
Abstract
Background
Vitellogenin (Vg), a key molecule for oocyte development synthesized in the fat body during blood-feeding, is released into the hemolymph and then taken into the oocytes via Vg receptor (VgR) in ticks. Previously, we showed that VgR mRNA is expressed in the ovary at the adult stage of parthenogenetic Haemaphysalis longicornis ticks and its expression increases after blood-feeding. However, intracellular localization of VgR mRNA and protein at each developmental stage of oocytes during oogenesis remains largely unclear.
Methods
mRNA and protein expression profiles of H. longicornis VgR (HlVgR) in the oocytes from the unfed to oviposition periods were analyzed by real-time PCR, in situ hybridization, and immunostaining. To elucidate the timing of the onset of Vg uptake, RNA interference (RNAi)-mediated gene silencing of HlVgR was performed.
Results
In situ hybridization revealed that HlVgR mRNA was detected in the cytoplasm of stage I-III oocytes, and weaker positive signals for HlVgR mRNA were found in the cell periphery of stage IV and V oocytes. Likewise, HlVgR protein was detected by immunostaining in the cytoplasm of stage I-III oocytes and in the cell periphery of stage IV and V oocytes. Each developmental stage of the oocytes showed distinct patterns of mRNA and protein expression of HlVgR. Moreover, RNAi of HlVgR caused delayed or arrested development in the oocytes. The ovaries of control ticks showed all developmental stages of oocytes, whereas stage I-III oocytes were found in the ovaries of HlVgR-RNAi ticks at 5 days after engorgement.
Conclusions
These results suggest that active uptake of Vg is required for development from stage III to stage IV during oogenesis. Our data clearly revealed an apparent shift in the intracellular localization of VgR for both mRNA and protein level in oocytes during oogenesis.
Similar content being viewed by others

Background
Ticks, vectors of harmful pathogens for human and animals, absolutely require a blood meal for their development and reproduction. Nutrients derived from a blood meal allow the synthesis of vitellogenin (Vg), an essential molecule for oocyte development, in the fat body of female ticks, being stimulated by ecdysteroid hormones [1,2,3,4,5,6,7,8,9,10]. The regulatory network underlying the synthesis of Vg (vitellogenesis) in ticks is being clarified by RNA interference (RNAi)-mediated gene knockdown experiments and inhibitor assays. A GATA factor was found to be a transcriptional activator of the Vg gene in the female Haemaphysalis longicornis [11]. Subsequent studies revealed that vitellogenesis is controlled by the activation of the target of rapamycin (TOR), a key molecule of a nutrient-sensing pathway in eukaryotic cells, through ecdysteroid hormone stimulation [12]. TOR phosphorylates S6 kinase and regulates the transcription and intracellular localization of a GATA factor in the fat body. Our previous data suggested that the serine/threonine protein kinase Akt is an upstream factor of TOR and essential not only for controlling gene transcription but also for regulating vitellogenesis [13].
Following vitellogenesis, Vg is released into the hemocoel filled with hemolymph and then is taken into developing oocytes in ticks [7, 10]. The absorption of Vg into the oocytes occurs via receptor-mediated endocytosis, and the Vg receptor (VgR) is a member of the low-density lipoprotein receptor (LDLR) superfamily [14]. In hard ticks, homologues of VgR have been identified from Amblyomma hebraeum (AhVgR) [15], Dermacentor variabilis (DvVgR) [16], H. longicornis (HlVgR) [17], Rhipicephalus microplus (RmVgR) and R. appendiculatus (RaVgR) [18]. These tick VgRs have common structural elements similar to those in insects: LDLR class A repeats, LDLR class B repeats, epidermal growth factor (EGF) precursor homology domains containing YWXD repeats, an O-linked carbohydrate domain, a transmembrane domain and a cytoplasmic domain [14,15,16,17,18]. The predicted molecular weight of tick VgR based on the amino acid sequences is approximately 200 kDa [15,16,17,18]. These studies showed that knockdown of VgR in female ticks led to the failure of Vg uptake in the oocytes and the subsequent oviposition, even though the ticks engorged completely. Immunoelectron microscopy revealed that VgR is localized on the external surface of the plasma membrane in developing tick oocytes [17]. In addition, whole tissue in situ hybridization using the ovaries of engorged females at 10 days following engorgement revealed that VgR mRNA was detected in the smallest oocytes, which had not yet begun to fill with yolk particles [15]. However, little is known about intracellular localization of VgR mRNA and protein at each developmental stage, namely stages I to V, of oocytes throughout oogenesis.
Transmission of some pathogens is tightly linked to the reproduction system in female ticks. After ingesting a blood meal containing some pathogens, ticks may give rise to progeny with these pathogens. An ixodid female tick generally lays a few thousand eggs [19]; therefore, the basis of oogenesis is an important key for the development of novel control strategies for pathogen transmission by ticks. For instance, it has been reported that VgR might be involved in transovarial transmission of Babesia parasites by a vector tick H. longicornis [17]. This finding led to the hypothesis that Babesia parasites bind to tick VgR and invade the developing oocytes of ticks and that some surface molecules of parasites have ligand-binding activity for VgR. Therefore, it is important to determine the expression pattern and intracellular localization of VgR to elucidate the interactions between Babesia parasites and tick VgR during oogenesis.
In the present study, we examined mRNA and protein expression profiles of HlVgR in the oocytes from the unfed to the oviposition periods by real-time PCR and in situ hybridization and immunostaining using ovarian tissue sections. Furthermore, to determine the timing of the onset of Vg uptake during oogenesis, oocytes of HlVgR-RNAi ticks were observed histologically on the basis of the classification criteria [20] for developing oocytes in the parthenogenetic H. longicornis.
Results
Expression of HlVgR mRNA in the ovary
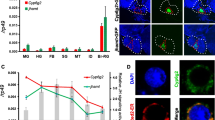
Real-time PCR with specific primers was conducted to determine the relative expression level of HlVgR in the ovary from the unfed to the oviposition periods. Average value of HlVgR mRNA expression was 0.005 in the unfed period. By contrast, the expression at the slow feeding was more than 1000-fold higher than that at the unfed period (t(5) = − 6.957, P = 0.0009) (Fig. 1). The expression levels of HlVgR in the rapid feeding period were higher than those at previous periods (t(5) = − 8.798, P = 0.0003) and were the highest at engorgement (t(5) = − 13.761, P < 0.0001). These results suggest that HlVgR was upregulated in the ovary during blood-feeding. In addition, expression levels of HlVgR at 4 days after engorgement (4dAE) (t(10) = 4.487, P = 0.0012) and at the beginning of the oviposition period (0dAO) (t(10) = 4.707, P = 0.0008) decreased significantly compare with those at engorgement. At 10 days after the beginning of oviposition (10dAO), HlVgR expression appeared to be increased again.

mRNA expression of HlVgR in the ovaries of H. longicornis from unfed to oviposition periods. Total RNA was extracted from each ovary sample. DNase-treated RNA was subjected to cDNA synthesis. Data were normalized by a real-time PCR analysis of HlP0 mRNA levels in the cDNA samples. Data represent the mean ± SD of triplicate samples. Asterisk and hash symbols indicate significant downregulation (Student t-test) and upregulation (Welchʼs t-test), respectively, at P < 0.01. Abbreviations: 4dAE, 4 days after engorgement; 0dAO, the beginning of oviposition; 10dAO, 10 days after the beginning of oviposition
Localization of HlVgR mRNA in the ovary
HlVgR mRNA was undetected by in situ hybridization in the ovaries in the unfed period (Fig. 2). The cytoplasm of oocytes at stages I and II was stained with the antisense probe of HlVgR in the slow feeding, rapid feeding, and engorgement periods. The expression of HlVgR mRNA in oocytes of all stages (stages I to V) was detected at 4dAE, 0dAO, and 10dAO. HlVgR mRNA was localized throughout the cytoplasm in the oocytes of stages I to III. Positive signals for HlVgR mRNA were found in the external region of the oocytes that were at stages IV and V, but the signals seemed to be weaker than those of the former stages. The expression of HlVgR mRNA was also detected in pedicel cells and the ovarian wall (Fig. 2).

Expression sites of HlVgR mRNA in H. longicornis ovary from unfed to oviposition periods. Paraffin sections of the ovaries were treated with sense probe (S) or antisense probe (AS). Arrowheads show pedicel cells. Abbreviations: W, ovarian wall; I, stage I oocyte; II, stage II oocyte; III, stage III oocyte; IV, stage IV oocyte; V, stage V oocyte. Scale-bars: 50 µm
Localization of HlVgR protein in the ovary
Positive signals of HlVgR were not detected by immunofluorescence staining in the ovary in the unfed period (Fig. 3a) but were found in the cytoplasm of oocytes in the slow feeding period. In the rapid feeding and engorgement periods, HlVgR signals were detected throughout the cytoplasm of stage I and II oocytes. The HlVgR signals were also detected in stage II to V oocytes at 4dAE, 0dAO, and 10dAO (Fig. 3b). HlVgR signals were scarcely observed in stage I oocytes during these periods. HlVgR signals were detected throughout the cytoplasm of the oocytes of stage II. Notably, the signals were localized throughout the cytoplasm as well as external region of the stage III oocytes, whereas the signals were detected at the chorion and the cell periphery in the oocytes of stages IV and V. In addition, HlVgR signals were localized in the cytoplasm surrounding the yolk granules. HlVgR signals were not detected in the pedicel cells and ovarian wall at any period examined in our study.

Localization of HlVgR protein in the ovary of H. longicornis. Positive signals are displayed in yellow color in the ovarian sections from unfed to engorgement periods (a) and from 4 days after engorgement to oviposition periods (b). Stage I or II oocytes at unfed to engorgement periods were observed in areas surrounded by dashed circles. Abbreviations: PC, phase contrast; I, stage I oocyte; II, stage II oocyte; III, stage III oocyte; IV, stage IV oocyte; V, stage V oocyte. Scale-bars: 50 µm
Oocyte development in HlVgR-RNAi ticks
To clarify the timing of the onset of Vg uptake during oogenesis, oocytes in the ovaries were observed after RNAi of the HlVgR gene. Real-time PCR revealed decreased expression levels of HlVgR mRNA at engorgement (t(2) = 16.327, P = 0.0037) and at 5 days after engorgement (t(2) = 12.749, P = 0.0061) compared with that in the control ticks (Fig. 4a, c). There was no difference in the body weight of engorged ticks between the HlVgR-RNAi and control groups (Table 1). The oocytes were typically colorless and white in color in both the HlVgR-RNAi and control ticks at engorgement (Fig. 4b). At this period, oocytes of stages I and II were observed in both ovaries on the histological sections. Control ticks had amber-colored oocytes in the ovaries and began oviposition at 5 days after engorgement, whereas the ovaries in the HlVgR-RNAi ticks possessed amberlite and white colored oocytes, which were classified into stages I to III (Fig. 4d). No yolk granules were found in the cytoplasm of any of the oocytes of the HlVgR-RNAi ticks. Although HlVgR-RNAi ticks began oviposition at 12 days after engorgement (Table 1), the eggs were not in amber color and not coated with waxy components as shown previously [17].

HlVgR-knockdown by RNA interference (RNAi) in H. longicornis. a, c Total RNA was purified from engorged ticks in each group. The gene silencing of HlVgR was assessed by real-time PCR. The HlVgR expression was normalized to the HlP0 mRNA expression. Data represent the mean ± SD of triplicate samples. HlVgR mRNA expressions of RNAi-treated ticks (HlVgR RNAi) were significantly lower than those of control ticks (Control) at both engorgement (a) and at 5 days after engorgement (c) (*P < 0.01; Welchʼs t-test). b, d Phenotypes of the ovaries of HlVgR RNAi-treated and control ticks. Note that HlVgR-knockdown caused delayed development in the oocytes at both engorgement (b) and at 5 days after engorgement (d). Abbreviations: I, stage I oocyte; II, stage II oocyte; III, stage III oocyte; IV, stage IV oocyte; V, stage V oocyte. Scale-bars: b, 1 mm for ovaries and 100 µm for ovarian sections; d, 2 mm for ovaries, and 200 µm for ovarian sections
Discussion
Expression levels of HlVgR mRNA in the ovary of H. longicornis increased with the progress of blood-feeding and reached the highest level at engorgement, suggesting that blood-feeding is a triggering process for the upregulation of HlVgR mRNA (Fig. 1). Because ticks have a panoistic ovary, the ratio of each oocyte stage is asynchronous during oogenesis [20]. Subsequently, the spatial patterning of HlVgR mRNA expression in each developmental stage of the oocytes was examined by in situ hybridization. The cytoplasm of oocytes at stages I and II was intensely stained with the antisense probe of HlVgR at the slow feeding, rapid feeding, and engorgement periods (Fig. 2). At these periods, the ovaries of parthenogenetic H. longicornis possess stage I and II oocytes, and the ratio of stage II to all oocytes in the rapid feeding period increased at engorgement [20]. In addition, the HlVgR mRNA level was significantly decreased at 4dAE and at 0dAO compared with that at engorgement (Fig. 1). At 4dAE and 0dAO, more than half of all oocytes were classified into stages III, IV or V [20]. By linking mRNA expression patterns with morphological features of oocytes, it was obvious that HlVgR was highly expressed in stage I and II oocytes compared to the other stages (Figs. 1 and 2 and [20]). These findings substantially correspond with the findings that a high degree of positive staining was detected in the smallest oocytes of A. hebraeum by whole tissue in situ hybridization [15]. Moreover, HlVgR mRNA was detected in the whole cytoplasm of stage I to III oocytes and under the oocyte membrane of oocytes at stages IV and V in our study (Fig. 2). The positive signals of HlVgR mRNA in mature oocytes seemed to be weaker than those of stage I-III oocytes. Taken together, real-time PCR and in situ hybridization showed that the transcription of HlVgR was activated in stages I and II of the oocytes, namely before the formation of yolk granules, regardless of the number of days after blood-feeding.
Immunostaining revealed that HlVgR was localized throughout the cytoplasm of the stage I–III oocytes and in the peripheral region of the stage IV and V oocytes (Fig. 3). As with the results of in situ hybridization, the intracellular localization of HlVgR changed as oocyte development progressed. Drosophila melanogaster VgR is uniformly distributed throughout the oocyte during the pre-vitellogenic stages of oogenesis, indicating that such oocytes do not take in Vg in the presence of VgR [21]. After the transition to the vitellogenic stages, VgR levels increase markedly at the cell periphery in the oocyte. By the end of vitellogenesis, the receptor localizes predominantly in the external region of the oocyte in the fruit fly. In the yellow fever mosquito Aedes aegypti, VgR is internalized, dissociated, and sorted and then recycled to the plasma membrane during Vg uptake [22]. In our study, we found that HlVgR was distributed throughout the cytoplasm of stage I and II oocytes (Fig. 3), suggesting that such oocytes are unlikely to take up Vg from hemolymph. In the stage III oocytes, positive signals were detected throughout the cytoplasm and in the external region of the oocytes. HlVgR was localized in the cytoplasm, especially in the peripheral region of the stage IV–V oocytes, which possess yolk granules in the cytoplasm [20].
Knockdown of HlVgR caused the delay or arrest of oogenesis in our study (Fig. 4), resulting in the inhibition of oocyte development from stage III to the subsequent stage. Control ticks started to lay eggs at 5 days after engorgement, whereas the oocytes of the RNAi-treated ticks at that timing were still developing to stage III. The ovaries of the RNAi-treated ticks showed an amberlite color, implying that Vg uptake in the oocytes starts from stage III. Seixas et al. [18] similarly reported that RmVgR-silenced female ticks showed a small degree of yolk uptake in the oocytes classified into the ovarian growth phase scores/indexes 2 and 3. They also showed a clear correlation between the amount of RmVgR mRNA and protein in the ovaries: RmVgR protein level was increased after the upregulation of mRNA expression in the ovaries of partially fed female ticks, and the level peaked before engorgement. Moreover, they described that when female ticks drop from the host animal, VgR mRNA expression levels decrease because the receptor is no longer important once Vg has already been taken up by the oocytes. However, the Vg concentration in the hemolymph reaches its highest level at 2–5 days after the beginning of oviposition in hard ticks [23,24,25]. As shown in our study, the expression level of VgR mRNA depends on the developmental stages of the oocytes. Therefore, the decrease of the VgR mRNA level was considered to be attributed to the decrease in the ratio of stage I and II oocytes to all oocytes. Oogenesis continues to progress throughout the oviposition period of around 1 month until the female ticks die.
Nurse or follicle cells, the providers of nutrients for insect oocytes, are non-existent in the ovary of ticks. Instead of such nurse or follicle cells, pedicel cells derived from ovarian wall epithelial cells are present in ticks. These pedicel cells are known to attach the oocytes to the ovarian wall [26, 27]. In addition to their structurally supportive role, it was speculated that tick pedicel cells have similar functions of nurse or follicle cells in insects. Ultrastructural observations led to the idea that yolk precursors, i.e. Vg, might be synthesized in pedicel cells of A. triste ticks and would then be transported into stage II oocytes [26]. In D. melanogaster, VgR mRNA is transported to the oocyte after synthesis in the nurse cells [21]. We found positive signals for HlVgR mRNA in both pedicel cells and the ovarian wall at the slow feeding, pre-oviposition, and oviposition periods (Fig. 2). HlVgR mRNA could be synthesized in the pedicel cells; however, further experiments are needed to clarify this point because HlVgR protein was not detected in pedicel cells and the ovarian wall by immunostaining.
Conclusions
Our study revealed that an apparent shift in the intracellular localization of HlVgR mRNA and protein was associated with the developmental stages of oocytes in parthenogenetic H. longicornis ticks. This study showed that Vg uptake via VgR is a critical process for the transition of oocytes from stage III to IV in the tick. Taken together with findings from our previous studies [11,12,13, 17, 20, 28], we propose a model of Vg uptake during oogenesis in parthenogenetic H. longicornis (Fig. 5). To date, multiple Vgs (HlVg-1, HlVg-2, and HlVg-3) were identified from the parthenogenetic H. longicornis [28]. This study revealed that the HlVg-1 transcript was detected only in the midgut of this tick species. The HlVg-2 was expressed in the ovary and fat body, while the HlVg-3 was found only in the fat body. “Vgs” in Fig. 5 includes HlVg-2 and HlVg-3 proteins based on our previous results. In other tick species, DvVg1 mRNA was detected in the midgut, fat body, and ovary of D. variabilis [29, 30]. However, DvVg2 mRNA was detected in both fat body and midgut but not found in the ovary. mRNA expression of AhVg1was observed in the fat body, midgut, and ovary, while AhVg2 was detected in both fat body and midgut in A. hebraeum [31]. In R. microplus, Vg-1 and Vg-2 are synthesized in the ovary [32]. The contribution of each Vg to vitellogenesis remains unclear; however, ovarian molecules are considered as targets for anti-tick vaccine development as they can affect tick biology by decreasing oogenesis and embryogenesis [33]. After blood-feeding, immunoglobulins pass through the midgut epithelium and then arrive in the hemolymph of arthropods while retaining their immunological characters [34]. It is believed that cell membrane receptors serve as vaccine candidate molecules in blood-feeding arthropods. Further understanding of the molecular mechanisms regulating oogenesis will provide valuable information for the control of ticks. Moreover, our current findings may help to clarify the interaction between transovarially transmitted pathogens and ovarian molecules, especially Vg or VgR, in ticks. The potential of HlVgR as an antigen for the development of an anti-tick vaccine could induce infertility in ticks and thus inhibit the transmission of tick-borne diseases.

A schema of Vg uptake during oogenesis of parthenogenetic H. longicornis. Synthesized Vgs (Vg-2 and Vg-3) are released from the fat body into the hemocoel filled with hemolymph. The Vgs are taken up by oocytes of stages III, IV and V. Abbreviations: I, stage I oocyte; II, stage II oocyte; III, stage III oocyte; IV, stage IV oocyte; V, stage V oocyte; Vgs, vitellogenins; VgR, vitellogenin receptor
Methods
Ticks and animals
The parthenogenetic H. longicornis ticks (Okayama strain) were maintained at the National Research Center for Protozoan Diseases, Obihiro University of Agriculture and Veterinary Medicine, Obihiro, Japan. The female ticks were fed on Japanese white rabbits (Japan SLC, Shizuoka, Japan) as described previously [35]. Eight-week-old female BALB/c mice (CLEA Japan, Tokyo) were used to produce anti-HlVgR polyclonal antibody. The rabbits were reared in a temperature- and humidity-regulated room under controlled lighting, water and commercial regular chow. The mice were maintained in a temperature- and humidity-regulated P2 room under same conditions as rabbits.
Reverse transcription and real-time PCR
For collection of the ovaries, 30 ticks at the unfed, 20 ticks at slow feeding (3 days after attachment), 5 ticks at rapid feeding (4–6 days after attachment), and 3 ticks at engorgement (5–7 days after attachment) periods were dissected. Twelve engorged females in individual glass tubes were placed in a container. The container was then placed in dark incubator at 25 °C and saturated humidity. The ovaries were also sampled from 4 ticks at 4 days (4dAE; pre-oviposition), 4 ticks at 7 days (0dAO; beginning of oviposition) and 4 ticks at 17 days (10dAO) after engorgement. Total RNA was extracted from the collected ovaries using TRI® reagent (Sigma-Aldrich, St. Louis, MO, USA) and incubated with DNase (TURBO DNA-free™ Kit; Life Technologies, Frederick, MD, USA) to remove genomic DNA. Single-strand cDNA was synthesized using purified total RNA and a ReverTra Ace® qPCR RT Kit (Toyobo, Osaka, Japan) according to a previous report [36]. Subsequently, real-time PCR was conducted using a 7300 Real-Time PCR System (Life Technologies) and THUNDERBIRD® SYBR qPCR Mix (Toyobo) according to the manufacturer’s protocol. The primers used in our study are listed in Table 2. Standard curves used for the calculation of relative gene expression were created from five-fold serial dilutions of cDNA derived from the ovaries of engorged females. The PCR conditions were as follows: 95 °C for 10 min, 40 cycles of denaturation at 95 °C for 15 s, and annealing/extension at 60 °C for 60 s. Dissociation curves for individual wells were created to confirm the single PCR product. Data were analyzed by the 7300 system SDS software version 1.4 for Windows 7. The control amplification was carried out using the primers designed for H. longicornis ribosomal protein P0 (HlP0) (GenBank: EU048401). All reactions were run in duplicate, and data obtained were used to calculate the mean ± standard deviation (SD). The differences in relative gene expression levels of HlVgR between each sample were compared using the Student t-test (vs Unfed) or Welchʼs t-test (vs Engorgement), depending on the data distribution. Statistical significance was set at P < 0.01.
Preparation of histological sections of ovary
Thirty-two ovaries in total collected at the same periods as described in the previous section were fixed with 4% paraformaldehyde-0.1% glutaraldehyde in phosphate-buffered saline (PBS) at 4 °C, overnight. The ovaries (10 µm thick) were sectioned with a cryostat (CM3050 S, Leica Biosystems, Wetzlar, Germany) as described previously [37]. To prepare paraffin sections, a total of 55 ovaries were immersed in 4% paraformaldehyde-2.5% glutaraldehyde in PBS at 4 °C, overnight. After washing with PBS, the ovaries were pre-embedded in 3% agarose (Sigma-Aldrich) in PBS and then embedded in paraffin (Sakura Finetek Japan, Tokyo, Japan). The sections (5 µm thick) were cut on a REM-700 microtome (Yamato Kohki Industrial, Saitama, Japan).
In situ hybridization
In situ hybridization was carried out according to the manufacturer’s protocol of the in situ hybridization reagents starting kit (Nippon Gene, Tokyo, Japan). RNA probes were generated from PCR products with T7 or SP6 ends produced by amplification of ovary cDNA using gene-specific primers (Table 2). The PCR was performed by a GeneAmp® PCR System 9700 (Life Technologies) and PrimeSTAR® MAX DNA Polymerase (Takara Bio, Shiga, Japan). The cycling conditions were as follows: 30 cycles of denaturation at 98 °C for 10 s and annealing/extension at 68 °C for 60 s. The PCR products (534 bp) were purified using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Sense and antisense RNA probes were generated using purified template DNA, T7, and SP6 RNA polymerase, respectively, and digoxigenin-11-UTP (Roche Diagnostics GmbH, Mannheim, Germany) for 60 min at 37 °C (for T7 RNA polymerase) and 40 °C (for SP6 RNA polymerase). Synthesized RNA probes were confirmed by electrophoresis on a 1.5% agarose gel and were then treated with alkali hydrolysis buffer for 44 min at 60 °C.
Paraffin sections of the ovaries were deparaffinized and treated with 5–8 µg/ml proteinase K (Nippon Gene) for 20 min at 37 °C. They were acetylated for 15 min at room temperature by adding 1 ml of acetic anhydride into 400 ml of acetylation buffer and then pre-hybridized in hybridization solution for 30 min at 42 °C. Hybridization was performed for 16 h at 42 °C after adding 1 µg/ml of sense or antisense probe per section. Hybridized sections were treated with blocking reagent (Roche Diagnostics GmbH) for 30 min at room temperature, treated with anti-digoxigenin-AP Fab fragments (Roche Diagnostics GmbH) for 1 h at room temperature, and then reacted with BCIP®/NBT liquid substrate system (Sigma-Aldrich) for 24 h at room temperature. Following staining of hybridized probes, nucleus staining was performed with 1% methyl green (Muto Pure Chemicals, Tokyo, Japan) for 2 min at room temperature. The sections were then observed under a BZ-9000 light microscope (Keyence, Osaka, Japan). To determine expression sites of HlVgR mRNA in the ovary, the developmental stages of each oocyte were classified into five stages as described previously [20].
Expression of recombinant HlVgR protein and production of mouse anti-HlVgR polyclonal antibody
A partial fragment (936 bp) corresponding to second ligand-binding domain with class A repeats of HlVgR (GenBank: AB299015) [17] was amplified by PCR with the forward primer containing a recognition site for XhoI and the reverse primer with a recognition site for EcoRI (Table 2). The PCR was conducted by a GeneAmp® PCR System 9700 (Life Technologies) and PrimeSTAR® MAX DNA Polymerase (Takara Bio). The cycling conditions were as follows: 30 cycles of denaturation at 98 °C for 10 s and annealing/extension at 68 °C for 60 s. The PCR product (952 bp) containing partial HlVgR and restriction enzyme sites purified by Wizard® SV Gel and PCR Clean-up System (Promega, WI, USA) was inserted into the pCold™ ProS2 DNA plasmid vector (Takara Bio), which had been digested with XhoI and EcoRI, with a Ligation-Convenience Kit (Nippon Gene) according to the manufacturer’s protocol. Recombinant plasmids were transformed into ECOS™-competent Escherichia coli DH5α (Nippon Gene). After transformation, the plasmids were confirmed by sequencing and used to transform ECOS™-competent E. coli BL21 (Nippon Gene). Expression of recombinant histidine-tagged HlVgR (rHlVgR; approximately 60 kDa) was induced with 0.1 mM isopropyl-β-D (-)-thiogalactopyranoside at 15 °C overnight. The rHlVgR was solubilized with binding buffer (pH 8.0) containing 8 M urea. The purification of rHlVgR was performed by His GraviTrap (GE Healthcare, Chicago, IL, USA) using elution buffer (pH 8.0) containing 8M urea under the denaturing conditions. After concentration of the purified rHlVgR using Pierce® Concentrator 9K MWCO (Thermo Fisher Scientific, Waltham, MA, USA), urea in the rHlVgR solution was removed using Slide-A-Lyzer™ G2 Dialysis Cassette 7K MWCO (Thermo Fisher Scientific). Fifty micrograms of soluble rHlVgR or PBS in Freund’s complete adjuvant (Sigma-Aldrich) was intraperitoneally injected into 8-week-old female BALB/c mice. The same antigen or PBS in Freund’s incomplete adjuvant (Sigma-Aldrich) was repeatedly injected intraperitoneally into the mice on days 14, 28 and 42 after the first inoculation. At 7 days after the last immunization, sera against rHlVgR were collected from mice. Anti-rHlVgR IgG in the sera was purified using the Melon™ Gel IgG Spin Purification Kit (Thermo Fisher Scientific) and was stored at − 30 °C until use.
Immunostaining
Frozen sections were incubated with Liberate Antibody Binding solution (Polysciences, Warrington, PA, USA) for 5 min at room temperature for antigen retrieval. After washing with PBS, the sections were blocked in 10% goat serum in PBS at 4 °C overnight. The sections were then incubated with anti-rHlVgR IgG that had been diluted in 5% goat serum in PBS (1:10) for 1 h at 37 °C. Subsequently, the sections were washed with PBS and then reacted with Alexa Fluor® 594 goat anti-mouse IgG (H + L) (1:4000; Life Technologies) for 1 h at 37 °C. The sections were then mounted in ProLong® Gold Antifade Mountant with DAPI (Life Technologies) after washing with PBS. Fluorescent signals were observed under a TCS SP5 confocal laser microscope (Leica Microsystems, Wetzlar, Germany), and the signals were displayed in yellow color to allow clearer detection of signals.
Histological observation of ovary in HlVgR-RNAi ticks
A 631-bp HlVgR fragment was amplified by PCR using T7 forward (5’-TAA TAC GAC TCA CTA TAG GCT GTC CCT CGG ATG ACT TCT CCT G-3’) and T7 reverse (5’-TAA TAC GAC TCA CTA TAG GTG CAG TTG CTC TCG TCC CGG CCG-3’) primers as described previously [17]. The PCR products were purified as described above and were then used to generate double-stranded RNA (dsRNA) using the T7 RiboMax™ Express RNAi System (Promega). Thirty-five female ticks fixed on a glass slide with adhesive tape were injected with 0.5 μl of 2 μg/μl (1 μg/tick) of the dsRNA of HlVgR or firefly luciferase as described previously [12]. Injected ticks were kept at 25 °C for 18 h in an incubator and then fed on rabbits. Twenty and fifteen engorged ticks of control and HlVgR RNAi groups, respectively, were collected from the rabbits and their body weight was measured. Statistical significance was assessed by the Student t-test. The ticks were then incubated in a chamber at 25 °C for monitoring of oviposition. The female ticks were dissected to collect 6 ovaries at the engorgement (5–7 days after attachment) and 3 ovaries at 5 days after engorgement. The ovaries immersed in PBS were morphologically observed under a SZX16 stereomicroscope (Olympus, Tokyo, Japan) with a DP21 camera (Olympus). After observation, the ovaries were immersed into 4% paraformaldehyde-0.1% glutaraldehyde in PBS at 4 °C, overnight, and then subjected to the preparation of frozen sections as described above. The frozen sections were stained with hematoxylin and eosin (Muto Pure Chemicals). The stained sections were then observed under a BZ-9000 microscope (Keyence). Moreover, total RNA was purified from the remaining ticks in each group and used to synthesize cDNA as described above. The gene silencing of HlVgR was assessed by real-time PCR using a 7300 Real-Time PCR System (Life Technologies) and THUNDERBIRD® SYBR qPCR Mix (Toyobo) as described above. The HlVgR expression was normalized to the HlP0 mRNA expression. The expressions for control group were set to 1.0, and relative expressions for HlVgR RNAi group were calculated. The data represented the mean ± SD of triplicate samples. The differences between control and HlVgR RNAi groups were compared using the Welchʼs t-test. Statistical significance was set at P < 0.01.
Abbreviations
- AhVgR:
-
Amblyomma hebraeum vitellogenin receptor
- dsRNA:
-
double-stranded RNA
- DvVgR:
-
Dermacentor variabilis vitellogenin receptor
- HlVgR:
-
Haemaphysalis longicornis vitellogenin receptor
- LDLR:
-
low-density lipoprotein receptor
- PBS:
-
phosphate-buffered saline
- RaVgR:
-
Rhipicephalus appendiculatus vitellogenin receptor
- RmVgR:
-
Rhipicephalus microplus vitellogenin receptor
- RNAi:
-
RNA interference
- SD:
-
standard deviation
- TOR:
-
target of rapamycin
- Vg:
-
Vitellogenin
- VgR:
-
Vitellogenin receptor
- 4dAE:
-
4 days after engorgement
- 0dAO:
-
the beginning of oviposition
- 10dAO:
-
10 days after the beginning of oviposition
References
Thompson DM, Khalil SM, Jeffers LA, Ananthapadmanaban U, Sonenshine DE, Mitchell RD, et al. In vivo role of 20-hydroxyecdysone in the regulation of the vitellogenin mRNA and egg development in the American dog tick, Dermacentor variabilis (Say). J Insect Physiol. 2005;51:1105–16.
Friesen KJ, Reuben Kaufman W. Quantification of vitellogenesis and its control by 20-hydroxyecdysone in the ixodid tick, Amblyomma hebraeum. J Insect Physiol. 2002;48:773–82.
James AM, Zhu XX, Oliver JH. Vitellogenin and ecdysteroid titers in Ixodes scapularis during vitellogenesis. J Parasitol. 1997;83:559–63.
Sankhon N, Lockey T, Rosell RC, Rothschild M, Coons L. Effect of methoprene and 20-hydroxyecdysone on vitellogenin production in cultured fat bodies and backless explants from unfed female Dermacentor variabilis. J Insect Physiol. 1999;45:755–61.
Ogihara K, Horigane M, Nakajima Y, Moribayashi A, Taylor D. Ecdysteroid hormone titer and its relationship to vitellogenesis in the soft tick, Ornithodoros moubata (Acari: Argasidae). Gen Comp Endocrinol. 2007;150:371–80.
Friesen KJ, Kaufman WR. Effects of 20-hydroxyecdysone and other hormones on egg development, and identification of a vitellin-binding protein in the ovary of the tick, Amblyomma hebraeum. J Insect Physiol. 2004;50:519–29.
Balashov YS. Bloodsucking ticks (Ixodoidea)-vectors of disease in man and animals (English translation). Misc Publ Entomol Soc Am. 1972;8:161–376.
Coons LB, Tarnowski B, Ourth DD. Rhipicephalus sanguinius: localization of vitellogenin synthesis by immunological methods and electron microscopy. Exp Parasitol. 1982;54:331–9.
Rosell-Davis R, Coons LB. Relationship between feeding, mating, vitellogenin production and vitellogenesis in the tick Dermacentor variabilis. Exp Appl Acarol. 1989;7:95–105.
Coons LB, Lamoreaux WJ, Rosell-Davis R, Tarnowski BI. Onset of vitellogenin production and vitellogenesis, and their relationship to changes in the midgut epithelium and oocytes in the tick Dermacentor variabilis. Exp Appl Acarol. 1989;6:291–305.
Boldbaatar D, Battur B, Umemiya-Shirafuji R, Liao M, Tanaka T, Fujisaki K. GATA transcription, translation and regulation in Haemaphysalis longicornis tick: analysis of the cDNA and an essential role for vitellogenesis. Insect Biochem Mol Biol. 2010;40:49–57.
Umemiya-Shirafuji R, Boldbaatar D, Liao M, Battur B, Rahman MM, Kuboki T, et al. Target of rapamycin (TOR) controls vitellogenesis via activation of the S6 kinase in the fat body of the tick, Haemaphysalis longicornis. Int J Parasitol. 2012;42:991–8.
Umemiya-Shirafuji R, Tanaka T, Boldbaatar D, Tanaka T, Fujisaki K. Akt is an essential player in regulating cell/organ growth at the adult stage in the hard tick Haemaphysalis longicornis. Insect Biochem Mol Biol. 2012;42:164–73.
Ogihara MH, Taylor D. Female reproductive system. In: Sonenshine DE, Roe RM, editors. Biology of ticks, vol. 1. 2nd ed. New York: Oxford University Press; 2014. p. 449–83.
Smith AD, Reuben Kaufman W. Molecular characterization of the vitellogenin receptor from the tick, Amblyomma hebraeum (Acari: Ixodidae). Insect Biochem Mol Biol. 2013;43:1133–41.
Mitchell RD, Ross E, Osgood C, Sonenshine DE, Donohue KV, Khalil SM, et al. Molecular characterization, tissue-specific expression and RNAi knockdown of the first vitellogenin receptor from a tick. Insect Biochem Mol Biol. 2007;37:375–88.
Boldbaatar D, Battsetseg B, Matsuo T, Hatta T, Umemiya-Shirafuji R, Xuan X, et al. Tick vitellogenin receptor reveals critical role in oocyte development and transovarial transmission of Babesia parasite. Biochem Cell Biol. 2008;86:331–44.
Seixas A, Alzugaray MF, Tirloni L, Parizi LF, Pinto AFM, Githaka NW, et al. Expression profile of Rhipicephalus microplus vitellogenin receptor during oogenesis. Ticks Tick Borne Dis. 2018;9:72–81.
Apanaskevich DA, Oliver JH. Life cycles and natural history of ticks. In: Sonenshine DE, Roe RM, editors. Biology of ticks, vol. 1. 2nd ed. New York: Oxford University Press; 2014. p. 59–73.
Mihara R, Umemiya-Shirafuji R, Abe Y, Matsuo T, Horiuchi N, Kawano S, et al. The development of oocytes in the ovary of a parthenogenetic tick, Haemaphysalis longicornis. Parasitol Int. 2018;67:465–71.
Schonbaum CP, Perrino JJ, Mahowald AP. Regulation of the vitellogenin receptor during Drosophila melanogaster oogenesis. Mol Biol Cell. 2000;11:511–21.
Snigirevskaya ES, Sappington TW, Raikhel AS. Internalization and recycling of vitellogenin receptor in the mosquito oocyte. Cell Tissue Res. 1997;290:175–83.
James AM, Oliver JH. Vitellogenin concentrations in the haemolymph and ovaries of Ixodes scapularis ticks during vitellogenesis. Exp Appl Acarol. 1996;20:639–47.
Yang X, Yu Z, He Y, Xu X, Gao Z, Wang H, et al. Purification of vitellin and dynamics of vitellogenesis in the parthenogenetic tick Haemaphysalis longicornis (Acari: Ixodidae). Exp Appl Acarol. 2015;65:377–88.
Rosell R, Coons LB. Determination of vitellogenin titer in the hemolymph of Dermacentor variabilis (Acarina: Ixodidae) using an indirect enzyme-linked immunosorbent assay. J Med Entomol. 1991;28:41–4.
de Oliveira PR, Mathias MI, Bechara GH. Vitellogenesis in the tick Amblyomma triste (Koch, 1844) (Acari: Ixodidae) role for pedicel cells. Vet Parasitol. 2007;143:134–9.
Denardi SE, Bechara GH, Oliveira PR, Nunes ET, Saito KC, Mathias MI. Morphological characterization of the ovary and vitellogenesis dynamics in the tick Amblyomma cajennense (Acari: Ixodidae). Vet Parasitol. 2004;125:379–95.
Boldbaatar D, Umemiya-Shirafuji R, Liao M, Tanaka T, Xuan X, Fujisaki K. Multiple vitellogenins from the Haemaphysalis longicornis tick are crucial for ovarian development. J Insect Physiol. 2010;56:1587–98.
Thompson DM, Khalil SM, Jeffers LA, Sonenshine DE, Mitchell RD, Osgood CJ, et al. Sequence and the developmental and tissue-specific regulation of the first complete vitellogenin messenger RNA from ticks responsible for heme sequestration. Insect Biochem Mol Biol. 2007;37:363–74.
Khalil SM, Donohue KV, Thompson DM, Jeffers LA, Ananthapadmanaban U, Sonenshine DE, et al. Full-length sequence, regulation and developmental studies of a second vitellogenin gene from the American dog tick, Dermacentor variabilis. J Insect Physiol. 2011;57:400–8.
Smith AD, Kaufman WR. Molecular characterization of two vitellogenin genes from the tick, Amblyomma hebraeum (Acari: Ixodidae). Ticks Tick Borne Dis. 2014;5:821–33.
Ramírez Rodríguez PB, Rosario Cruz R, Domínguez García DI, Hernández Gutiérrez R, Lagunes Quintanilla RE, Ortuño Sahagún D, et al. Identification of immunogenic proteins from ovarian tissue and recognized in larval extracts of Rhipicephalus (Boophilus) microplus, through an immunoproteomic approach. Exp Parasitol. 2016;170:227–35.
Antunes S, Rosa C, Couto J, Ferrolho J, Domingos A. Deciphering Babesia-vector interactions. Front Cell Infect Microbiol. 2017;7:1–8.
Sauer JR, McSwain JL, Essenberg RC. Cell membrane receptors and regulation of cell function in ticks and blood-sucking insects. Int J Parasitol. 1994;24:33–52.
Fujisaki K. Development of acquired resistance precipitating antibody in rabbits experimentally infested with females of Haemaphysalis longicornis (Ixodoidea: Ixodidae). Natl Inst Anim Health Q (Tokyo). 1978;18:27–38.
Umemiya-Shirafuji R, Galay RL, Maeda H, Kawano S, Tanaka T, Fukumoto S, et al. Expression analysis of autophagy-related genes in the hard tick Haemaphysalis longicornis. Vet Parasitol. 2014;201:169–75.
Umemiya R, Matsuo T, Hatta T, Sakakibara S, Boldbaatar D, Fujisaki K. Cloning and characterization of an autophagy-related gene, ATG12, from the three-host tick Haemaphysalis longicornis. Insect Biochem Mol Biol. 2007;37:975–84.
Acknowledgements
We thank Dr. Yasuyuki Abe (Prefectural University of Hiroshima) for assistance with methodology and Dr. Damdinsuren Boldbaatar (Mongolian University of Life Sciences) for advice on the manuscript.
Funding
This work was supported by a Grant-in-Aid for Young Scientists (B) from the Japan Society for the Promotion of Science (16K18794) and the Akiyama Life Science Foundation. This work was partially supported by The Japan Agency for Medical Research and Development (AMED)/The Japan International Cooperation Agency (JICA), the Science and Technology Research Partnership for Sustainable Development (SATREPS), and the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan as a project for Joint Usage/Research Center.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors’ contributions
RUS, RM, and KF designed this study. RUS and RM performed the experiments, data analysis, and drafted the manuscript. HS participated in the design, coordination, and revision of the manuscript. All authors actively contributed to the interpretation of the findings. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The care and use of experimental animals in this study were approved by the Animal Care and Use Committee of Obihiro University of Agriculture and Veterinary Medicine (approval no. 28-35).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Umemiya-Shirafuji, R., Mihara, R., Fujisaki, K. et al. Intracellular localization of vitellogenin receptor mRNA and protein during oogenesis of a parthenogenetic tick, Haemaphysalis longicornis. Parasites Vectors 12, 205 (2019). https://doi.org/10.1186/s13071-019-3469-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3469-9