95 complex is required for proper telomere length maintenance and micronuclear genome stability. Mol Cell. 2000;6:827–37.
Serradell MC, Saura A, Rupil LL, Gargantini PR, Faya MI, Furlan PJ, et al. Vaccination of domestic animals with a novel oral vaccine prevents Giardia infections, alleviates signs of giardiasis and reduces transmission to humans. NPJ Vaccines. 2016;1:16018.
He P, Wang X, Zhang X, Jiang Y, Tian W, Zhang X, et al. Short and narrow flag leaf1, a GATA zinc finger domain-containing protein, regulates flag leaf size in rice (Oryza sativa). BMC Plant Biol. 2018;18:273.
Liu Z, Zhang F, Zhao X, Bai F. Effects of zinc-finger proteins and artificial zinc-finger proteins on microbial metabolisms—a review. Sheng Wu Gong Cheng Xue Bao. 2014;30:331–40.
Hoovers JM, Mannens M, John R, Bliek J, van Heyningen V, Porteous DJ, et al. High-resolution localization of 69 potential human zinc finger protein genes: a number are clustered. Genomics. 1992;12:254–63.
Casoni F, Croci L, Bosone C, DʼAmbrosio R, Badaloni A, Gaudesi D, et al. Zfp423/ZNF423 regulates cell cycle progression, the mode of cell division and the DNA-damage response in Purkinje neuron progenitors. Development. 2017;144:3686–97.
Striedner Y, Schwarz T, Welte T, Futschik A, Rant U, Tiemann-Boege I. The long zinc finger domain of PRDM9 forms a highly stable and long-lived complex with its DNA recognition sequence. Chrom Res. 2017;25:155–72.
Yu LZ, Birky CW Jr, Adam RD. The two nuclei of Giardia each have complete copies of the genome and are partitioned equationally at cytokinesis. Eukaryot Cell. 2002;1:191–9.
Nicolle O, Rouillon A, Guyodo H, Tamanai-Shacoori Z, Chandad F, Meuric V, et al. Development of SNAP-tag-mediated live cell labeling as an alternative to GFP in Porphyromonas gingivalis. FEMS Immunol Med Microbiol. 2010;59:357–63.
Regoes A, Hehl AB. SNAP-tag mediated live cell labeling as an alternative to GFP in anaerobic organisms. Biotechniques. 2005;39(809–10):812.
Sagolla MS, Dawson SC, Mancuso JJ, Cande WZ. Three-dimensional analysis of mitosis and cytokinesis in the binucleate parasite Giardia intestinalis. J Cell Sci. 2006;119:4889–900.
Mie M, Naoki T, Uchida K, Kobatake E. Development of a split SNAP-tag protein complementation assay for visualization of protein-protein interactions in living cells. Analyst. 2012;137:4760–5.
Chen M, Li W, Zhang Z, Liu S, Zhang X, Zhang XE, et al. Novel near-infrared BiFC systems from a bacterial phytochrome for imaging protein interactions and drug evaluation under physiological conditions. Biomaterials. 2015;48:97–107.
Miller KE, Kim Y, Huh WK, Park HO. Bimolecular fluorescence complementation (BiFC) analysis: advances and recent applications for genome–wide interaction studies. J Mol Biol. 2015;427:2039–55.
Weber-Boyvat M, Li S, Skarp KP, Olkkonen VM, Yan D, Jantti J. Bimolecular fluorescence complementation (BiFC) technique in yeast Saccharomyces cerevisiae and mammalian cells. Methods Mol Biol. 2015;1270:277–88.
Mie M, Naoki T, Kobatake E. Development of a split SNAP-CLIP double labeling system for tracking proteins following dissociation from protein–protein complexes in living cells. Anal Chem. 2016;88:8166–71.
Ramunas J, Yakubov E, Brady JJ, Corbel SY, Holbrook C, Brandt M, et al. Transient delivery of modified mRNA encoding TERT rapidly extends telomeres in human cells. FASEB J. 2015;29:1930–9.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation (NSFC) of China (grant numbers 31672288, 31772732 and 31272550) and a grant from Jilin Provincial Science and Technology Department (No. 20190103075JH).
Author information
Authors and Affiliations
Contributions
JTZ and NZ performed most of the laboratory work and data analyses and helped draft the manuscript. YHY, PTG, XHL and NW performed some of the laboratory work. CW, XCW and XL analyzed and interpreted the raw data. XCZ and JHL conceived and coordinated the study and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Yeast screening results for the mating reaction between pGBKT7-TRBD-transformed Y187 and AH109. a Screening results for Y187 and AH109. Positive control: yeast screening positive control; negative control: yeast screening negative control; ZFD: zinc finger domain. b Autoactivation detection of pGBKT7-TRBD. Positive control: positive control plasmid pCL; bait plasmid: bait plasmid pGBKT7-TRBD. c Toxicity test for pGBKT7-TRBD. Positive control: empty vector; TRBD plasmid: pGBKT7-TRBD.
Additional file 2: Figure S2.
The expression of recombinant proteins on a protein gel. a The SDS-PAGE analysis of purified C-terminal part protein. b The SDS-PAGE analysis of purified His-TRBD protein. c The SDS-PAGE analysis of purified GST-ZFD protein. d The SDS-PAGE analysis of purified whole TERT protein.
Additional file 3: Figure S3.
Cleavage activity of the hammerhead ribozyme in vitro. a Standard curve for pC631-SNAP-Ham-ZFD. b Melting curve for pC631-SNAP-Ham-ZFD.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zheng, JT., Zhang, N., Yu, YH. et al. Identification of a TRBD zinc finger-interacting protein in Giardia duodenalis and its regulation of telomerase. Parasites Vectors 12, 568 (2019). https://doi.org/10.1186/s13071-019-3821-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3821-0