
Abstract
Background
Some researchers have reported that Toxoplasma gondii can cause serious reproductive impairment in male animals. Specifically, T. gondii destroy the quality of sperm in the epididymis, which affects their sexual ability. However, among such studies, none have investigated the male reproductive transcriptome. Therefore, to investigate the relationship between T. gondii and sperm maturation, we infected mice with T. gondii prugniaud (PRU) strain and performed transcriptome sequencing of the epididymis.
Results
Compared with the control group, 431 upregulated and 229 downregulated differentially expressed genes (DEGs) were found (P-value < 0.05, false discovery rate (FDR) < 0.05 and |log2 (fold change)| ≥ 1). According to results of a bioinformatics analysis, Gene Ontology (GO) function is divided into three categories: cellular component, molecular function and biological process. Upon performing GO analysis, we found that some DEGs correlated with an integral part of membrane, protein complex, cell surface, ATP binding, immune system process, signal transduction and metabolic process which are responsible for the epididymal injury. DEGs were mapped to 101 unique KEGG pathways. Pathways such as cytokine-cytokine receptor interaction, glycolysis/gluconeogenesis and apoptosis are closely related to sperm quality. Moreover, Tnfsf10 and spata18 can damage the mitochondria in sperm, which decreases sperm motility and morphology.
Conclusions
We sequenced the reproductive system of male mice chronically infected with T. gondii, which provides a new direction for research into male sterility caused by Toxoplasma infection. This work provides valuable information and a comprehensive database for future studies of the interaction between T. gondii infection and the male reproductive system.
Similar content being viewed by others


Background
Toxoplasma gondii is an intracellular parasite that infects almost all warm-blooded animals [1, 2]. Pregnant women infected with T. gondii can transmit the infection to their fetus through vertical transmission resulting in abortion, fetal abnormalities and death [3]. Additionally, Toxoplasma infection can cause damage to the male reproductive system, such as sexual dysfunction and infertility. The epididymis is an important accessory organ of the male reproductive system, where sperm maturation and storage take place. Sperm maturation in the epididymis is a highly programmed process which may be affected by the epididymis microenvironment. Therefore, studying the characteristics of the epididymis is important for identification of factors affecting sperm maturation. Previous studies have shown that reproductive pipeline damage [4] and hypogonadism [5] are associated with T. gondii infection. Furthermore, a large number of tachyzoites in semen of infertile patients and anti-sperm antibodies (AsAb) were detected in T. gondii infection [6]. Other studies have reported more advanced pathological changes in T. gondii infection cases, such as granular degeneration of vas deferens epithelial cells, luminal stenosis and interstitium infiltration with inflammatory cells [7]. The density and motility rate of sperm in epididymis infected with T. gondii were significantly lower than the control group; however, the rate of sperm deformity was increased [8, 9].
Recently, various -omics technologies, including transcriptomics, proteomics and metabolomics, have been developed [10]. Transcriptomic analysis is one of the most common high-throughput techniques, which identify the types and copy numbers of mRNAs, while the cells are living in a functional state [11]. Genetic studies have shown that mRNA act as a “bridge” for genetic information transmission between DNA and protein. Hence, it is a valuable source to identify the expression of all genes with a specific time and space in the cell.
Previous studies have investigated the association between T. gondii and male infertility using epidemiology, pathology and serology methods and techniques [12], detection of Toxoplasma DNA in semen [13] and observation of the tissue and cell damage [14]. To our knowledge, studies investigating the differentially expressed genes of host and T. gondii by transcriptome sequencing of the male reproductive system are limited. The main objective of this study was to examine differential gene expression by RNA sequencing (RNA-seq) technology in order to identify key genes associated with T. gondii (PRU strain) chronic infection of the epididymis in male Kunming mice.
Methods
Study population and experiment set-up
Thirty specific-pathogen-free eight-week-old Kunming male mice were purchased from the Laboratory Animal Center of Guangdong Province. Fifteen mice were subjected as the experiment groups, and 15 mice were subjected as the control group (to reduce individual differences, we set the epididymal tissue of 5 mice as a biological replicate, the experiment group and the control group were repeated three times). Mice in the experimental group were inoculated with four cysts of T. gondii PRU strain diluted with normal saline to 0.5 ml through the intragastric administration route (we have previously studied the optimal number of PRU strains infections per mice, which tried to minimize the case damage caused by T. gondii infection in mice). Meanwhile, the control groups were given the same amount of normal saline only. The male mice were sacrificed at 35 days post-infection. Under sterile conditions, the epididymides were harvested. Under the microscope, the peripheral adipose tissue and blood vessels of the harvested epididymides were carefully removed. The processed epididymides were subjected to quick freezing by storing them in liquid nitrogen at – 80 °C for subsequent analysis.
Transcriptome sequencing, data analysis and verification
At present, transcriptome sequencing is a mature high-throughput second-generation sequencing method. A brief description of the experimental process is provided in Table 1; detailed methods of transcriptome sequencing, data analysis and verification can be found in Additional file 1: Text S1.
Results
Pre-experiment
Before the histological analysis, we conducted a preliminary study on the sperm quality of T. gondii infection, including the statistics of sperm survival rate and total sperm count. The sperm survival rate was calculated according to the formula: Sperm survival rate = (Total sperm count - Dead sperm count)/Total sperm count × 100%; three experiments were carried out in each group. The results showed that the sperm survival rate of the experimental group (32%) was significantly lower than that of the control group (74.67%).
The sperm count per milliliter semen was calculated according to the formula: Sperm count per milliliter semen = (5 medium square count/80) × 400 × dilution multiple × 10,000. We diluted sperm 10 times for microscopic examination. The experimental data are shown in Additional file 2: Figure S1; there were significant differences in total sperm count between the experimental group and the control group (P = 0.0058). The data showed that T. gondii infection had a negative effect on sperm quality.
Identification of infection in mice
The outward appearance changes of infected mice comprised mainly a stress reaction about a week after infection, which included depression and the fur is not smooth. Thirty days after infection, 20 mice were selected for tail vein blood collection and qPCR was used to verify the infection of mice (T. gondii ROP18 virulence gene was detected and using the β-actin as a reference); the result is shown in Additional file 3: Figure S2. All of the 20 mice were infected. Fifteen mice were randomly selected for dissection and epididymis tissue was quickly removed. Meanwhile, the mouse brain was grinded and diluted with 100 µl saline solution; 5 µl of brain homogenate was taken for microscopic examination and the number of cysts was counted. Cysts from each mouse were counted three times and averaged. Statistical results showed that 5 µl brain homogenate contained three or more cysts. Therefore, we determined that all of the experimental mice were infected with T. gondii.
Quality analysis of RNA
The purity of total RNA was high, and the ratios OD260/OD280 and OD260/OD230 were both higher than 2.0 (Table 2). The RNA integrity number (RIN) was higher than 7, and the ratio of 28S/18S rRNA was higher than 0.7. This indicated that the RNA had good integrity and met the requirements of the subsequent experiments.
Sequencing data preprocessing
The sample library constructed by Illumina Hiseq 2500 sequencing platform. We obtained 321,183,388 raw reads (48.17G). After trimming, we obtained 314,632,244 clean reads (47.21G). Among them, the amounts of sequencing data of each sample were more than 6G. In high throughput sequencing, each measurement of a basic group gives a corresponding value, which is a measure of the accuracy (Table 3).
Analysis of gene expression
RPKM/FPKM was used to measure the abundance of gene expression in the transcriptome. We used FPKM to estimate the abundance of known genes expresses in different samples. After the construction of new transcripts, 69,907 transcripts and 29,128 unigenes were obtained. The five-demographic data (distribution) of experimental and control groups are shown in Fig. 1.

Statistics of gene expression distribution. Infection group: the maximum, upper quartile, median, lower quartile and the minimum values are 92252.6, 1.84, 0.03, 0 and 0, respectively. Control group: the maximum, upper quartile, median, lower quartile and minimum values are 107446, 1.7, 0.02, 0 and 0, respectively
Analysis of DEGs
Significant DEGs from the differential genes with P < 0.05, |log2 (fold change)| ≥ 1 and the FDR corrected P-value < 0.05 were selected. The overall distribution of DEGs was observed by drawing a volcano plot (Fig. 2). Compared with the control group, the number of up- and downregulated genes was 431 and 229, respectively. Partial DEGs are listed in Tables 4 and 5.

Distribution map of significant differentially expressed genes. Bright red dots indicate significantly upregulated genes, while blue dots indicate significantly downregulated genes
Bioinformatics analysis for the DEGs
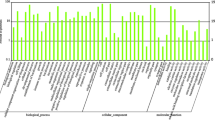
GO database analysis was conducted on the DEGs, including biological processes (1746), cellular components (1275) and molecular functions (785); the distribution diagram of GO terms with significant changes in classification is shown in Fig. 3. The main categories in the biological processes include immune system process, response to stress and signal transduction. As for the cellular components, the GO terms with the most differentially expressed genes were cell, intracellular and organelle. The molecular functions were involved mainly in the protein binding followed by ion binding, and transmembrane transporter activity.

GO analysis of the significant DEGs
KEGG database analysis showed that the DEGs were enriched in 101 pathways. The most enriched pathways of DEGs are exhibited in Fig. 4. Markedly, cell adhesion (31), phagosome (27) and natural killer cell mediated cytotoxicity (17) were the major three enriched pathways (Fig. 4). However, the pathway directly related to male reproduction was not found.

The most enriched pathways of DEGs
Verification results by qPCR analyzing and western blotting
The eight genes related to reproduction were identified by qPCR (Table 6); these expression trends were coincident with RNA-seq results (Fig. 5). The result of qPCR did not coincide with the RNA-seq perfectly; however, it is still possible to increase the reliability of these genes to a certain extent. DEGs including Piwil2, Spata18 and Tnfsf10 were selected for Western blot analysis. The results of the Western blot showed the same changing trend as the results of RNA-seq, although the specific amount of expression was different (Fig. 6).

Results of qPCR and compared with RNA-seq

Results of Western blot compared with RNA-seq. Lane 1: control group; Lane 2: infection group; from top to bottom, represent the β-actin internal reference, spata18, tnfsf10 and piwil2 protein
Discussion
We investigated the pathogenesis of T. gondii in the male reproductive tract of mice at the stage of sperm maturation using RNA-Seq technology. Additionally, we performed enrichment methodologies analysis to screen the DEGs that might be related to the reproductive functions. Results were confirmed by using qPCR and Western blot methods. Our findings showed that the RNA-Seq is accurate.
The analysis of DEGs suggested that some genes were interrelated with the infection of T. gondii, such as Iigp1 (interferon-inducible GTPase 1), Tgtp1 (T-cell-specific guanine nucleotide triphosphate-binding protein 1) and Tgtp2. Iigp1 found in the parasitophorous vacuoles (PV) plays a role in the destruction of PV and death of the parasite, when the Iigp1 gene is overexpressed [15]. We found that Iigp1 was in regulation of autophagy in the GO classification. Hence, it is possible that Iigp1 is involved in autophagy, although the specific mechanism is not clear. During infection of the type II of T. gondii, Tgtp1 and Tgtp2 were recruited to the PV membrane, PV vesiculation, rupture and digestion of the parasite within the cytosol [15, 16]. In our study, the Iigp1, Tgtp1 and Tgtp2 were significantly upregulated by 94-fold, 48-fold and 23-fold, respectively. This could be the result of host resistance to T. gondii. However, after T. gondii infection, Tgtp1 and Tgtp2 had low levels of gene expression abundance. The average abundance of Tgtp1 was 13.7 for the experimental group and 0.58 for the control group and the average abundances of Tgtp 2 were 15.39 and 0.31, respectively. Tgtp1 and Tgtp2 are mainly expressed in the thymus and lymph nodes, predominantly T-cells and IFNG-stimulated macrophages [17]. In other words, these genes are tissue-specific. After T. gondii infection, the immune system was stimulated, and the expression level of these genes was greatly increased.
Glycolytic pathway and sperm motility
There are several enriched pathways in our data analysis. Glycolysis was thought to be the oldest and most primitive way of biological energy harvesting. We focused on DEGs that affect glycolysis such as phosphoglycerate kinase 2 (Pgk2), phosphoglycerate mutase (Pgam) and lactate dehydrogenase C (LDHC). Phosphoglycerate kinase is the first regulation factor of ATP generated in the glycolysis pathway. In the reproductive system, the examination of sperm in elderly and young male patients with asthenospermia showed that the level of Pgk2 was reduced. Pgk2, which is related to the quality of sperm, is mainly expressed in able-bodied spermatids [18]. Pgk2 deficiency leads to a decrease in sperm motility and ATP levels [19]. Pgam is an important enzyme of glycolysis and gluconeogenesis pathway, catalyzes the 3-phosphoglycerate (3-PGA) into 2-phosphoglycerate (2-PGA), which dominates the balance between glycolysis and gluconeogenesis. Moreover, Pgam2 is related to the fibrous sheath of the main segment in the sperm flagellum [20]. The fibrous sheath on the sperm flagellum is the source of ATP and an essential structure of sperm motility [21]. Therefore, Pgam2 is the key determinant to maintaining sperm motility and morphology. LDHC is an enzyme that controls the conversion of pyruvate to L-lactic acid, which plays a key role in the process of spermatogenesis and capacitation by participating in cellular energy metabolism [22]. LDHC was found in the human spermatozoa and spermatogenic cells and was increased gradually with meiosis. LDHC in mature sperm is mainly located in the fibrous sheath of the main segment of the tail of sperm, which is involved in the regulation of sperm motility. In addition, LDHC is also participates in the homeostasis of ATP. Deficiency/destruction of germline-specific LDHC leads to infertility, including a reduction in sperm ATP content and reduced sperm motility [22, 23]. Conclusively, the enzymes are closely linked with sperm activity and morphology.
In the present study, the DEGs of glycolysis indicated that T. gondii infection might also result in dysregulation of those enzymes in humans, thus promoting a negative influence on the quality of male sperm. Normally, the energy production efficiency of oxidative phosphorylation is much higher than that of glycolysis, but glycolytic pathways have their particular importance in sperm energy sources. Mukai & Okuno [24] noticed that when the oxidative phosphorylation was blocked by rotenone of mitochondrial respiratory chain inhibitors, the sperm keeps moving. Therefore, glycolysis is not only an important source of energy for sperm but also plays a role in sperm capacitation [25] and acrosome reaction [26, 27].
Mitochondrial damage and apoptosis
Spermatogenesis associated 18 (Spata18) is a key regulator of mitochondrial quality that mediates repair or degradation of unhealthy mitochondria [28]. There is no pathway for Spata18, but previous research has shown that it is a monitor of cell differentiation process, which is involved in the maturation of spermatids into spermatozoa [29].
In our study, the expression of Tnfsf10 gene was upregulated to 2.89-fold in the pathway of cytokine-cytokine receptor interaction, apoptosis and natural killer cell mediated cytotoxicity. Tnfsf10 (Trail) is a member of the tumor necrosis factor (TNF) superfamily, and is an important signal molecule to maintain the stability of germ cells and the functional spermatogenesis in testis [30]. Apoptosis, or programmed cell death (PCD), is a biological process controlled by genes and plays a vital role in maintaining the morphology of normal tissues and organs. So far, the mitochondrial pathway is considered to be the classical pathway of apoptotic signal pathways. Mitochondria control the life and death of cells in aerobic environments, which is the main place of ATP production, and the major regulatory site of cell apoptosis. Mitochondrial permeability transition pores (MPTP) exist in the inner and outer membranes of mitochondria, and the permeability transition plays a vital role in cell apoptosis [31]. The pathway of cytokine-cytokine receptor interaction shows Tnfsf10 as a cytokine and interacts with the cytokine receptor Tnfsf10b. In the apoptosis pathway, Tnfsf10b (2.3-fold upregulation) as a death receptor, interacts with Tnfsf10 causing production of the death inducing signaling complex (DISC), which initiates the caspaseprotease family (aspartate-specific cysteine proteases) and activates apoptosis through DNA fragmentation, low synthesis of ploy, and destruction of nuclear membrane integrity [32]. At the same time, the translocation and permeabilization of mitochondrial membrane could induce apoptosis through Bax/Bak [33]. MPTP opening resulting from exogenous injury factors (such as Ca2+ overload) would lead to necrosis and apoptosis of cells. Some scholars have found that the synergism of melittin with Trail could increase the number of apoptosis in hepatoma cell through activating Ca2+/calmodulin-dependent protein kinase [34]. This is consistent with the finding of Chen et al. [35] who stated that the Ca2+ dependent calpain and/or Ca2+ independent cathepsins participate in apoptosis-like PCD and necrosis-like PCD. Trail gene-deficient (Trail2/2) mice were significantly reduced by 54% compared to the wild type mice, which explains the negative effects on sperm maturation in the reproductive system [30]. A high concentration of soluble Tnfsf10 was detected in the seminal plasma, which revealed a variable expression of Trail receptors in the sperm cellular fraction among different participants [36]. Those results suggest that Tnfsf10 may play an important role in sperm maturation.
The cytotoxicity of natural killer (NK) cells is motivated by the activating cell receptors. It can switch on the cascade reaction of downstream signal transduction. In our study, the Src family kinase (Fcgr4) was related to the cytotoxic pathway which is mediated by NK cells. The Fcgr4 also could active the mitogen activated protein kinase (MAPK) pathway, which could produce the cytotoxicity and secret proinflammatory cytokines (such as IFN-γ, TNF-α and GMCS). The upregulation of piwi and Itgb2 genes mediates the target cell apoptosis by Tnfsf10, which is caused by perforin and granzyme. The Gzmb (Granzyme B) gene is responsible for aspartate-specific cysteine proteases (DNA fragmentation, low synthesis of ploy and destruction of nuclear membrane integrity) which cause apoptosis.
From these pathways, we consider that Tnfsf10 is directly or indirectly connected with the male reproductive system. Chronic infection of T. gondii gives rise to dysregulation genes in these pathways, which in turn have a negative consequence on the integrity and motility of sperm. At the stage where spermatids mature into spermatozoa, mitochondria formed the mitochondrial sheath by gathering around the tail and wrapping around the shaft spirally. Finally, it can provide the energy for rotational motion of the tail in the way of making full use of energy in plasma drops. The fertility was influenced by various channels to create the apoptosis of germ cells.
Testis-specific genes in the epididymis
Our data analysis revealed that many DEGs were upregulated in the epididymis; however, previous studies have shown that they are testicular-specific genes, such as Piwil2 and RGS22 (regulator of G-protein signaling 22) genes. Piwil2 is a member of the Argonaute family and PIWI (the P-element-induced wimpy testis) subfamily. PIWI family proteins are only expressed in animal gonad tissues and have tissue specificity, these are closely related to the maintenance of germ stem cells and spermatogenesis [37]. The Piwil2 gene, expressed in tumor stem cells and germ cells, plays an important role in the self-renewal of stem cells, embryo formation and transcriptional regulation [38]. Similarly, Rgs22 is a specific gene in the male testis, which can be expressed simultaneously in a variety of epithelial tumors. In testis, Rgs22 protein is associated with spermatogenesis, which can be expressed in spermatogenic cells involved in the spermatogonia differentiation into sperm cells and Sertoli cells. Hu et al. [39] found that the expression of Rgs22 was downregulated in patients with azoospermia. Proteins are located in spermatogenic cells and Leydig cells, which interact with the guanine-nucleotide-binding proteins [39]. Thus, we consider that Rgs22 may be related to the meiotic stage of spermatogenesis.
In summary, Piwil2 and Rgs22 genes are testis-specific genes, mainly expressed in spermatogenic cells and play a key role in the spermatogenesis process. Other genes that were identified (see Table 7) are testis-specific and highly expressed in the epididymis. One possible reason for the latter observation is the invasion of T. gondii, which contributes to the destruction of the blood-testis barrier of the testis, and disruption of the spermatogenesis homeostasis [40]. Furthermore, spermatogenic cells in the seminiferous tubules were disordered, resulting in the increase of testis-specific gene expression in the epididymis. However, the epididymis has its tissue-specific microenvironment [41, 42] which is different from that in the testis. The microenvironment in the epididymis is not appropriate to regulate the cells in the testis and therefore, the specific mechanism is unclear and needs further investigation.
Conclusions
We sequenced the reproductive system of male mice chronically infected with T. gondii, which provides a new direction for research into male sterility caused by Toxoplasma infection. This work provides valuable information and a comprehensive database for future studies of the interaction between T. gondii infection and the male reproductive system.
Availability of data and materials
We have uploaded the transcriptome data to the Sequence Read Archive (SRA) database, submission: SUB5863608, accession ID: PRJNA552423
Abbreviations
- PRU:
-
Prugniaud
- DEGs:
-
differentially expressed genes
- FDR:
-
false discovery rate
- FC:
-
fold change
- GO:
-
Gene Ontology
- AsAb:
-
anti-sperm antibodies
- RNA-seq:
-
RNA sequencing
- Gb:
-
gigabases
- FPKM:
-
fragment per kilobase of exon model per million mapped reads
- SAM:
-
sequence alignment/map format
- GTF:
-
gene transfer format
- UCSC:
-
University of California Santa Cruz
- FRP:
-
horseradish peroxidase
- DAB:
-
diaminobenzidine
- ECL:
-
enhanced chemiluminescence
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MGI:
-
mouse genome informatics
- RIN:
-
RNA integrity number
- PV:
-
parasitophorous vacuoles
References
Jones JL, Dubey JP. Foodborne toxoplasmosis. Clin Infect Dis. 2012;55:845–51.
Zhou P, Chen Z, Li HL, Zheng H, He S, Lin RQ, Zhu XQ. Toxoplasma gondii infection in humans in China. Parasites Vectors. 2011;4:165.
Guha R, Miley L, Aspiri M, Dhamija R. Congenital toxoplasmosis. Pediatr Neurol. 2017;70:81–2.
Barreto F, Hering F, DallʼOglio MF, Martini FD, Campagnari JC, Srougi M. Testicular toxoplasmosis: a rare case of a testicular mass. Actas Urol Esp. 2008;32:666–8.
Suresh BP, Nagendra K, Navaz RS, Ravindranath HM. Congenital toxoplasmosis presenting as hypogonadotropic hypogonadism. Indian J Pediatr. 2007;74:577–9.
Bohring C, Skrzypek J, Krause W. Influence of antisperm antibodies on the acrosome reaction as determined by flow cytometry. Fertil Steril. 2001;76:275–80.
Oktenli C, Doganci L, Ozgurtas T, Araz RE, Tanyuksel M, Musabak U, et al. Transient hypogonadotrophic hypogonadism in males with acute toxoplasmosis: suppressive effect of interleukin-1 beta on the secretion of GnRH. Hum Reprod. 2004;19:859–66.
Terpsidis KI, Papazahariadou MG, Taitzoglou IA, Papaioannou NG, Georgiadis MP, Theodoridis IT. Toxoplasma gondii: reproductive parameters in experimentally infected male rats. Exp Parasitol. 2009;121:238–41.
Abdoli A, Dalimi A, Movahedin M. Impaired reproductive function of male rats infected with Toxoplasma gondii. Andrologia. 2012;44(Suppl. 1):679–87.
Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–36.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63.
Lopes WD, Costa AJ, Souza FA, Rodrigues JD, Costa GH, Soares VE, et al. Semen variables of sheep (Ovis aries) experimentally infected with Toxoplasma gondii. Anim Reprod Sci. 2009;111:312–9.
Teixeira W, Tozato M, Pierucci JC, Vital GP, Cruz AC, Lopes W, et al. Investigation of Toxoplasma gondii in semen, testicle and epididymis tissues of primo-infected cats (Felis catus). Vet Parasitol. 2017;238:90–3.
Colosi HA, Jalali-Zadeh B, Colosi IA, Simon LM, Costache CA. Influence of Toxoplasma gondii infection on male fertility: a pilot study on immunocompetent human volunteers. Iran J Parasitol. 2015;10:402–9.
Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, et al. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 2005;1:e24.
Zhao Y, Ferguson DJ, Wilson DC, Howard JC, Sibley LD, Yap GS. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J Immunol. 2009;182:3775–81.
Lafuse WP, Brown D, Castle L, Zwilling BS. Cloning and characterization of a novel cDNA that is IFN-gamma-induced in mouse peritoneal macrophages and encodes a putative GTP-binding protein. J Leukoc Biol. 1995;57:477–83.
Liu XX, Zhang H, Shen XF, Liu FJ, Liu J, Wang WJ. Characteristics of testis-specific phosphoglycerate kinase 2 and its association with human sperm quality. Hum Reprod. 2016;31:273–9.
Danshina PV, Geyer CB, Dai Q, Goulding EH, Willis WD, Kitto GB, et al. Phosphoglycerate kinase 2 (PGK2) is essential for sperm function and male fertility in mice. Biol Reprod. 2010;82:136–45.
Nakamura N, Dai Q, Williams J, Goulding EH, Willis WD, Brown PR, et al. Disruption of a spermatogenic cell-specific mouse enolase 4 (eno4) gene causes sperm structural defects and male infertility. Biol Reprod. 2013;88:90.
Krisfalusi M, Miki K, Magyar PL, OʼBrien DA. Multiple glycolytic enzymes are tightly bound to the fibrous sheath of mouse spermatozoa. Biol Reprod. 2006;75:270–8.
Goldberg E, Eddy EM, Duan C, Odet F. LDHC: the ultimate testis-specific gene. J Androl. 2010;31:86–94.
Odet F, Gabel SA, Williams J, London RE, Goldberg E, Eddy EM. Lactate dehydrogenase C and energy metabolism in mouse sperm. Biol Reprod. 2011;85:556–64.
Mukai C, Okuno M. Glycolysis plays a major role for adenosine triphosphate supplementation in mouse sperm flagellar movement. Biol Reprod. 2004;71:540–7.
Ickowicz D, Finkelstein M, Breitbart H. Mechanism of sperm capacitation and the acrosome reaction: role of protein kinases. Asian J Androl. 2012;14:816–21.
Hung P, Miller MG, Meyers SA, VandeVoort CA. Sperm mitochondrial integrity is not required for hyperactivated motility, zona binding, or acrosome reaction in the rhesus macaque. Biol Reprod. 2008;79:367–75.
Fraser LR, Quinn PJ. A glycolytic product is obligatory for initiation of the sperm acrosome reaction and whiplash motility required for fertilization in the mouse. J Reprod Fertil. 1981;61:25–35.
Kitamura N, Nakamura Y, Miyamoto Y, Miyamoto T, Kabu K, Yoshida M, et al. Mieap, a p53-inducible protein, controls mitochondrial quality by repairing or eliminating unhealthy mitochondria. PLoS ONE. 2011;6:e16060.
Bornstein C, Brosh R, Molchadsky A, Madar S, Kogan-Sakin I, Goldstein I, et al. SPATA18, a spermatogenesis-associated gene, is a novel transcriptional target of p53 and p63. Mol Cell Biol. 2011;31:1679–89.
Lin YC, Richburg JH. Characterization of the role of tumor necrosis factor apoptosis inducing ligand (TRAIL) in spermatogenesis through the evaluation of trail gene-deficient mice. PLoS ONE. 2014;9:e93926.
Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90:1281–9.
Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–27.
Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol. 2001;153:1265–76.
Wang C, Chen T, Zhang N, Yang M, Li B, Lü X, et al. Melittin, a major component of bee venom, sensitizes human hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating CaMKII-TAK1-JNK/p38 and inhibiting IκBα kinase-NFκB. J Biol Chem. 2009;284:3804–13.
Chen J, Mehta JL, Haider N, Zhang X, Narula J, Li D. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ Res. 2004;94:370–6.
Zauli G, Celeghini C, Monasta L, Martinelli M, Luppi S, Gonelli A, et al. Soluble TRAIL is present at high concentrations in seminal plasma and promotes spermatozoa survival. Reproduction. 2014;148:191–8.
Kuramochi-Miyagawa S, Kimura T, Ijiri TW, Isobe T, Asada N, Fujita Y, et al. Mili, a mammalian member of piwi family gene, is essential for spermatogenesis. Development. 2004;131:839–49.
Ling W, Zhigang H, Tian H, Bin Z, Xiaolin X, Hongxiu Z. HPV 16 infection up-regulates Piwil2, which affects cell proliferation and invasion in cervical cancer by regulating MMP-9 via the MAPK pathway. Eur J Gynaecol Oncol. 2015;36:647–54.
Hu Y, Xing J, Chen L, Guo X, Du Y, Zhao C, et al. RGS22, a novel testis-specific regulator of G-protein signaling involved in human and mouse spermiogenesis along with GNA12/13 subunits. Biol Reprod. 2008;79:1021–9.
Yang R. Effect of Toxoplasma gondii infection on cytokines and spermatogenic cells in rats. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 2011;29:274–8.
Dacheux JL, Belleannee C, Jones R, Labas V, Belghazi M, Guyonnet B, et al. Mammalian epididymal proteome. Mol Cell Endocrinol. 2009;306:45–50.
Gatti JL, Castella S, Dacheux F, Ecroyd H, Metayer S, Thimon V, et al. Post-testicular sperm environment and fertility. Anim Reprod Sci. 2004;82–83:321–39.
Acknowledgements
Not applicable.
Funding
This work was supported, in part, by grants from the National Key Research and Development Programme of China (2017YFD0501305), National Natural Science Foundation of China (31972707), Major Scientific Research Projects (Characteristic Innovation) of Guangdong Province (2017KTSCX018), the Key Research and Development Programme of Guangdong Province (2019B020218004), the Guangzhou Zoo Project (h2016141), the Natural Science Foundation of Guangdong Province (2019A1515011534, 2016A030313396), and Huizhou Science and Technology Projects (2015B040009001).
Author information
Authors and Affiliations
Contributions
YXZ contributed to the data analysis and the writing of the manuscript. XXZ contributed to the drafting of the manuscript. JAH and YSM contributed to the revising of the manuscript. WYH, GFL, YPW and XZ contributed to the data collection and the laboratory work. YXZ and XXZ contributed to the animal experiment. ZGY and XML contributed to the conception of the idea and design. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was carried out in accordance with the principles of the Basel Declaration and recommendations of the People’s Republic of China, the Animal Ethics Committee of South China Agricultural University. The protocol was approved by the South China Agricultural University Experimental Animal Welfare Ethics Committee (Approval No. SCXK 2013-0002).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Text S1.
Detailed methods of transcriptome sequencing, data analysis and verification.
Additional file 2: Figure S1.
Effect of T. gondii infection on total sperm count.
Additional file 3: Figure S2.
Identification of infection in experimental mice.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zheng, YX., Zhang, XX., Hernandez, J.A. et al. Transcriptomic analysis of reproductive damage in the epididymis of male Kunming mice induced by chronic infection of Toxoplasma gondii PRU strain. Parasites Vectors 12, 529 (2019). https://doi.org/10.1186/s13071-019-3783-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3783-2