
Abstract
Background
Aedes aegypti is associated with dengue, yellow fever, chikungunya and Zika viruses. This vector is widespread in tropical and subtropical areas, and can also occur in temperate areas at higher latitudes. The geographical distribution of Ae. aegypti continues to spread due to human activities. This is the first study to examine the population genetic structure of this insect in El Salvador, Central America.
Methods
Aedes aegypti larvae were collected from six geographical regions of El Salvador: Sonsonate, San Salvador, Chalatenango, Usulután, San Miguel and Morazán. Larvae were raised into adults, identified and preserved. Two molecular markers, amplified fragment length polymorphism (AFLP) genotyping and mitochondrial DNA (mtDNA) cytochrome c oxidase subunit 1 (cox1) sequencing, were used to investigate population genetic structure.
Results
Structure analysis found two genetically distinct populations; one occurs predominantly in the north and west, and a mix of two populations occurs in the southeast of the country. Genetic distances ranged from 0.028 (2.8%) to 0.091 (9%), and an AMOVA analysis found 11% variation between populations. Mitochondrial DNA cox1 sequences produced a haplotype network which consisted of 3 haplogroups and 10 haplotypes. Haplogroup 1 had low haplotype and nucleotide diversity and was found in all six regions. Haplogroups 2 and 3 had higher haplotype and nucleotide diversity, and were less abundant; haplogroup 3 was found in only 3 of the six regions studied. Bottleneck tests were significant, suggesting that populations had undergone a recent bottleneck. A maximum likelihood tree, which combined samples from this study with available sequences in GenBank, suggested that two genetically divergent lineages had been introduced.
Conclusions
Relatively high genetic diversity was found in Ae. aegypti in El Salvador. The mtDNA sequences clustered into two lineages, as found in previous studies. Samples in El Salvador may be introduced from regions in North and South America where past eradication was not complete. Future study of genotypes in surrounding countries would provide a more complete picture of the movement and potential source of introductions of this vector. The distribution of the lineages and haplogroups may further our understanding of the epidemiology of Ae. aegypti associated vector borne diseases.
Similar content being viewed by others


Background
The mosquito Aedes aegypti is associated with yellow fever, dengue, chikungunya and Zika viruses. These diseases impact the health of millions globally each year. It is estimated that at least 50 million per year are infected with dengue, with inhabitants of 100 or more countries at risk of infection [1]. Aedes aegypti typically occurs in tropical and subtropical regions of the world, but can also occur in temperate regions at higher latitudes [2]. This mosquito continues to be transported around the globe with human movement and global commerce, exerting a major impact on public health.
Studies have examined genetic diversity of Ae. aegypti on a global scale [3,4,5,6,7]. In Africa, the region of origin, two forms of Ae. aegypti vary in their ecology and are considered subspecies. The sylvatic ancestral form of Ae. aegypti, Ae. aegypti formosus, resides primarily in forested environments and tree holes, and a second anthropophilic form, Ae. aegypti aegypti, occurs in urban areas in homes where water is stored [3, 7,8,9]. The evolutionary history of the vector is complex, yet studies suggest that mosquitoes have been moved from Africa to America and then from America to Asia, an idea supported by the finding that populations in America have higher genetic diversity than those in Asia [2, 5]. In addition, evidence suggests that different lineages have been transported out of East and West Africa [6, 10].
Regional studies have investigated the genetic diversity of introduced Ae. aegypti in numerous countries in the Western Hemisphere, Asia and Australia. A variety of molecular markers have been utilized in these studies including microsatellites and mitochondrial DNA. Studies have found evidence for one, two or three introduced clades of Ae. aegypti outside of their region of origin of Africa [11,12,13]. For example, collections of Ae. aegypti from the northeast coast of Mexico suggest two divergent lineages and two introductions [14]. In Brazil, evidence was found for at least two introduced lineages [15, 16]. Results from Argentina suggested three lineages of Ae. aegypti; low nucleotide diversity suggested passive dispersal between Argentine populations and those from adjacent countries [12]. In Bolivia, two divergent populations were found with low nucleotide diversity, suggesting a recent introduction or a small founding population [13]. Another source of population divergence in Ae. aegypti in the Western Hemisphere could be populations that persisted through eradication programs, which might reinvade surrounding regions [16,17,18]. In Brazil, population genetic studies suggested that after eradication, Ae. aegypti was later reintroduced [16]. In northern Brazil, Ae. aegypti were more closely related to those from Venezuela, where eradication did not occur. The former study along with a subsequent study in Brazil suggested that the Ae. aegypti populations currently found in Brazil are populations which were reintroduced in the country after eradication was achieved [16, 17]. Other studies found that once Ae. aegypti are introduced into a region, they could undergo genetic divergence due to environmental factors and geographical barriers [19, 20]. For example, populations of Ae. aegypti in Peru were found to be divergent on different sides of the Andes, possibly due to climatic differences and geographical isolation [19, 20].
In El Salvador, Central America, Ae. aegypti is the primary vector of dengue, chikungunya and Zika virus [21]. El Salvador is a relatively small country, with a Pacific coast and varied topography and climate. Aedes aegypti was previously eradicated from El Salvador, and from much of the Western Hemisphere, in the 1950s and 1960s [18, 22]. Similarly, in the countries surrounding El Salvador, eradication was also achieved: in Nicaragua by 1958, and in Guatemala and Honduras by 1959. El Salvador was declared eradicated by 1960, and Costa Rica was free of Ae. aegypti by 1961. However, in El Salvador, Ae. aegypti was reintroduced in a shipment of tires from the USA to San Salvador in 1965 [23]. By 1982, an epidemic of dengue was reported, and in 1983, 2867 cases were reported from throughout the country [24, 25]. The mosquito may additionally have been reintroduced through ports, human movement, or along transportation routes from nearby countries, or small pockets of mosquitoes may have survived in isolated refugia. Aedes aegypti is currently widespread through the country. In 2014, chikungunya cases were first reported in El Salvador, followed by the first reports of Zika in 2015 [21]. Previous studies have suggested that knowledge of the genetic variability of a vector could contribute to vector control and reducing cases of vector-borne disease [26]. For example, strains of Ae. aegypti can vary in vector competence and insecticide resistance, and thus respond differentially to control [26]. Little is known about the population genetic structure of Ae. aegypti in Central America. The objectives of this study were to examine the genetic variability of populations of Ae. aegypti in six regions of El Salvador, and to investigate the number of possible introductions and lineages of this insect in the country.
Methods
Study sites
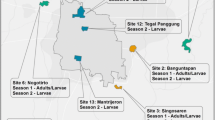
The study was conducted between May-August 2014 in El Salvador during the rainy season. Larvae of Ae. aegypti were collected in six departments of the country: Sonsonate, San Salvador, Chalatenango, Usulután, San Miguel and Morazán (Fig. 1). These six departments were located in three different regions of El Salvador. The western zone of the country (Sonsonate) has a port on the Pacific coast in Acajutla. The central zone consists of San Salvador and Chalatenango; San Salvador is the capital and Chalatenango is a sparsely populated mountainous region which borders Honduras. In the south and eastern parts of the country, collections were made in Usulután, San Miguel and Morazán (Fig. 1). Usulután borders the Pacific coast and includes the Bay of Jiquilisco and nature reserves. San Miguel is a hot, dry interior region of the country, located on the Pan American Highway. Finally, Morazán is a department located between San Miguel and the border of Honduras (Fig. 1). In each department, one or two neighborhoods were visited to collect samples, with at least two or three sites per neighborhood used to collect larvae (Table 1). For example, the samples collected in Sonsonate were from two neighborhoods, San Antonio and El Carmen, with three sampling sites within each neighborhood (Table 1). Sample sites within each neighborhood included barrels of standing water and outdoor sinks, commonly called ‘pilas’. The samples collected from each site in this study typically consisted of a few of the numerous larvae which were present in a pila or barrel. In addition, findings of a previous study found that the mean number of families represented per oviposition site for Ae. aegypti was 4.7 [27]. This previous research, along with the sampling design in the present study, reduced the chance that samples collected were siblings. After collection, larvae were placed in a glass jar for transport to the laboratory for rearing at the University of El Salvador.

Collection locations including the municipality and department where samples of Ae. aegypti were obtained in El Salvador
Rearing larvae and adult identification
Larvae were transported to the Center for Health Research (CENSALUD) at the University of El Salvador, in San Salvador. In the laboratory, larvae from each collection site were placed into plastic cups with distilled water and maintained in wire mesh cages (20 × 10 × 10 cm) until adults emerged. Each day, cages were checked for newly emerged adults, which were frozen and later identified to the species level based on morphology. Only adult females were used for this study.
DNA extraction
Adult females were used for DNA extraction, with at least 30 adult females individually extracted from each of the six regions. DNA extraction was completed with the Qiagen DNEasy® Blood and Tissue Extraction Kit (Qiagen, Venlo, Netherlands) following standard protocols [28], using an overnight incubation of the samples at 65 °C. The quantity of DNA in each sample was measured using the Qubit 2.0 fluorimeter Hs DNA kit (ThermoFisher, Waltham, MA, USA) and averaged 50 ng/μl.
Population genetic structure: amplified fragment length polymorphisms (AFLPs)
Amplified fragment length polymorphisms were produced for each mosquito using three primer combinations with procedures described by [29] and modified by Joyce et al. [30]. The three primer combinations used were the following: (i) MseI-CAT/EcoRI-ACG; (ii) MseI-CAC/EcoRI-ACT; and (iii) MseI-CAC/EcoRI-ACA. Details of AFLP reactions are described in Joyce et al. [30]. Initial DNA template for reactions used 5 μl (~200 ng DNA). Prior to capillary electrophoresis, 0.4 μl of GeneScan Liz 500 size standard and 0.9 μl of HiDi formamide (ThermoFisher, Waltham, MA, USA) were added to 1 μl of the final product of each sample. Samples were run on an Applied Biosystems 3730 Genetic Analyzer (Thermo Fisher, Waltham, MA, USA). Genemapper 5.0 software was used to determine the presence or absence of each allele. The peak detection threshold was set for each primer combination and was typically 100 luminescent units. Phylip 3.695 was used to calculate Nei’s pairwise genetic distance and to generate a neighbor-joining tree used to visualize genetic similarity of individuals. Structure 2.3.4 [31] was run using the following parameters: a ‘burn-in’ of 50,000 iterations, followed by 50,000 iterations, an admixture model, and independent loci. The maximum number of potential populations for K was set as the number of geographic sampling locations plus 4 (K = 6 locations + 4 = 10) as suggested by Pritchard et al. [32], and each iteration was run 20 times. The Structure output was used as input for Structure Harvester [33] using the method of Evanno et al. [34] to determine the most likely value for K. Clumpak software was used to permutate runs of K = 2, with Distruct was used visualize results [35].
A Mantel test was used to examine the relationship between genetic distance and geographical distance (isolation by distance) using GenAlex 6.5 [36]. An analysis of molecular variation was run to examine genetic variation among the six regions, using GenAlex 6.5 and 999 permutations. A principal components analysis was also performed with GenAlex 6.5.
Bottleneck version 1.2.02 was used to test whether there was a significant departure from an equilibrium expectation of heterozygotes in each population [37]. We used polymorphic loci from the AFLP data for each population in the six regions, and for individuals in each of the three haplogroups. The infinite allele model (IAM) was run for 1000 iterations, and results were examined with the Wilcoxon sign rank test.
Mitochondrial DNA cox1
For each insect, DNA was used to sequence a ~650 bp region of mitochondrial DNA cytochrome c oxidase subunit 1 (cox1) (known as the ‘barcode’) using the universal forward primer LCO1490 (5'-GGT CAA CAA ATC ATA AAG ATA TTG G-3') and the reverse primer HCO2198 (5'-TAA ACT TCA GGG TGA CCA AAA AAT CA-3') [38, 39]. A polymerase chain reaction (PCR) mix for six samples consisted of the following: 195.6 μl sterile ultra-pure water; 2.4 μl Taq polymerase (Clonetech, Mountainview, CA, USA); 30 μl Taq 10× buffer; 24 μl dNTPs; 6 μl forward primer; and 6 μl reverse primer and 2 μl template DNA (~100 ng template). For each reaction, 2 μl template DNA was added to each vial and the contents were vortexed and spun down. The PCR program was as follows: an initial 1 min warm-up at 95 °C; then 40 cycles of a touchdown program consisting of 92 °C for 30 s, 43–52 °C for 30 s (with a 0.3 °C temperature increase each s), and 72 °C for 60 s; after 40 cycles, a 68 °C final extension for 10 min and then a hold at 4 °C. PCR products were run on a 1.5% agarose gel to visualize the amplification of products of ~650 bp. Samples were cleaned-up using the Exo-sap-it (Affymetrix, Inc, Santa Clara, CA, USA) cleanup kit and run on a 3730 Genetic Analyzer. Resulting sequences were analyzed using Geneious 7 (Biomatters, Aukland, New Zealand) software to produce consensus sequences [40]. Sequences were trimmed, forward and reverse sequences were aligned, and a consensus sequence was produced. Sequences were aligned in Geneious 7.0 using the Clustal W alignment function and used to produce an unrooted neighbor-joining (NJ) tree. We also generated a phylogenetic tree using model-based maximum likelihood (ML) analysis for the same dataset [41]. Using the model selection option in MEGA 7.0, we found that the Tamura 3 parameter with Gamma distribution (G) was the best-fit model to our dataset based on the lowest BIC (Bayesian Information Criterion) value [41]. ML analysis used this best-fit model and clade support was assessed via 1000 bootstrap replicates [41].
The mitochondrial DNA cox1 sequences were used to determine the overall number of haplotypes, haplotype diversity, nucleotide diversity and Tajima’s D using DNAsp 5.10 [42]; Tajima’s D was calculated to test whether there was a departure from neutrality, such as a population expansion or contraction. Subsequently, these same parameters were determined for each of the six regions of the country, and for the mitochondrial DNA haplogroups. Results were used to construct a haplotype network using PopArt 1.7 and selecting the TCS option [43].
Additionally, a combined maximum likelihood phylogenetic tree was constructed using samples from El Salvador and individuals of Ae. aegypti previously sequenced which were available in the GenBank database. Samples included in the tree were selected in the following manner. First, one individual from each of the three most common mitochondrial haplogroups in El Salvador was used for a blast search. Resulting sequences which were 99–100% similar were retrieved, and those from North and South America were selected for inclusion in the tree to compare with those from El Salvador [44]. Several samples from East and West Africa were also included for comparison [45]. In addition, several GenBank accessions from Bennett et al. [6] were included, as they were known to be free of nuclear mitochondrial DNA (NUMTs). Concerns have been raised about whether nuclear mitochondrial DNA (NUMTs) may contribute to the presence of multiple lineages when using mitochondrial DNA sequences for phylogenetic studies of Ae. aegypti [46]. In this study, we attempted to overcome this issue by including sequences from GenBank in the combined phylogenetic tree from previous studies where samples were known to be free of NUMTs [6]. From El Salvador, at least two individuals from each of the three common haplogroups, and at least 2 individuals from each of the 6 regions were included in the tree. This resulted in 14 samples from El Salvador and 16 sequences from GenBank for the combined phylogenetic analysis. Trees were constructed in a manner similar to those described previously. Sequences were aligned in Geneious 7.0 using the Clustal W alignment function, and a phylogenetic tree was produced using model-based maximum likelihood (ML) analysis [41]. The maximum likelihood tree used the Hasegawa-Kishino-Yano model (H-K-Y) with Gamma distribution with invariant sites (G + I) and clade support was assessed via 1000 bootstrap replicates.
Results
Amplified fragment length polymorphisms (AFLPs)
From collections in the six regions, 137 samples were used for DNA extraction (Tables 1 and 2). The three AFLP primer combinations used produced 90, 138 and 97 alleles. Structure analysis and subsequent Structure Harvester analysis using the Evanno method found that K = 2 as the mostly likely number of genetically distinct populations (Fig. 2, Additional file 1: Figure S1, Additional file 2: Figure S2). The first group shown in green (Fig. 2) consisted of the individuals from Sonsonate, San Salvador and Chalatenango, the departments located in the northwestern and central part of the country. Samples from the south and east of the country from two departments, Usulután and Morazán, had individuals that clustered into two separate groups (green and red, Fig. 2), while all individuals from San Miguel clustered in the red group (Fig. 2).

Analysis of population structure for Ae. aegypti with the software Structure 2.2.3. Parameters selected were for diploid individuals, 50,000 iterations, admixed data, and independent loci. Each vertical bar represents an individual mosquito. The y-axis shows the probability of an individual being assigned to one of the two genetic clusters. Structure Harvester found that K = 2; there were two genetically distinct populations
Nei’s genetic distance between the populations ranged from 0.028 to 0.091 (Table 3). Smaller genetic distances were found between Morazán and Chalatenango (0.028), Morazán and Jiquilisco (0.028), Sonsonate and San Salvador (0.032), and San Miguel and Jiquilisco (0.035) (Table 3, Fig. 2). The largest genetic distance was between San Miguel and San Ignacio (Chalatenango) (0.091) followed by that between San Miguel and Sonsonate (0.067) (Table 3). The Mantel test to examine the relationship between genetic distance and geographical distance was significant (r = 0.976, P = 0.010) (Additional file 3: Figure S3). The AMOVA analysis was significant (P = 0.001), and found 11% genetic variation among the six populations (Table 4). Bottleneck analyses for individuals from each region, and from the three mtDNA haplogroups (below) found a significant excess of heterozygotes (P < 0.001), indicating each population had undergone a recent bottleneck. Results from the IAM model were significant for each of these populations at P < 0.001.
The principal components analysis found that the first axis accounted for 44.75% of the variation, while the second and third axes explained 26.20% and 13.43% of the variation, respectively (Additional file 4: Figure S4). Examining axis 1 (the x-axis) from left to right, there was a separation of the populations from the northwest to the southeast part of the country; samples from Chalatenango, San Salvador and Sonsonate were on the left side of axis 1 (x-axis), while those from Jiquilisco, Morazán and San Miguel (which are in the southeast) located on the right side of axis 1. The left of the second axis (y-axis) separated inland mountainous Chalatenango from coastal Sonsonate and nearby San Salvador, while the top right quadrant (y-axis) had the three populations found in the southeastern portion of the country; Morazán and Jiquilisco clustered together, with San Miguel located at the edge of the same cluster. The six populations clustered into three groups.
Mitochondrial DNA cox1
In total 84 individuals were sequenced from the six regions, with numbers sequenced: Sonsonate (n = 12); San Salvador (n = 11); San Ignacio (n = 11); Jiquilisco (n = 14); San Miguel (n = 16); and Morazán (n = 20). The neighbor-joining consensus tree of individuals from El Salvador produced 3 groups, with 0.01 (1%) genetic distance between groups 1 and 2, and 1% between groups 2 and 3. Between groups 1 and 3, the genetic distance was ~2%. A maximum likelihood tree also indicated three groups (Fig. 3). The largest group in the tree consisted of 49 individuals from all six departments, while the second largest group consisted of 23 individuals from all six departments. Finally, the third main group of the tree consisted of 10 individuals from San Salvador, San Miguel and Morazán (Fig. 3).

Mitochondrial DNA cox1 sequences from 82 Ae. aegypti collected in El Salvador. Maximum likekihood tree, Tamura 3 model, 1000 replicates were run and nodes with support above 65% are indicated. Haplogroup 1 (HG1) and haplogroup 2 (HG2): individuals from all six departments; haplogroup 3 (HG3): collections from San Salvador, San Miguel and Morazán. Abbreviations: Son, Sonsonate; SS, San Salvador; SI, San Ignacio, Chalatenango; JC, Jiquilisco, Usulután; SM, San Miguel; Mz, Morazán
A haplotype network was also constructed using the 84 mtDNA sequences, and was found to consist of 3 branches (Fig. 4), which included a total of 10 haplotypes. The haplotypes are indicated on the haplotype network and three haplogroups are indicated in blue (HG1), green (HG2) and red (HG3) and correspond to the haplogroups labeled on the maximum likelihood tree (Figs. 3 and 4). The overall haplotype diversity for all samples was 0.610, Tajima’s D was 1.99 (ns, P > 0.05), and the overall nucleotide diversity was 0.016 (Table 5).

Haplotype network based on 84 mitochondrial DNA cox1 sequences of Ae. aegypti collected in six regions of El Salvador. Ten haplotypes were found in three haplogroups with an overall haplotype diversity of 0.610
Analyses of haplotype diversity, nucleotide diversity and Tajima’s D were also determined for each of the 6 regions (Table 5). Sonsonate on the Pacific coast had the smallest number of haplotypes (2) and lowest nucleotide diversity (0.003). Tajima’s D was only significant for Sonsonate (-2.043, P < 0.01) and not for the other five regions (P > 0.10, Table 5). San Salvador (the capital) had 4 haplotypes, a haplotype diversity of 0.764 and a nucleotide diversity of 0.017, while Morazán had 7 haplotypes, a haplotype diversity of 0.774 and nucleotide diversity of 0.011 (Table 5). The three haplogroups found mtDNA sequences varied in the number of haplotypes, haplotype diversity and nucleotide diversity. The first haplogroup (indicated in blue) had 2 haplotypes, a haplotype diversity of 0.041 and a nucleotide diversity of 0.00014, and the lowest genetic diversity of the three haplogroups (Fig. 4). The second haplogroup (indicated in green) had 4 haplotypes, a haplotype diversity of 0.577, and a nucleotide diversity of 0.002. The third haplogroup (indicated in red) consisted of 4 haplotypes, and had the highest haplotype and nucleotide diversity, 0.682 and 0.0075, respectively (Fig. 4).
For each of the six regions, the proportion of samples belonging to each haplogroup was found and mapped onto its corresponding region of the country (Fig. 5). For Sonsonate, the majority of sequences were in haplogroup 1 (blue) (Fig. 5). San Salvador had a mix of the three haplogroups. Of the six regions sampled, Chalatenango had the largest number of sequences in haplogroup 2 (73%) (green). Jiquilisco, Usulután 69% of samples in haplogroup 1 (blue) and 31% in haplogroup 2. San Miguel and Morazán samples had the largest number of sequences which belonged to haplogroup 3, shown in red (Fig. 5). San Miguel had 69% of samples in haplogroup 1 (blue), and 25% in haplogroup 3 (red, Fig. 5), while Morazán had ~50% of samples in haplogroup 1, 30% in haplogroup 2 and 20% in haplogroup 3 (red, Fig. 5).

Map of collection locations showing the proportion of each haplogroup in the mtDNA sequences from each region
The combined phylogenetic tree consisted of 14 individuals from El Salvador and 16 sequences from GenBank produced a tree with two main lineages (Fig. 6). The first lineage contained El Salvador samples from haplogroup 1 (JC 1, Son 23, SM 23 and Son 22) clustered with GenBank accessions MF172260 from French Guyana, MF 371161 Washington DC, USA, KM203140.1 Colombia H1, KM203143.1 Colombia H4 and MF371161.1 Georgia, USA. This lineage also had a subtree that consisted of three individuals from haplogroup 3 (red) from El Salvador (SS 3, MZ 16, SM 4) (Fig. 6). The second major lineage in the combined maximum likelihood tree consisted of samples from El Salvador haplogroup 2 (green) from this study (JC 22, SS 1, SI 4, SI 1, SS 9) which grouped together with GenBank accessions MF371164 from Florida USA, KM203146 Colombia H7, KM203142.2 Colombia H3, and JX456414.1 Brazil, among others. A subtree contained MF371168.1 from Georgia, which grouped closely with samples from El Salvador SS 9 (Fig. 6).

Maximum likelihood tree based on the H-K-Y model, of samples from each region of El Salvador and each haplogroup, combined with Ae. aegypti sequences from GenBank. Abbreviations: Son, Sonsonate; SS, San Salvador; SI, San Ignacio, Chalatenango; JC, Jiquilisco, Usulután; SM, San Miguel; Mz, Morazán
Discussion
In this study, two molecular markers were used to examine the genetic diversity of Ae. aegypti in El Salvador, Central America. Data from this study found a high level of genetic diversity among samples and suggest at least two lineages have been introduced to El Salvador. The previous eradication program of Ae. aegypti in much of the Western Hemisphere was followed by a reemergence of this insect in most of its previous range, including in El Salvador. This may be due to the ease of movement of insect eggs and increased trade and migration. Few data from Central America on the genetic diversity of Ae. aegypti were available for comparison, but samples were compared with others from North and South America to consider the source of the introductions.
The data from the Structure Harvester analysis of the AFLP data found there were two genetically divergent groups of Ae. aegypti in El Salvador. The northern region of the country including Sonsonate, San Salvador and Chalatenango consisted of one genetically distinct group, while individuals from San Miguel in the eastern and interior region of the country, consisted of a second genetically divergent group. Samples from Jiquilisco (Usulután) and Morazán, also in the south and eastern region, consisted of a mix of the two groups. Genetic distances were largest between San Miguel and Chalatenango (nearly 10%) and the overall AMOVA of the six populations found 11% genetic variation. These values are similar to those found in other studies; for example, 15% in Colombia [47], 19% in Argentina [12] and 20% in Mexico [11]. The principal components analysis found that the six populations located in three quadrants, and isolation by distance was significant. The largest variation was explained by the first axis (44.75%), with the departments in the north and west (Chalatenango, San Salvador, Sonsonate) separating from those in the southeast (Jiquiliscco, San Miguel and Morazan). The combined results from the PCA analysis and the Structure Harvester analysis suggest that individuals from the northwest region of the country, Chalatenango, San Salvador and Sonsonate, have some degree of genetic isolation from the three populations in the south.
There are several explanations for the two genetically distinct groups found in the Structure Harvester analysis. Studies have suggested at least two subspecies of Ae. aegypti [3, 7, 48] which occur in different habitats. However, in this study, larvae were collected from the same type of habitat, from barrels and wash basins (‘pilas’) outside homes in neighborhoods, and all individuals in this study are believed to be Ae. aegypti aegypti. Other studies have suggested that divergence may also occur between populations from the rainy season and the dry season [9]. Seasonal divergence is not likely to explain the genetic divergence observed in this study, since all collections were made from May to August during the rainy season. Rather, the genetic divergence appears to follow a spatial pattern along a northwest to southeast gradient.
A large degree of genetic diversity was also found in the mitochondrial DNA sequences in this study. There were three haplogroups with a total of 10 haplotypes, with an overall haplotype diversity of 0.610 and a nucleotide diversity of 0.015. These values are relatively high, and are similar to those found by Bennett et al. [6] for samples from America (11 haplotypes and a nucleotide diversity of 0.013). Samples from El Salvador also had a nucleotide diversity similar to that found in studies for Ae. aegypti in Colombia and Venezuela [47, 49]. In El Salvador, the areas with the most haplotypes occurring were Morazán and San Miguel. These regions are close to the Gulf of Fonseca, which borders El Salvador, Honduras and Nicaragua on the Pacific coast. The higher genetic diversity in this region may represent introduced populations from regions which were not subject to eradication programs.
The most abundant haplotype sequenced in the present study was haplotype 1, which was found in all six regions of the country. In Sonsonate, on the Pacific coast of El Salvador, over 90% of individuals sequenced were haplogroup 1, and the nucleotide diversity in Sonsonate was the lowest of all regions (0.003) (Fig. 5). Low nucleotide diversity could indicate a bottleneck after a small number of individuals were introduced, or perhaps a population which was reduced in size due to insecticides [11]; these hypotheses require testing. The Tajima’s D value for Sonsonate was significant (-2.043), suggesting a population expansion in this region. This haplogroup was the most widespread, suggesting it has been in El Salvador longer than the other two haplogroups. It may have been moved into other departments by passive transport [50].
In El Salvador, the second most abundant haplotype observed (h2, green) occurred more frequently in San Ignacio, Chalatenango than in the other five departments (Fig. 5). San Ignacio is a cool (~25 °C), high elevation (~2500 m above sea level) mountainous area on the northeastern border of El Salvador and Honduras. San Ignacio, Chalatenango had four haplotypes, a haplotype diversity of 0.577 and a nucleotide diversity of 0.009. The bottleneck test found this group also had a significant departure from an equilibrium of heterozygotes, indicating a recent population bottleneck. Individuals in this haplogroup clustered in the phylogenetic tree (Fig. 6) with those from Brazil and Florida, as well as Colombia; the combined maximum likelihood tree had two main lineages with low to moderate bootstrap support. Previous studies of Ae. aegypti have similarly found phylogentic trees with two lineages and varying levels of support [44, 48, 49]. Haplogroup two may have been introduced in El Salvador in the post-eradication era; it has a higher nucleotide and haplotype diversity than haplogroup 1, and a more limited distribution. However, further work would be needed to test this hypothesis.
A third haplogroup (red, Fig. 6) was found in the mtDNA cox1 sequences from El Salvador. These individuals formed a small subgroup of the first lineage of the combined phylogenetic tree. While several previous studies of Ae. aegypti have suggested that three lineages have been introduced in the Western Hemisphere [12, 15], more recent studies and this one suggest that two divergent groups of Ae. aegypti have been introduced into El Salvador. The third haplogroup in our study was found in the capital city San Salvador, and in the south and east portion of El Salvador, in San Miguel and Morazán. The third (red) haplogroup clustered in the first lineage with the blue haplogroup from El Salvador, yet had much higher haplotype and nucleotide diversity. This group had no match to sequences in GenBank which was 99–100% similar, suggesting it is from an area which has yet to be included in global level population analyses.
Conclusions
This study of Ae. aegypti genetic variability in El Salvador agrees with other studies in the Western Hemisphere in finding at that at least two genetically divergent groups have been introduced. Introductions may have occurred through ports with international cargo or via transport along major corridors such as the Pan American Highway between adjacent Central American countries. The genetic variability of the populations of Ae. aegypti in Central America has only recently been investigated. Future work should expand this study to examine genetic diversity of Ae. aegypti in surrounding countries which will contribute to our understanding of the reintroductions of this invasive species in Central America.
References
San Martín JL, Brathwaite O, Zambrano B, Solórzano JO, Bouckenooghe A, Dayan G, Guzman MG. The epidemiology of dengue in the Americas over the last three decades: a worrisome reality. Am J Trop Med Hyg. 2010;82:128–35.
Gloria-Soria A, Lima A, Lovin DD, Cunningham JM, Severson DW, Powell JR. Origin of a high-latitude population of Aedes aegypti in Washington DC. Am J Trop Med Hyg. 2018;98:445–52.
Tabachnick WJ, Powell JR. A world-wide survey of genetic variation in the yellow fever mosquito, Aedes aegypti. Genet Res. 1979;34:215–29.
Brown JE, McBride CS, Johnson P, Ritchie S, Paupy C, Bossin H, et al. Worldwide patterns of genetic differentiation imply multiple ‘domestications’ of Aedes aegypti, a major vector of human diseases. Proc R Soc B. 2011;278:2446–54.
Brown JE, Evans BR, Zheng W, Obas V, Barrera-Martinez L, Egizi A, et al. Human impacts have shaped historical and recent evolution in Aedes aegypti, the dengue and yellow fever mosquito. Evolution. 2014;68:514–25.
Bennett KL, Shida F, Linton YM, Misinzo G, Kaddumukasa M, Djouaka R, et al. Historical environmental change in Africa drives divergence and admixture of Aedes aegypti mosquitoes: a precursor to successful worldwide colonization? Mol Ecol. 2016;25:4337–54.
Gloria-Soria A, Ayala D, Bheecarry A, Calderon-Arguedas O, Chadee DD, Chiappero M, et al. Global genetic diversity of Aedes aegypti. Mol Ecol. 2016;25:5377–95.
Tabachnick WJ, Munstermann LE, Powell JR. Genetic distinctness of sympatric forms of Aedes aegypti in East Africa. Evolution. 1979;33:287–95.
Paupy C, Brengues C, Ndiath O, Toty C, Hervé JP, Simard F. Morphological and genetic variability within Aedes aegypti in Niakhar, Senegal. Infect Genet Evol. 2010;10:473–80.
Moore M, Sylla M, Goss L, Burugu MW, Sang R, Kamau LW, et al. Dual African origins of global Aedes aegypti s.l. populations revealed by Mitochondrial DNA. PLoS Negl Trop Dis. 2013;7:e2175.
Gorrochotegui-Escalante N, Munoz MDL, Fernandez-Salas I, Beaty BJ, Black WC. Genetic isolation by distance among Aedes aegypti populations along the northeast coast of Mexico. Am J Trop Med Hyg. 2002;62:200–9.
Rondon, Duenas JC, Albrieu Llinas G, Panzetta-Dutari GM, Gardenal CN. Two different routes of colonization of Aedes aegypti in Argentina from neighboring countries. J Med Entomol. 2009;46:1344–54.
Paupy C, Le Goff G, Brengues C, Guerra M, Revolla J, Barja Simon Z, et al. Genetic structure and phylogeography of Aedes aegypti, the dengue and yellow-fever mosquito vector in Bolivia. Infect Genet Evol. 2012;12:1260–9.
Ravel S, Monteny N, Olmos DV, Verdugo JE, Cuny G. A preliminary study of the population genetics of Aedes aegypti (Diptera: Culicidae) from Mexico using microsatellite and AFLP markers. Acta Trop. 2001;78:241–50.
Scarpassa VM, Cardoza TB, Cardoso RP. Population genetics and phylogeography of Aedes aegypti (Diptera: Culicidae) from Brazil. Amer J Trop Med Hyg. 2008;78:895–903.
Monteiro FA, Shama R, Martins AJ, Gloria-Soria A, Brown JE, Powell JR. Genetic diversity of Brazilian Aedes aegypti: patterns following an eradication program. PLoS Negl Trop Dis. 2014;8:e3167.
Kotsakiozi P, Gloria-Soria A, Caccone A, Evans B, Schama R, Martins AJ, et al. Tracking the return of Aedes aegypti to Brazil, the major vector of the dengue, chikungunya and Zika viruses. PLoS Negl Trop Dis. 2017;11:e0005653.
Organización Panamericana de la Salud (OPS). Dengue y dengue hemorrágico en las Américas: guías para su prevención y control. In: Publicación Científica, vol. 548. Washington: Organización Panamericana de la Salud; 1995.
da Costa da Silva AL, Capurro ML, Bracco JE. Genetic lineages in the yellow fever mosquito Aedes (Stegomyia) aegypti (Diptera: Culicidae) from Peru. Mem Inst Oswaldo Cruz. 2005;100:539–44.
Cáceres OA, León W. Variantes genéticas de Aedes aegypti y su asociación con el serotipo del virus dengue en una área endémica del Perú. Ministerio de Salud, Serie Informes Técnicos. 2007;96:1–14.
Organización Panamericana de la Salud (OPS). Alerta epidemiológica: Síndrome neurológico, anomalías congénitas e infección por virus Zika. In: Implicaciones para la salud publica en las Américas. Washington: OPS; 2015.
Slosek J. Aedes aegypti in the Americas: review of their interactions with the human population. Soc Sci Med. 1986;23:249–57.
Soper FL. Aedes aegypti and yellow fever. Bull World Health Organ. 1967;36:521–7.
CDC. Dengue: Mexico, El Salvador, Honduras. MMWR. 1983;32:586–8.
Hayes JM, Garcia-Rivera E, Flores-Reyna R, Suarez-Rangel G, Rodriguez-Mata T, Coto-Portillo R, et al. Risk factors for infection during a severe dengue outbreak in El Salvador in 2000. Am J Trop Med Hyg. 2003;69:629–33.
Ocampo CB, Wesson DM. Population dynamics of Aedes aegypti from a dengue hyperendemic urban setting in Colombia. Am J Trop Med Hyg. 2004;71:506–13.
Apostol BL, Black WC, Reiter P, Miller BR. Use of randomly amplified polymorphic DNA amplified by polymerase chain reaction markers to estimate the number of Aedes aegypti families at oviposition sites in San Juan, Puerto Rico. Am J Trop Med Hyg. 1994;51:89–97.
Qiagen. DNeasy Tissue Handbook. Valencia: Qiagen; 2006.
Vos PR, Hogers M, Bleeker M, Reijans T, Van de Lee M, Hornes A, et al. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 1995;23:4407–14.
Joyce AL, Melese E, Ha PT, Inman A. Population genetic structure of the Culex pipiens (Diptera: Culicidae) complex, vectors of West Nile virus, in five habitats. Parasit Vectors. 2018;11:10.
Pritchard JK, Wen X, Falush D. Documentation for structure software: version 2.2.2007. https://web.stanford.edu/group/pritchardlab/software/structure22/readme.pdf.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Earl DA, von Holdt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resources. 2012;4:359–61.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I. Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Res. 2015;15:1179–91.
Peakall R, Smouse PE. GenAlEx 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006;6:288–95.
Cornuet JM, Luikhart G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1996;144:2001–14.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Cywinska A, Hunter FF, Hebert PDN. Identifying Canadian mosquito species through DNA barcodes. Med Vet Entomol. 2006;20:413–24.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Librado P, Rozas J. Dnasp v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Leigh JW, Bryant D. POPART: full feature software for haplotype network construction. Methods Ecol Evol. 2015;6:1110–6.
Jaimes-Duenez J, Arboleda S, Triana Chavez O, Gomez-Palacio A. Spatio-temporal distribution of Aedes aegypti (Diptera: Culicidae) mitocondrial lineages in cities with distinct dengue incidence rates suggests complex population dynamics of the dengue vector in Colombia. PLoS Negl Trop Dis. 2015;9:e0003553.
Morlais I, Severson DW. Complete mitochondrial DNA sequence and amino acid analysis of the cytochrome c oxidase subunit I (COI) from Aedes aegypti. DNA Seq. 2002;13:123–7.
Hlaing T, Willoughby TL, Somboon P, Socheat D, Setha T, Min S, et al. Mitochondrial pseudogenes in the nuclear genome of Aedes aegypti mosquitoes: implications for past and future population genetic studies. BMC Genetics. 2009;10:11.
Caldera SM, Jaramillo MC, Cochero S, Pérez-Doria A, Bejarano EE. Diferencias genéticas entre poblaciones de Aedes aegypti de municipios del norte de Colombia, con baja y alta incidencia de dengue. Biomédica. 2013;33:89–98.
Bracco JE, Capurro ML, Lourenço-de-Oliveira R, Sallum MAM. Genetic variability of Aedes aegypti in the Americas using a mitochondrial gene: evidence of multiple introductions. Mem Inst Oswaldo Cruz. 2007;102:573–80.
Herrera F, Urdaneta L, Rivero J, Zoghbi J, Ruiz J, Carrasquel G, et al. Population genetic structure of the dengue mosquito Aedes aegypti in Venezuela. Mem Inst Oswaldo Cruz. 2006;101:625–33.
Huber K, Loan LL, Chantha N, Failloux AB. Human transportation influences Aedes aegypti gene flow in Southeast Asia. Acta Trop. 2004;90:23–9.
Acknowledgements
We would like to acknowledge Ashley Valle Arevalo, a student research assistant at the University of California Merced, who assisted with the molecular work. Roberto Guillen Paredes and the Center for Health Research (CENSALUD) at the University of El Salvador contributed equipment, and Eduardo Romero Chevez at the Ministry of Health Vector Control El Salvador for mosquito collections.
Funding
The project was supported by the University of California Merced, which contributed to the cost of research supplies and supporting undergraduate student assistants.
Availability of data and materials
The data supporting the conclusions of this article are included within the article and its additional files. The datasets used and analyzed during the present study are available from the corresponding author upon reasonable request. GenBank accessions numbers for the mitochondrial DNA sequences are the following:
JC1(MK028219); JC2(MK028220); JC4(MK028221); JC7(MK028222); JC8(MK028223); JC9(MK028224); JC14(MK028225); JC15(MK028226); JC17(MK028227); JC20(MK028228); JC21(MK028229); JC22(MK028230); JC23(MK028231); JC24(MK028232); MZ1(MK028233); MZ2(MK028234); MZ3(MK028235); MZ4(MK028236); MZ5(MK028237); MZ9(MK028238); MZ10(MK028239); MZ11(MK028240); MZ12(MK028241); MZ13(MK028242); MZ16(MK028243); MZ17(MK028244); MZ18(MK028245); MZ19(MK028246); MZ20(MK028247); MZ22(MK028248); MZ23(MK028249); MZ24(MK028250); MZ25(MK028251); MZ26(MK028252); SI1(MK028253); SI2(MK028254); SI3(MK028255); SI4(MK028256); SI6(MK028257); SI7(MK028258); SI20(MK028259); SI25(MK028260); SI26(MK028261); SI28(MK028262); SM1(MK028263); SM2(MK028264); SM3(MK028265); SM4(MK028266); SM6( MK028267); SM7(MK028268); SM8(MK028269); SM9(MK028270); SM10(MK028271); SM11(MK028272); SM18(MK028273); SM19(MK028274); SM20(MK028275); SM21(MK028276); SM22(MK028277); SM23(MK028278); Son4(MK028279); Son5(MK028280; Son6(MK028281); Son7(MK028282); Son8(MK028283); Son9(MK028284); Son13(MK028285); Son17(MK028286); Son22(MK028287); Son23(MK028288); Son25(MK028289); Son26(MK028290); SS1(MK028291); SS3(MK028292); SS4(MK028293); SS5(MK028294); SS9(MK028295); SS10(MK028296); SS11(MK028297); SS16(MK028298); SS25(MK028299); SS26(MK028300).
Author information
Authors and Affiliations
Contributions
Study design: ALJ and MM. Data collection: ALJ, RT and MMT. Data analysis: ALJ, MMT, MM and RT. Manuscript preparation: ALJ, MMT and MM. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Results from a Structure Harvester analysis of all six populations of Ae. aegypti. Structure Harvester uses the results from Structure to calculate the Delta K value, the change in likelihood, for the number of potential clusters. Structure Harvester calculated the most likely number of clusters was 2 (K = 2) (TIFF 4202 kb)
Additional file 2:
Figure S2. Results from a Structure Harvester analysis of all six populations of Ae. aegypti in this study. Each row shows the probability of K populations and delta K. The most likely number of populations was K = 2. (TIFF 5816 kb)
Additional file 3:
Figure S3. Mantel test to examine the relationship between genetic distance and geographical distance for six populations of Ae. aegypti (r = 0.976, P = 0.010). (TIFF 1791 kb)
Additional file 4:
Figure S4. Principal components analysis using genetic distance output of AMOVA of the six populations of Ae. aegypti in the study. (TIFF 1744 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Joyce, A.L., Torres, M.M., Torres, R. et al. Genetic variability of the Aedes aegypti (Diptera: Culicidae) mosquito in El Salvador, vector of dengue, yellow fever, chikungunya and Zika. Parasites Vectors 11, 637 (2018). https://doi.org/10.1186/s13071-018-3226-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-018-3226-5