
Abstract
Background
Schistosomiasis is neglected tropical parasitic disease affecting both humans and animals. Due to the human health impact, population genetic studies have focused on the three main human-infecting schistosome species: Schistosoma mansoni, S. haematobium and S. japonicum. Here we present novel data on the population genetic structure of Schistosoma bovis, a highly widespread and prevalent schistosome infecting ruminants, and therefore of veterinary importance.
Methods
Adult S. bovis were sampled in the two main abattoirs of Cameroon (Yaoundé and Douala). Twenty-two cows originating from four distinct localities were sampled and a total of 218 parasites were recovered. All parasites were genotyped using a panel of 14 microsatellite markers and a sub-sample of 91 parasites were sequenced and characterized with the mitochondrial (cox1) and nuclear (ITS) genetic markers.
Results
No significant difference in allelic richness, heterozygosity, nucleotide diversity and haplotype diversity was observed between the populations. Additionally, no strong genetic structure was observed at the country scale. Our data also show that S. bovis is more polymorphic than its sister species, S. haematobium, and that the haplotype diversity is similar to that of S. mansoni while the nucleotide diversity does not significantly differ from that of S. haematobium. The resulting negative Tajima’s D* and Fu and Li’s D* indices could be a signature of population demographic expansion. No S. haematobium/S. bovis hybrids were observed in our populations, thus all samples were considered as pure S. bovis.
Conclusions
This study provides novel insights into genetic diversity and population genetic structure of S. bovis. No strong genetic structure was observed at the country scale but some genetic indices could be associated as a signature of population demographic expansion.
Similar content being viewed by others
Background
Schistosomiasis is a chronic and debilitating water-borne parasitic disease of humans and animals. It poses serious public health and veterinary concerns worldwide particularly in tropical and subtropical areas. In humans, the disease affects nearly 218 million people in 78 countries with an estimated 200,000 deaths per year. Almost 95% of the infected people live in Africa [1]. Six species of schistosome are known to infect humans: Schistosoma mansoni, S. haematobium, S. intercalatum and S. guineensis in Africa, and S. mekongi and S. japonicum in Asia [2]. Animals can be infected by 19 species of schistosomes of which five receive particular attention because of the associated infection severity observed in domestic ruminants in Africa (S. mattheei, S. bovis, S. curassoni) and in Asia (S. japonicum and S. mekongi) [3]. Regarding livestock infections, it has been estimated that 165 million cattle are infected worldwide [4]. Although human schistosomiasis is well monitored in most countries, bovine schistosomiasis receives negligible attention in comparison. In particular, the repartition, prevalence, intensity and transmission dynamics of livestock infections are poorly documented.
Environmental changes due to natural phenomena or human activities particularly agricultural practices associated with dam construction, can have a significant impact on the epidemiology and distribution of human and animal schistosomiasis and may also lead to interspecies hybridization events [5]. For instance, the creation of the Diama Dam on the Senegal River has created a more stable flow of water, creating irrigation canals, rice-growing areas and water points for animals, leading to the emergence of new human and livestock schistosomiasis outbreaks [6, 7]. In Cameroon, creation of the Lagdo Dam on the Benoue River also increased the prevalence of human schistosomiasis [8] and in the same way, the deforestation of the Loum rainforest allowed the establishment of B. truncatus, the intermediate host of S. haematobium thus allowing its introgressive hybridization with S. guineensis (formerly known as S. intercalatum) [9, 10]. Seasonal changes in cattle rearing methods in Africa (transhumance, nomadism) are important in the movement of domestic livestock, so animals can come into contact with multiple potential schistosomiasis transmission sites. Hybrids are known to exist between species that infect humans but also between those that have different hosts such as animals and humans [3]. The latter hybrid forms are particularly worrying because they theoretically could be enabling zoonotic transmission.
In Cameroon, out of 23 million inhabitants, there are five million people at risk of schistosomiasis, of which two million are infected [11] with three schistosome species [S. haematobium, S. mansoni and S. guineensis (formerly known as S. intercalatum)] involved in human infections, creating a considerable human health concern [11]. Urogenital schistosomiasis, caused by S. haematobium, is endemic in northern Cameroon and other foci are located in the south of the country in Kékem, Loum, Barombi-kotto, Kumba and Marumba [12,13,14]. Limited data are available concerning bovine schistosomiasis. In 1990, the prevalence of ruminant schistosomiasis in northern Cameroon (Garoua and Maroua cities), was recorded using egg morphological diagnostics, as 79.5 and 10.03% for S. bovis and S. curassoni, respectively [15]. Since several Schistosoma species are endemic to Cameroon, interspecific mating between different species have been reported between S. haematobium × S. guineensis and also between S. haematobium × S. mansoni [16, 17] but there are no data on S. haematobium × S. bovis hybrids. Schistosoma haematobium is a human parasite living in the veins around the urinary bladder while S. bovis is a livestock parasite living in the mesenteric veins around the gut. S. haematobium × S. bovis hybrids are largely present in West Africa, including Senegal, Mali, Benin and Niger [3, 18]. This latter hybrid has also recently emerged in the south of Europe [19,20,21].
Here, our aim is to document how populations of S. bovis are structured in Cameroon at a large geographical scale. We used genetic approaches, including both mitochondrial (cox1) and nuclear markers (ITS and microsatellites), first to characterize the population genetic structure of S. bovis and secondly to identify potential S. haematobium × S. bovis hybrids in Cameroon. Additionally, we took advantage of the fact that the microsatellite markers used in this study are informative for both S. haematobium and S. bovis to compare the genetic diversity between these two sister species.
Methods
Parasite sampling from cattle
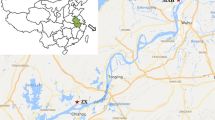
Authorization for parasite sampling was obtained from the SODEPA in Cameroon (Société de Développement et d’Exploitation des Productions Agricoles). In Cameroon the majority of cattle breeding areas are situated in the north of the country (Fig. 1) and animals are transported to the main cities to be slaughtered. The sampling was done in the two main abattoirs situated in the south of Cameroon (i.e. Yaoundé and Douala). Before sampling, the stockbreeder was questioned to identify the provenance of the cattle. Only those which had their origin identified were included in the present study. In total, 480 cows (Table 1) originating from four distinct localities along a linear north-to-south transect in the eastern part of the country were checked for S. bovis infection (Fig. 1). Maroua is at the extreme north of the country and Garoua, Ngaoundéré and Bertoua are 175, 377 and 672 km south of Maroua, respectively.

Map of the areas from where the cows originated: Maroua, Garoua, Ngaoundéré and Bertoua. The different proportions of cattle breeding in each area are shown as a percentage [35]
Upon slaughter, the mesenteric vessels of each cow were visually inspected for schistosome parasites. The observed adult schistosomes were recovered using forceps and washed using saline solution. Adult pairs were gently separated into males and females while alive before being individually stored in 95% ethanol for subsequent genetic analyses.
Molecular methods
Total genomic DNA from a total of 218 individual schistosomes (111 males and 107 females) from 22 cows were individually extracted using the E.Z.N.A. tissue DNA extraction kit (Omega Bio-tek, Norcross, USA). All specimens were genotyped using a panel of microsatellites markers for population genetic analysis. A subsample of 91 individuals (46 males and 45 females) were further molecularly characterized using both the nuclear ITS and mitochondrial cox1 markers to clarify species and hybrid status and investigate mitochondrial genetic diversity. The details of this subsample is presented in Additional file 1: Table S1.
Microsatellite genotyping
Individual samples were genotyped with a set of microsatellites initially developed for S. haematobium by Webster et al. [22]; however, S. haematobium and S. bovis are closely related sister species so the microsatellites were known to cross-react with S. bovis (Webster B, personal communication). Of the 18 microsatellites tested, 14 successfully amplified on our S. bovis samples and were thus used in the present study. The remaining four markers (C131, Sh7, Sh10 and Sh8) succeeded on less than 20% of the specimens and were thus discarded from our dataset. Microsatellite amplifications were performed using the Qiagen® Multiplex PCR kit, (Qiagen, Hilden, Germany). The forward primers were fluorescently labeled using 6-FAM, VIC, NED and PET dyes (Applied Biosystems, Foster City, USA) as per [22]. PCRs were carried out according to the manufacturer’s standard microsatellite amplification protocol except for the final volume of 10 μl including 2.5 μl of DNA template. Thermal cycling was performed with an initial hot-start activation of 15 min at 95 °C, followed by 30 cycles of 94 °C for 30 s, 56 °C for 90 s and 72 °C for 60 s, with a final extension at 60 °C for 30 min. Microsatellite reactions were performed using a TECNE TC-Plus Thermal Cycler (Cole-Palmer, Stone, UK). The microsatellite reactions were sent to Genoscreen (Lille, France) for genotype determination. Peak calling and genotype determination was done using GeneMarker Software. We used automatic size determination of the allele size using the Fragment Animal Analysis option of Genemarker Software with the GS500 size standard. All of the allele determination was double checked by visual inspection and the microsatellite matrix was exported as a data spreadsheet (Additional file 2: Table S2).
cox1 and ITS amplification and sequencing
The partial cox1 and ITS gene regions were amplified from the DNA samples using primers defined by Lockyer et al. [23] and Barber et al. [24], respectively. PCRs were carried out in a final reaction volume of 25 μl, comprising 2 μl of DNA template, 5 μl 5× Colorless GoTaq® Flexi Buffer, 1.5 μl of MgCl2, 5 μl of dNTP (10 mM), 1 μl of each primer (10 μM) and 0.2 μl of Taq polymerase (Go taq®G2 Hot Start Polymerase). Cycling conditions were the same for both genes: 3 min at 95 °C followed by 35 cycles of 95 °C for 40 s, 52 °C for 40 s and 72 °C for 70 s, with an extension of 4 min at 72 °C. After amplification, 4.5 μl of the individual PCR products were stained with ethidium bromide, loaded on a 1.5% agarose gel and visually inspected to check the size and the quality of the amplicons (1100–1200 bp). All positive DNA products were then purified and sequenced in both directions (i.e. using both the forward and the reverse primers) on an Applied Biosystems Genetic Analyser at Genoscreen.
Population structure and genetic diversity
Microsatellites
The genetic diversity of the schistosomes from each origin, defined as populations, was assessed by computing the expected heterozygosity (He), number of alleles (A), allelic richness (Ar) and Fis at each microsatellite locus using FSTAT v.2.9.3.2. [25]. These parameters were compared between the populations using the pairwise Friedman rank tests. The genetic differentiation between sites was assessed using pairwise Fst according to Weir & Cockerham [26] using FSTAT v.2.9.3.2. Next, we checked for potential genetic structure among individuals using a principal components analysis (PCA) as implemented in the Genetix software. We also performed an analysis of molecular variance (AMOVA) to evaluate the partitioning of the overall genetic variance among individuals according to three hierarchical levels: (i) within host; (ii) among hosts within the site of origin; and (iii) among sites. The AMOVA was performed using Arlequin software v.3.5 [27]. Finally, we determined the uppermost level of genetic structure of all individuals using the Bayesian clustering approach implemented in the Structure software [28]. In particular, we tested the number of clusters from K = 1 to K = 10. Three runs were computed for each K each of which consisted of 106 iterations after a ‘burn-in’ period of 250,000 iterations, using an admixture model and the other parameters were set by default. The log likelihood for each K was then averaged over the three runs using the corrsieve package in R. The ΔK-values were then computed in R to determine the most likely K number among the K tested as described in Evanno et al. [29]. Two K-values (K = 2 and K = 4) were identified as the most likely number of genetic clusters. For each of these two K-values, an additional 10 runs were computed using 106 iterations and setting the same initial parameters as those described previously. The probability for each individual to belong to each K was then averaged over the 10 runs (Q values). These Q values were then graphically represented using Clumpp [30] and the Distruct [31] software.
Finally, we took advantage of the fact that the microsatellite markers used in this study are informative for both S. haematobium and S. bovis to compare the genetic diversity between the two species based upon the 14 shared markers. To do so, we used a raw dataset from a previous article focusing on S. haematobium [22]. This raw dataset includes individual genotypes using 16 microsatellites (including the 14 markers used in this study) for 50 S. haematobium miracidia larvae from both Niger and Zanzibar [22]. Both, the allelic richness and the genetic diversity at each microsatellite locus were estimated using FSTAT v.2.9.3.2. for the two species and then compared using pairwise Wilcoxon rank tests.
ITS and cox1
The sequences were checked for possible sequencing errors and concatenated using Sequencher software v.4.5. The resulting cleaned sequences were next aligned using Bioedit v.7.0.9 and ClustalW software. Species assignation was done by comparing ITS and cox1 sequences to the GenBank nucleotide database. Concerning ITS, because this gene differs at 5 mutation points between S. haematobium and S. bovis, the sequence chromatograms were carefully checked at these mutation sites to identify possible heterogenous ITS sequences [32]. For the mitochondrial cox1 data, polymorphism indices (haplotype, nucleotide diversities, Tajima’s D* and Fu and Li’s D* statistics) were calculated using DnaSP v.6.0 software [33]. Significance of the genetic structuring patterns were tested using AMOVA [27]. The best-fit model of evolution (HKY+G) was determined using the maximum likelihood value implemented in MEGA v.6.0.6 [34]. A phylogenetic tree was constructed using MEGA v.6.0.6 software using a maximum likelihood method. The support for tree branching was based on 500 bootstrap replicates and rooted with Schistosoma haematobium (AY157209.1) as the outgroup. All sequences were uploaded to GenBank (accession numbers MH647122-MH647179).
Results
Parasitological survey
Among the 480 cows inspected, 94 were found to be positive (19.5%) for adult schistosome worms. Depending on the animals’ origin, the prevalence varied from 13.5 to 37.5% although there was no significant difference (χ2 = 4.8, df = 3, P = 0.18; Table 1).
Microsatellite analysis
All the computed genetic diversity indexes (He, A, Ar and Fis) are presented in Table 2. No difference between localities was observed for He (χ2 = 4.036, df = 3, P = 0.258), Ar (χ2 = 2.66, df = 3, P = 0.45) and Fis (χ2 = 3.6, df = 3, P = 0.308). The only statistical difference was observed for the numbers of allele A (χ2 = 17.65, df = 3, P = 0.001), which could be attributed to the sampling size disequilibrium between the sites, from n = 28 in Bertoua to n = 71 samples in Garoua. The AMOVA analysis revealed that most of the genetic variation (98.09%) is found within animals and not between animals sampled (Table 3).
Among the different K-values tested, K = 2 and K = 4 displayed maximal likelihood values suggesting that the individuals are grouped into 2 or 4 clusters (ΔK; Additional file 3: Figure S1). However, no apparent genetic structure was observed among the samples. Indeed, all individuals displayed a genetic background that could not have been clearly attributed to any individual genetic group (population) or to any of the sampling areas (Fig. 2).

Structure bar plots obtained showing the lack of population structure of S. bovis from the 4 sampling areas in Cameroon for K = 2 and K = 4
The data showed that despite the large geographical scale of the sampled cows there is no, or negligible, genetic structure between the different schistosome populations. This is also illustrated by the pairwise Fst values computed between sites that do not exceed 0.5% (Table 4). Similarly, the first two axes of the PCA (accounting for 38.77 and 32.29% of the total variation for principal components 1 and 2, respectively) showed very weak structuration among the four populations sampled (Fig. 3).

Principal components analysis (PCA) plot. Each parasite is represented by a dot and the color label corresponds to their population origin. A total of 28 variables (14 microsatellites × 2 alleles) were used in the analysis. The first and the second axis explained 38.77% and 32.29% of the genetic variation, respectively
Interspecific comparisons
Table 5 presents the allelic richness and gene diversity for S. bovis from this study and S. haematobium from [22]. The two S. haematobium populations (Niger and Zanzibar) presented both lower allelic richness and lower diversity than the whole S. bovis population from Cameroon. The difference was statistically significant for allelic richness only (Wilcoxon rank test, P < 0.05).
Sequence analyses
Only one ITS genotype was identified among the 91 screened schistosomes and this was identified as S. bovis. No double peak was observed in any sequence at the polymorphic sites between S. bovis and S. haematobium. All the cox1 sequences were also identified as S. bovis. Thus, according to these two markers all the schistosomes analyzed were considered as pure S. bovis. A total of 58 cox1 haplotypes (accession numbers MH647122-MH647179) were identified. The haplotype diversity (± SD) and nucleotide diversity (± SD) were 0.961 ± 0.012 and 0.00602 ± 0.00043, respectively. No difference in haplotype and nucleotide diversity was observed according to the sampling site (Table 6). Both Tajima’s D* (-2.24; P < 0.01) and Fu and Li’s D* (-4.61; P < 0.02) statistics were significantly negative.
AMOVA analysis revealed that 88.83% of the genetic variation can be explained by the variation within animals whereas the remaining is due to variation among animals within sites (8.81%) or among sites (2.36%) (Table 3), which corroborates the results obtained from the microsatellite dataset. The absence of any genetic structuring with regard to the area of origin is confirmed by no or very low branch support and significant mixing of the populations on the phylogenetic tree (Additional file 4: Figure S2).
Discussion
The four localities from which animals were screened in this study belong to the northern region of Cameroon which possesses 90% of Cameroon’s cattle herds (Fig. 1) [35]. Other intensive cattle breeding areas exist in the west and north-west regions; however, animals were not obtained from these regions. On average, we found that 19.5% of the inspected cows were infected (range 13.5–37.5%). This prevalence is much lower than that found by Ousseni in 1990 [15]. Such a difference might be at least partly explained by non-exclusive hypothetical factors including the slaughter season, or the age and welfare of the inspected animals. Indeed, all these parameters are known to influence infection prevalence in cows [36]. Unfortunately, we lack information to test these hypotheses.
The sequence data indicate that all the infections were of pure S. bovis and we did not find any evidence of interspecific hybridization, consistent with the fact that S. haematobium × S. bovis hybrids have yet to be found in cattle [32]. However, just like Webster et al. [32], we did not inspect the veins bordering the urinary bladder of the cows, and so any potential S. haematobium or hybrid schistosomes may have been missed. The presence of S. haematobium × S. bovis hybrids in humans have been suspected in the northern region of Cameroon [37]. These data were based on an ITS2-RFLP molecular approach to identify hybrid profiles between S. haematobium and other species from the S. haematobium group; however, the resolution of this method is not high enough to distinguish S. bovis, S. mattheei, S. curassoni and S. guineensis. The authors hypothesized that these hybrids were S. haematobium × S. bovis hybrids, but because S. guineensis is also present in Cameroon and the ITS is identical for S. guineensis and S. bovis, the nature of these hybrids remains unclear [10, 17].
Both the mitochondrial (cox1) and nuclear markers (microsatellites) indicated that most of the genetic variation observed among parasites is observed within and not between sampled animals. The factors influencing the genetic diversity of schistosomes were nicely depicted in the S. mansoni-Rattus rattus system on an island of Guadeloupe (West Indies) [38]. In particular, co-infections by multiple genotypes released by intermediate host snails, the dispersal ability of larval forms of different genotypes, and the movement/dispersion of the definitive host all result in important genetic diversity at the infrapopulation level. In this regard, it has been proposed that the definitive hosts act as “genetic mixing bowls” for the parasites [39]. Because this study is the first on S. bovis, only interspecific comparison with closely related species using relevant comparable markers can be performed. Previous studies using cox1 gene sequencing show a distinct pattern of genetic diversity according to the schistosome species concerned. At the scale of the African continent, the haplotype diversity (h) and the nucleotide diversity (Π) were 0.94 ± 0.0067 and 0.02553 [40] and 0.36 ± 0.014 and 0.00434 [41] for S. mansoni and S. haematobium, respectively. Thus the two parameters converge with S. mansoni having both higher haplotype and nucleotide diversity than S. haematobium. Our study shows that S. bovis is more polymorphic than its sister species S. haematobium (based on both nuclear and mitochondrial markers) but also shows that the haplotype diversity is similar to that of S. mansoni (h = 0.961 ± 0.012) while the nucleotide diversity does not significantly differ from that of S. haematobium (Π = 0.00602). Moreover, we found that the Tajima’s D* and Fu and Li’s D* computed on our S. bovis dataset were significantly negative, which could constitute a signature of population demographic expansion [42]. These results contrast with Tajima’s D* estimates recently computed on both S. mansoni and S. haematobium in Yemen (both estimates did not differ from zero [43]). This major demographic difference between schistosomes associated with cattle or humans might be explained by the absence of or very few anthelminthic treatments in animals. Indeed, the increasing pressure of praziquantel treatments fortunately strongly affects the demographic dynamics and the genetic composition of human schistosome populations [44, 45].
Whatever the method or the marker (mitochondrial or nuclear) used, we did not find any evidence for inter- or intrapopulation structuring despite considerable geographical distances separating the populations. This pattern clearly differs from that of S. mansoni for which populations are generally strongly genetically structured with high Fst values even at small geographical scales. Such strong genetic structuring has been evidenced in several countries including Kenya [46], Brazil [39], Uganda [47], Ethiopia [48] and Senegal [49]. Conversely, little is known on the genetic structuring of S. haematobium populations [50, 51]. Based on a phylogeographic approach performed at the African continent scale, Webster et al. [41] revealed that S. haematobium [52] is poorly structured compared to S. mansoni [51]. Several factors might explain the absence of genetic structure in S. bovis populations. First, as previously mentioned, the absence of anthelminthic treatments in cattle might foster the expansion of S. bovis populations. The effective population size for S. bovis is expected to be large enough to limit the effect of possible genetic drift and population differentiation. Secondly, host dispersal is known to promote parasite dispersion, which in turn might homogenize populations at the genetic level. Dispersion of S. bovis is mainly driven by cattle movements across the country through transhumance practices. Transhumance is a common practice where pastoralists move their livestock seasonally across different grazing lands [53]. While these movements are beneficial in terms of optimizing the use of land and other natural resources [54], they also may play a role in disease transmission [55] and have other significant regional or local social/economic impacts [56]. The specific movement patterns of animals in conjunction with environmental conditions (wet versus dry and cold versus hot) could result in significant changes in whether transmission of infectious diseases is prevented or facilitated by movements [57]. In Cameroon, transhumance occurs frequently between the drier northern and the humid southern regions. Indeed, livestock in this country are mainly assured by a group of pastoralists collectively called the Fulanis who practice transhumance as the pendular and seasonal movement of the herds and as their lifestyle. For that purpose, during the long dry seasons, these nomads move south with their herds to find more grazing sites and also for economic reasons. In fact, livestock farming in not routinely practiced in the south but is more common in the West and North West regions.
Conclusions
This study provides novel insights into the population genetics of S. bovis. No strong genetic structure was observed at the country scale but some genetic indices could be associated as a signature of population demographic expansion. More studies on genetic diversity and population genetic structuring of animal schistosomes are needed to infer the role of the definitive hosts on parasite gene flow.
Abbreviations
- ITS:
-
Internal transcribed spacer
- cox1:
-
Cytochrome c oxidase subunit 1
References
GBD 2016 Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the global burden of disease study 2016. Lancet. 2017;390:1151–210.
Rollinson D, Simpson AJG. The Biology of Schistosomes. From Genes to Latrines. London: Academic Press; 1987.
Leger E, Webster JP. Hybridizations within the genus Schistosoma: implications for evolution, epidemiology and control. Parasitology. 2017;144:65–80.
De Bont J, Vercruysse J. The epidemiology and control of cattle schistosomiasis. Parasitol Today. 1997;13:255–62.
King KC, Stelkens RB, Webster JP, Smith DF, Brockhurst MA. Hybridization in parasites: consequences for adaptive evolution, pathogenesis, and public health in a changing world. PLoS Pathog. 2015;11:e1005098.
Vercruysse J, Southgate VR, Rollinson D, De Clercq D, Sacko M, De Bont J, et al. Studies on transmission and schistosome interactions in Senegal, Mali and Zambia. Trop Geogr Med. 1994;46:220–6.
Diaw O, Vassiliades G, Thiongane Y, Seye M, Sarr Y, Diouf A. Extension des trématodoses du bétail après la construction des barrages dans le bassin du fleuve Sénégal. Rev Elev Med Vet Pays Trop. 1998;51:113–20.
Robert CF, Bouvier S, Rougemont A. Epidemiology of schistosomiasis in the riverine population of Lagdo Lake, northern Cameroon: mixed infections and ethnic factors. Trop Med Parasitol. 1989;40:153–8.
Southgate VR, van Wijk HB, Wright CA. Schistosomiasis at Loum, Cameroun; Schistosoma haematobium, S. intercalatum and their natural hybrid. Z Parasitenkd. 1976;49:145–59.
Webster BL, Tchuem Tchuente LA, Jourdane J, Southgate VR. The interaction of Schistosoma haematobium and S. guineensis in Cameroon. J Helminthol. 2005;79:193–7.
Tchuem Tchuente LA, Momo SC, Stothard JR, Rollinson D. Efficacy of praziquantel and reinfection patterns in single and mixed infection foci for intestinal and urogenital schistosomiasis in Cameroon. Acta Trop. 2013;128:275–83.
Tchuem Tchuente LA, Kamwa Ngassam RI, Sumo L, Ngassam P, Dongmo Noumedem C, Nzu DD, et al. Mapping of schistosomiasis and soil-transmitted helminthiasis in the regions of centre, east and west Cameroon. PLoS Negl Trop Dis. 2012;6:e1553.
Ratard R, Ekani MM, Greer GJ, Kouemeni L, Mimpfoundi R, Ngonseu E. Schistosomiasis in Cameroon. Parasitol Today. 1988;4:199.
Ratard RC, Kouemeni LE, Bessala MM, Ndamkou CN, Greer GJ, Spilsbury J, et al. Human schistosomiasis in Cameroon. I. Distribution of schistosomiasis. Am J Trop Med Hyg. 1990;42:561–72.
Ousseni S. Schistosomoses des ruminants domestiques au Cameroun septentrionale. Thèse medicine vétérinaire, University Cheikh Anta Diop, Dakar, Senegal; 1990.
Cunin P, Tchuem Tchuente LA, Poste B, Djibrilla K, Martin PM. Interactions between Schistosoma haematobium and Schistosoma mansoni in humans in north Cameroon. Tropical Med Int Health. 2003;8:1110–7.
Wright CA, Southgate VR, van Wijk HB, Moore PJ. Letter: Hybrids between Schistosoma haematobium and S. intercalatum in Cameroon. Trans R Soc Trop Med Hyg. 1974;68:413–4.
Huyse T, Van den Broeck F, Hellemans B, Volckaert FA, Polman K. Hybridisation between the two major African schistosome species of humans. Int J Parasitol. 2013;43:687–9.
Berry A, Paris L, Boissier J, Caumes E. Schistosomiasis screening of travelers to Corsica, France. Emerg Infect Dis. 2015;22:159.
Boissier J, Grech-Angelini S, Webster BL, Allienne JF, Huyse T, Mas-Coma S, et al. Outbreak of urogenital schistosomiasis in Corsica (France): an epidemiological case study. Lancet Infect Dis. 2016;16:971–9.
Ramalli L, Mulero S, Noel H, Chiappini JD, Vincent J, Barre-Cardi H, et al. Persistence of schistosomal transmission linked to the Cavu river in southern Corsica since 2013. Euro Surveill. 2018;23:1–4.
Webster BL, Rabone M, Pennance T, Emery AM, Allan F, Gouvras A, et al. Development of novel multiplex microsatellite polymerase chain reactions to enable high-throughput population genetic studies of Schistosoma haematobium. Parasit Vectors. 2015;8:432.
Lockyer AE, Olson PD, Ostergaard P, Rollinson D, Johnston DA, Attwood SW, et al. The phylogeny of the Schistosomatidae based on three genes with emphasis on the interrelationships of Schistosoma Weinland, 1858. Parasitology. 2003;126:203–24.
Barber KE, Mkoji GM, Loker ES. PCR-RFLP analysis of the ITS2 region to identify Schistosoma haematobium and S. bovis from Kenya. Am J Trop Med Hyg. 2000;62:434–40.
Goudet J, Perrin N, Waser P. Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol Ecol. 2002;11:1103–14.
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Francis RM. pophelper: an R package and web app to analyse and visualize population structure. Mol Ecol Resour. 2017;17:27–32.
Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–8.
Webster BL, Diaw OT, Seye MM, Webster JP, Rollinson D. Introgressive hybridization of Schistosoma haematobium group species in Senegal: species barrier breakdown between ruminant and human schistosomes. PLoS Negl Trop Dis. 2013;7:e2110.
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–302.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Bouba, M. Evolution et situation actuelle de l’élevage au Cameroun. Cameroon: Ministère de l’Elevage, des Pêches et des Industries Animales; 2005. www.oecd.org/fr/agriculture/politiques-agricoles/37472487.doc. Accessed 19 Jan 2019
Christensen NO, Mutani A, Frandsen F. A review of the biology and transmission ecology of African bovine species of the genus Schistosoma. Z Parasitenkd. 1983;69:551–70.
Luogbou Nzu DD, Netongo Masumbe P, Bayemi PH, Mbacham Fon W, Tchuem Tchuente LA. Detection of hybrid Schistosoma haematobium group species in Cameroon by PCR-RFLP of the second internal transcribed spacer (ITS-2). World J Pharmaceut Res. 2015;4:196–1980.
Theron A, Sire C, Rognon A, Prugnolle F, Durand P. Molecular ecology of Schistosoma mansoni transmission inferred from the genetic composition of larval and adult infrapopulations within intermediate and definitive hosts. Parasitology. 2004;129:571–85.
Thiele EA, Sorensen RE, Gazzinelli A, Minchella DJ. Genetic diversity and population structuring of Schistosoma mansoni in a Brazilian village. Int J Parasitol. 2008;38:389–99.
Webster BL, Culverwell CL, Khamis IS, Mohammed KA, Rollinson D, Stothard JR. DNA barcoding of Schistosoma haematobium on Zanzibar reveals substantial genetic diversity and two major phylogenetic groups. Acta Trop. 2013;128:206–17.
Webster BL, Emery AM, Webster JP, Gouvras A, Garba A, Diaw O, et al. Genetic diversity within Schistosoma haematobium: DNA barcoding reveals two distinct groups. PLoS Negl Trop Dis. 2012;6:e1882.
Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709.
Sady H, Al-Mekhlafi HM, Webster BL, Ngui R, Atroosh WM, Al-Delaimy AK, et al. New insights into the genetic diversity of Schistosoma mansoni and S. haematobium in Yemen. Parasit Vectors. 2015;8:544.
Gower CM, Gehre F, Marques SR, Lamberton PHL, Lwambo NJ, Webster JP. Phenotypic and genotypic monitoring of Schistosoma mansoni in Tanzanian schoolchildren five years into a preventative chemotherapy national control programme. Parasit Vectors. 2017;10:593.
French MD, Churcher TS, Basanez MG, Norton AJ, Lwambo NJ, Webster JP. Reductions in genetic diversity of Schistosoma mansoni populations under chemotherapeutic pressure: the effect of sampling approach and parasite population definition. Acta Trop. 2013;128:196–205.
Agola LE, Mburu DN, DeJong RJ, Mungai BN, Muluvi GM, Njagi EN, et al. Microsatellite typing reveals strong genetic structure of Schistosoma mansoni from localities in Kenya. Infect Genet Evol. 2006;6:484–90.
Stothard JR, Webster BL, Weber T, Nyakaana S, Webster JP, Kazibwe F, et al. Molecular epidemiology of Schistosoma mansoni in Uganda: DNA barcoding reveals substantial genetic diversity within Lake Albert and Lake Victoria populations. Parasitology. 2009;136:1813–24.
Aemero M, Boissier J, Climent D, Mone H, Mouahid G, Berhe N, et al. Genetic diversity, multiplicity of infection and population structure of Schistosoma mansoni isolates from human hosts in Ethiopia. BMC Genet. 2015;16:137.
Van den Broeck F, Maes GE, Larmuseau MH, Rollinson D, Sy I, Faye D, et al. Reconstructing colonization dynamics of the human parasite Schistosoma mansoni following anthropogenic environmental changes in northwest Senegal. PLoS Negl Trop Dis. 2015;9:e0003998.
Gower CM, Gabrielli AF, Sacko M, Dembele R, Golan R, Emery AM, et al. Population genetics of Schistosoma haematobium: development of novel microsatellite markers and their application to schistosomiasis control in Mali. Parasitology. 2011;138:978–94.
Gower CM, Gouvras AN, Lamberton PH, Deol A, Shrivastava J, Mutombo PN, et al. Population genetic structure of Schistosoma mansoni and Schistosoma haematobium from across six sub-Saharan African countries: implications for epidemiology, evolution and control. Acta Trop. 2013;128:261–74.
Webster BL, Webster JP, Gouvras AN, Garba A, Lamine MS, Diaw OT, et al. DNA ‘barcoding’ of Schistosoma mansoni across sub-Saharan Africa supports substantial within locality diversity and geographical separation of genotypes. Acta Trop. 2013;128:250–60.
Stenning DJ. Transhumance, migratory drift, migration; patterns of pastoral Fulani nomadism. J R Anthropol Inst Great Britain Ireland. 1957;87:57–73.
Moritz M, Hamilton IM, Chen YJ, Scholte P. Mobile pastoralists in the logone floodplain distribute themselves in an ideal free distribution. Curr Anthropol. 2014;55:115–22.
Spergser J, Macher K, Kargl M, Lysnyansky I, Rosengarten R. Emergence, re-emergence, spread and host species crossing of Mycoplasma bovis in the Austrian Alps caused by a single endemic strain. Vet Microbiol. 2013;164:299–306.
Perry BD, Rich KM. Poverty impacts of foot-and-mouth disease and the poverty reduction implications of its control. Vet Rec. 2007;160:238–41.
Rebaudet S, Sudre B, Faucher B, Piarroux R. Environmental determinants of cholera outbreaks in land Africa: a systematic review of main transmission foci and propagation routes. J Infect Dis. 2013;208:S46–54.
Acknowledgments
We thank Dr Bonnie Webster for constructive comments on the manuscript.
Funding
Not applicable.
Availability of data and materials
Data supporting the conclusions of this article are included within the article and its additional files. Sequence data are submitted to the GenBank database under the accession numbers MH647122-MH647179 and microsatellite data are provided in Additional file 2: Table S2. The datasets used in the present study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
JB and FFD-T conceived and designated the study. FFD-T, OR and JB wrote the manuscript. JB and OR performed computer analysis. AKS and AJ-F performed the molecular analyses. ANN and LAT-T improved the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Details of the schistosomes from different cows that were molecularly characterized using the nuclear ITS and mitochondrial cox1 markers. (DOCX 13 kb)
Additional file 2:
Table S2. Microsatellite dataset. (XLS 47 kb)
Additional file 3:
Figure S1. ΔK-values calculated by Evanno’s method detecting K = 2 and K = 4 subpopulations as the most genetically probable within the 14 loci analyzed. (DOCX 32 kb)
Additional file 4:
Figure S2. Mitochondrial cox1 maximum likelihood tree based of the S. bovis samples. The code for each sample corresponds to the sampling site-the animal number-the sex of the schistosome (M or F) and the parasite number. (DOCX 21 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Djuikwo-Teukeng, F.F., Kouam Simo, A., Allienne, JF. et al. Population genetic structure of Schistosoma bovis in Cameroon. Parasites Vectors 12, 56 (2019). https://doi.org/10.1186/s13071-019-3307-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3307-0