
Abstract
Background
Plasmodium merozoite surface protein-1 (MSP-1) is released into the bloodstream during merozoite invasion, and thus represents a crucial malarial vaccine target. Although substantial research effort has been devoted to uncovering the genetic diversity of MSP-1 for P. falciparum and P. vivax, there is minimal information available regarding the genetic profiles and structure of P. ovale. Therefore, the aim of the present study was to determine the extent of genetic variation among two subspecies of P. ovale by characterizing the MSP-1 N-terminal sequence at the nucleotide and protein levels.
Methods
N-terminal of MSP-1 gene were amplified from 126 clinical samples collected from imported cases of malaria in migrant workers returning to Jiangsu Province from Africa using a conventional polymerase chain reaction (PCR) assay. The PCR products were then sequenced and analyzed using the GeneDoc, MegAlign, MEGA7 and DnaSP v.6 programs.
Results
The average pairwise nucleotide diversities (π) of P. ovale curtisi and P. ovale wallikeri MSP-1 genes (pomsp1) were 0.01043 and 0.01974, respectively, and the haplotype diversity (Hd) were 0.746 and 0.598, respectively. Most of the nucleotide substitutions detected were non-synonymous, indicating that the genetic variations of pomsp1 were maintained by positive diversifying selection, thereby suggesting their role as a potential target of a protective immune response. Amino acid substitutions of P. ovale curtisi and P. ovale wallikeri MSP-1 could be categorized into five and three unique amino acid variants, respectively.
Conclusions
Low mutational diversity was observed in pomsp1 from the Jiangsu Province imported malaria cases; further studies will be developed such as immunogenicity and functional analysis.
Similar content being viewed by others
Background
Malaria is one of the most serious infectious diseases of humans worldwide. An estimated 216 million cases of malaria were reported in 2016 and the global tally of malaria-caused deaths reached 445,000 [1]. Five species in the genus Plasmodium (P. falciparum, P. vivax, P. malariae, P. ovale and P. knowlesi) are known to cause human malaria under natural transmission [2]. In China, the majority of malaria cases are caused by P. vivax and P. falciparum, most of which are imported from malaria-endemic areas. Jiangsu Province, located in eastern China, was an unstable malaria transmission area and there has been no local malaria infection report since 2012. However, the number of imported malaria cases in Jiangsu ranked in the top three provinces in China, with 1799 imported malaria cases reported from 2005 to 2014 [3, 4]. As a neglected human parasite causing infection, P. ovale was first reported and named by Stephens in 1922 as one of the major Plasmodium species infecting humans [5]. Plasmodium ovale has a wide geographic distribution, including the Middle East, Indonesia, and Southeast Asia [6,7,8]. In Africa, only 0.7–10% of human malaria cases are caused by P. ovale infections; thus, the diagnosis of P. ovale is often overlooked due to the low levels of parasitemia and mixed-species malaria infections [7, 9]. Notably, approximately 300 malaria cases in China imported from Africa annually are caused by P. ovale. There are two subspecies of P. ovale, P. ovale curtisi (classical type) and P. ovale wallikeri (variant type) [10], which show dimorphism of multiple genetic loci [2].
Merozoites surface proteins (MSPs) are released into the bloodstream of the host in extracellular forms, and are thus promising vaccine targets since they play a critical role in erythrocyte invasion [11]. As the predominant member of MSPs, MSP-1 has been detected in all examined Plasmodium species to date, and plays an important role during erythrocyte attachment [12]. Thus, naturally acquired antibodies to MSP-1 inhibit erythrocyte invasion and are associated with protection from clinical malaria in field studies [13]. However, MSP-1-based vaccines show low protective efficacy against clinical malaria, which may be attributed to the genetic diversity of MSP-1, leading to failure of anti-malaria parasite control measures. Moreover, antigenic diversity allows the parasite to evade natural immune responses, which may cause vaccines to lose efficacy [14]. The N-terminal fragments of the MSP-1 genes of P. falciparum and P. vivax (pfmsp1 and pvmsp1, respectively) show polymorphism due to selection pressure, which has hindered MSP-1-based vaccine development [15, 16]. Comparatively, the C-terminal of MSP-1 is a conserved sequence, which is carried into the infected erythrocytes during merozoite invasion [17]. A recent study demonstrated the low diversity of the pocmsp1 and powmsp1 gene in 10 P. ovale isolated from symptomatic malaria patients in Thailand, which may be related to a low transmission rate or repeated bottleneck effects [18]. However, there is limited evidence of the genetic diversity of pomsp1. Therefore, characterization of pomsp1 is necessary toward understanding the population genetic structure and finding a suitable candidate for vaccine development. Accordingly, in the present study, the pomsp1 N-terminal sequence was analyzed from the both subspecies of P. ovale obtained from infected migrant workers returning to China from Africa. We determined the levels of polymorphisms and nucleotide divergence of msp1 sequences to validate the classification of P. ovale curtisi and P. ovale wallikeri as distinct species or subspecies, and trace signatures of selection.
Methods
Study areas and sample collection
The samples of P. ovale curtisi and P. ovale wallikeri were obtained from febrile patients at local hospitals of Jiangsu Province in China between 2012 and 2016, who had recently returned from working in tropical regions of sub-Saharan Africa endemic for malaria. A total of 126 P. ovale-infected blood samples were collected. Identification of the isolates was confirmed using polymerase chain reaction (PCR) of specific gene sequences. Parasite species were distinguished by PCR amplification using the real-time TaqMan PCR [3].
PCR amplification and sequencing of the pomsp1 N-terminal
The N-terminal nucleotide sequences of MSP-1 from P. ovale curtisi and P. ovale wallikeri were amplified by PCR using the primers designed as pocmsp1-Forward (5'-GAA ACG CTC GAA AAT TAT A-3') and pocmsp1-Reverse (5'-ACA GGA TCA GTA AAC AGA CCT T-3'), and powmsp1-Forward (5'-GAA ACG CTC GAA AAT TAT A-3') and powmsp1-Reverse (5'-ATC GGT AAA CAG ACC TTC CAT-3'), respectively. The pocmsp1 (GenBank: KC137343) and powmsp1 (GenBank: KC137341) sequences from the GenBank database were used as reference gene sequences. The reactions were carried out in a volume of 20 μl including 1 μl genomic DNA, 7.4 μl double-distilled water, 0.8 μl of each primer, 0.5 units DNA polymerase, and 2 mM deoxynucleoside triphosphate within 10 μl premix (2× Phanta® Max Master Mix, Nanjing, China). The PCR amplification was performed in a Mastercycler (Eppendorf, Hamburg, Germany) under the following programme: denaturation at 95 °C for 3 min; followed by 35 cycles of 95 °C for 15 s, 51 °C for 15 s and 72 °C for 30 s; and a final extension at 72 °C for 5 min. The amplified products were analyzed by 1% agarose gel electrophoresis and visualized under an ultraviolet transilluminator (Bio-Rad ChemiDoc MP, Hercules, USA). The size of the PCR products was estimated based on the mobility relative to the standard DNA marker (TRANSGEN BIOTECH, Beijing, China). PCR products were cloned into pUC57 vector and the universal primers M13F (5'-TGT AAA ACG ACG GCC AGT-3') and M13R (5'-CAG GAA ACA GCT ATG AC-3') were used for sequencing, which was performed by GENEWIZ (Suzhou, China) on an ABI 3730xl DNA Analyzer (Thermo Fisher Scientific, Waltham, USA).
Sequence alignment and data analysis
The geographical distribution map of P. ovale curtisi and P. ovale wallikeri was constructed using Arcgis10.2 software [19]. The primary structure of the PoMSP-1 protein was predicted with a bioinformatics tool (http://smart.embl-heidelberg.de/). To evaluate the diversity of the two subspecies, the pocmsp1 (KC137343) and powmsp1 (KC137341) sequences were used as templates and aligned using GeneDoc2.7.0 [20]. The nucleotide sequences of pomsp1 were translated to deduced amino acid (aa) sequences using MegAlign module of Lasergene 7 software package DNASTAR [21] and then aligned with reference aa sequences. The codon-based test of purifying selection was conducted using the MEGA7 program [22]. The rates of non-synonymous mutations (dN) and synonymous mutations sites (dS) were computed by Z-test using the Nei & Gojobori method [23] with the Jukes and Cantor correction and 100 bootstrap replications.
The average pairwise nucleotide diversity (π), haplotype diversity (Hd), and number of haplotypes (H) were calculated by DnaSP v6 [24]. The nucleotide diversity was analyzed with a window length of 50 base pairs (bp) and a step size of 3 bp using DnaSP v6. Tajima’s D, Fu and Li’s D*, and Fu and Li’s F* tests were used to measure the degree of deviation from neutrality [25, 26]. Phylogenetic trees of the N-terminal of MSP-1 were constructed using the neighbor-joining method according to the nucleotide sequences. The MSP-1 sequences of other Plasmodium species included the MSP-1 haplotypes of malaria parasites from humans (P. falciparum, P. malariae, P. ovale and P. vivax), gorillas (P. praefalciparum, P. alderi and P. billcollinsi), chimpanzees (P. reichenowi, P. blacklocki and P. gaboni), macaques (P. knowlesi and P. cynomolgi), and murine infections (P. yoelii, P. chabaudi and P. berghei), which were collected from the NCBI and PlasmoDB databases. Evolutionary relationships of the aligned sequences were determined using neighbor-joining approaches in MEGA 7.0.
Results
Geographical origin of P. ovale curtisi and P. ovale wallikeri
The total of 126 P. ovale clinical isolates showed a geographical distribution across 15 countries of sub-Saharan Africa. The isolates were mainly derived from Equatorial Guinea (n = 37, 29.7%), Angola (n = 27, 21.4%), Nigeria (n = 19, 15.1%), and the Republic of Congo (n = 13, 10.3%) located on the west coast of Africa (Fig. 1). Comparatively, the P. ovale curtisi isolates spanned a wider range of countries (15 countries) than P. ovale wallikeri isolates (10 countries). Overall, 61 (48.4%) cases of P. ovale curtisi infection and 65 (51.6%) cases of P. ovale wallikeri infection were identified in this study (Table 1).

Map of Africa showing the countries of origin of P. ovale samples. a P. ovale. b P. ovale curtisi. c P. ovale wallikeri
Characterization of PoMSP-1
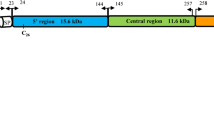
The lengths of MSP-1 encoded by the full-length P. ovale curtisi (GenBank: KC137343) and P. ovale wallikeri (GenBank: KC137341) genes were 1727 and 1672 (aa), respectively, each beginning with a predicted 19 aa signal peptide sequence (aa 1–19). Similar to PvMSP-1 and PfMSP-1, some other specific regions were identified in the P. ovale curtisi predicted protein primary structure, such as a coiled-coil region (aa 288–368 and 450–495), Pfam region (aa 1011–1546), and EGF domains (aa 1624–1660 and 1667–1705) (Fig. 2a). Similarly, the PowMSP-1 predicted protein primary structure contained a signal peptide (aa 1–19), coiled-coil region (aa 458–496), Pfam region (aa 950–1491 aa), and two EGF domains (aa 1569–1605 and 1612–1650) (Fig. 2b).

Predicted P. ovale MSP-1 protein primary structure and MSP-1 N-terminal fragment size. a P. ovale curtisi. b P. ovale wallikeri. c pomsp1 N-terminal fragment. Abbreviations: M, DNA marker; 1, pocmsp1 N-terminal fragment; 2, powmsp1 N-terminal fragment. Purple, blue, orange and green colors indicate signal peptide, coiled-coil, Pfam and EGF domains, respectively. Arrowheads indicate pomsp1 N- terminal for sequencing
Nucleotide polymorphism of pomsp1
The MSP-1 genes of the 126 P. ovale isolates were amplified corresponding to nucleotide positions 58–1125 (Fig. 2c). There were five genotypes of the pocmsp1 N-terminal, and 52 isolates (85.2%) showed a non-synonymous mutation compared with the reference GH01 strain, with 44 nucleotides (67.7%) showing a non-synonymous mutation in the powmsp1 N-terminal. Interestingly, 31 P. ovale curtisi isolates had 27 serine residues, while only 21 serine residues existed in the others. There were six more non-synonymous aa changes detected in P. ovale wallikeri isolates (Fig. 3).

Amino acid sequence alignment of P. ovale MSP-1 N-terminal fragment. a P. ovale curtisi. b P. ovale wallikeri
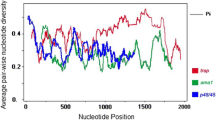
Overall, 26 single nucleotide polymorphisms (SNPs) were found among 61 samples with an average π value of 0.01043 in pocmsp1, and 42 SNPs were detected among 65 samples with an average π value of 0.01974 in powmsp1. A sliding method plot with a window length of 50 bp and a step size of 3 bp using DnaSP v6 revealed a π value in the range of 0–0.09688 and 0–0.19221 for pocmsp1 and powmsp1, respectively. The conserved region was observed from 0.2–0.7 kb length in pocmsp1 and before 0.6 kb in powmsp1 with approximate π values of 0 (Fig. 4). The haplotype (gene) diversity of pocmsp1 could be categorized into five distinct haplotypes with an estimated Hd of 0.746 and three distinct haplotypes with an estimated Hd of 0.598 in powmsp1 samples (Table 2). The average number of nucleotide differences (k) for pocmsp1 and powmsp1 was 11.139 and 21.081, respectively.

Sliding window plots of sequence diversity (π) and Tajima’s D. a Sequence diversity of pocmsp1. b Tajima’s D of pocmsp1. c Sequence diversity of powmsp1. d Tajima’s D of powmsp1
Genetic population structure based on the pomsp1 N-terminal
The population genetic structure of the P. ovale isolates was analyzed based on the MSP-1 N-terminal gene polymorphisms applied to the codon-based test of purifying selection according to average dS and dN values within each isolate. There was clear evidence of positive selection or diversifying selection in P. ovale MSP-1 [Prob = 1.000, dS - dN = -0.06 (pocmsp1), -0.478 (powmsp1)]. In addition, Tajima’s D, Fu and Li’s D* and F* tests rejected a neutral model of polymorphism occurrence with values for pocmsp1 (Tajima’s D = 3.22138, P < 0.01; Fu and Li’s D* = 1.88498, P < 0.02; Fu and Li’s F* = 2.77112, P < 0.02) and powmsp1 (Tajima’s D = 4.57287, P < 0.001; Fu and Li’s D* = 2.00379, P < 0.02; Fu and Li’s F* = 3.5575, P < 0.02), respectively (Table 2).
Phylogenetic analysis
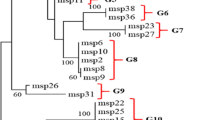
As predicted based on the low level of genetic diversity and signature of positive selection described above, a close phylogenetic relationship was detected in pomsp1 sequences between the subspecies based the branch lengths of pocmsp1 and powmsp1 with 100% bootstrap support (Fig. 5). Phylogenetic trees of 26 MSP-1 gene alleles from the 18 species of Plasmodium in human and non-human primates were constructed using the neighbor-joining method (Additional file 1: Figure S1).

Phylogenetic relationship of MSP-1 N-terminal genes within pomsp1 sequences based on neighbor-joining method
Discussion
The life-cycle of the malaria parasite alternates between the human host and the mosquito vector, which is complex with extensive genetic and antigenic diversity across different stages of the parasite’s life [14]. The genetic diversity of P. ovale might have impacted malaria transmission and the success of malaria control strategies. Gaining a deeper understanding of the mechanisms and patterns of genetic recombination and sequence variation may help in designing a vaccine that could represent the worldwide repertoire of polymorphic malaria surface antigens [27]. The sequences of pocmsp1 and powmsp1 showed a low level of diversity in a limited number of Asian isolates [18]. Hence, we analyzed the N-terminus of pocmsp1 (61 isolates, 48.4%) and powmsp1 (65, 51.6%), and found that pocmsp1 was more conserved than powmsp1 with 26 (14 synonymous, 12 non-synonymous) and 42 (26 synonymous, 16 non-synonymous) sites of nucleotide diversity, respectively.
Neutrality tests were further performed to determine the signatures of natural selection on the MSP-1 N-terminal fragment of P. ovale. Significantly positive values for these statistics reflect an excess of intermediate frequency alleles, which can result from population bottlenecks or balancing selection. The sequences for subspecies P. ovale curtisi and P. ovale wallikeri were further divided into five and three branches that were all within the same evolutionary branches. The MSP-1 N-terminal sequences placed the two subspecies in a distinct bifurcating branch, and the split of pocmsp1 and powmsp1 seems to be relatively more recent. Therefore, the MSP-1 N-terminal sequences of P. ovale curtisi and P. ovale wallikeri support the ancient divergence times of the malaria parasite lineage [28].
The Z-test (dS - dN < 0) indicated that strong positive or purifying selection within the parasite population. These results were in agreement with previous studies which suggested that such mechanisms might be in favour of parasites to evade targeted host immune responses [29]. In addition, the genetic diversities at the P. ovale MSP-1 N-terminal [π = 0.01043 ± SD 0.00061 (pocmsp1), π = 0.01974 ± SD 0.00055 (powmsp1)] were lower compared to that of P. falciparum and P. vivax [30], which may be related to the lower transmission rate of P. ovale from diverse geographical origins [8]. These findings were similar to previously published data demonstrating a low level of sequence diversity of the MSP-1 gene in P. ovale [18].
The intragenic recombination of MSP-1 gene is a major informative pattern at the level of population sequence diversity. The frequency of allelic recombination has important guiding significance for the population structure of parasites [31]. A previous study demonstrated that P. falciparum has a low level of genetic diversity in areas with low transmission rates and high level of sequence diversity in areas with high transmission rates [32]. High mutational diversity was observed in pvmsp1 isolated from Thailand northwestern region [33]. The level of nucleotide diversity in both pocmsp1 and powmsp1 N-terminal sequences detected in this study showed lower magnitude than that reported for pvmsp1 and pfmsp1 [34, 35].
Conclusions
This study provides valuable reference information on the genetic diversity of P. ovale curtisi and P. ovale wallikeri isolates imported from Africa to China based on analysis of the MSP-1 N-terminal sequence. To our knowledge, this is the first report of the genetic diversity, selection signature, and population structure of the N-terminal of pomsp1 gene from an African population. The low level of genetic diversity indicated that these genes are under purifying selection. Therefore, these sequences have potential for vaccine development, which requires further investigation of the immunogenicity and antigenicity of P. ovale MSP-1.
Abbreviations
- MSP-1:
-
Merozoite surface protein-1
- PCR:
-
Polymerase chain reaction
- aa:
-
Amino acid
- Hd :
-
Haplotype diversity
- SNP:
-
Single nucleotide polymorphism
- dN :
-
Rates of non-synonymous substitutions
- dS :
-
Rates of synonymous substitutions
- H:
-
Haplotype
- Hd :
-
Haplotype diversity
- k :
-
Average number of pairwise nucleotide differences within the population
- π :
-
Nucleotide diversity
References
WHO. World Malaria Report (2008–2017). Geneva: World Health Organization; 2017. http://www.who.int/malaria/publications/world_malaria_report/en/
Sutherland CJ, Polley SD. Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J Infect Dis. 2010;201:1544.
Cao Y, Wang W, Liu Y, Cotter C, Zhou H, Zhu G, et al. The increasing importance of Plasmodium ovale and Plasmodium malariae in a malaria elimination setting: an observational study of imported cases in Jiangsu Province, China, 2011–2014. Malar J. 2016;15:459.
Luo EP, Wang WM, Liu YB, Cao YY, Zhou HY, Xu T. Analysis of epidemic situation of malaria in Jiangsu Province from 2005 to 2014. Chin J Schisto Control. 2015;27:251–4 (In Chinese).
Stephens JWW. A new malaria parasite of man. Ann Trop Med Parasitol. 1922;16:383–8.
Collins WE, Jeffery GM. Plasmodium ovale: parasite and disease. Clin Microbiol Rev. 2005;18:570–81.
Win TT, Lin K, Mizuno S, Zhou M, Liu Q, Ferreira MU, et al. Wide distribution of Plasmodium ovale in Myanmar. Trop Med Int Health. 2010;7:231–9.
Kawamoto F, Liu Q, Ferreira MU, Tantular IS. How prevalent are Plasmodium ovale and P. malariae in East Asia? Parasitol Today. 1999;15:422–6.
Bichara C, Flahaut P, Costa D, Bienvenu AL, Picot S, Gargala G. Cryptic Plasmodium ovale concurrent with mixed Plasmodium falciparum and Plasmodium malariae infection in two children from Central African Republic. Malar J. 2017;16:339.
Win TT, Jalloh A, Tantular IS, Tsuboi T, Ferreira MU, Kimura M, et al. Molecular analysis of Plasmodium ovale variants. Emerg Infect Dis. 2004;10:1235–40.
Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124:755–6.
Holder AA, Blackman MJ, Burghaus PA, Chappel JA, Ling IT, Mccallumdeighton N, et al. A malaria merozoite surface protein (MSP1)-structure, processing and function. Mem Inst Oswaldo Cruz. 1992;87:37–42.
Egan AF, Morris J, Barnish G, Allen S, Greenwood BM, Kaslow DC, et al. Clinical immunity to Plasmodium falciparum malaria is associated with serum antibodies to the 19-kDa C-terminal fragment of the merozoite surface antigen, PfMSP-1. J Infect Dis. 1996;173:765–9.
Takala S, Branch O, Escalante AA, Kariuki S, Wootton J, Lal AA. Evidence for intragenic recombination in Plasmodium falciparum: identification of a novel allele family in block 2 of merozoite surface protein-1: asembo bay area cohort project XIV. Mol Biochem Parasitol. 2002;125:163–71.
Mayengue PI, Ndounga M, Malonga FV, Bitemo M, Ntoumi F. Genetic polymorphism of merozoite surface protein-1 and merozoite surface protein-2 in Plasmodium falciparum isolates from Brazzaville, Republic of Congo. Malar J. 2011;10:276.
Carmen FB, Sergi S, Marina B, Stanisic DI, Alves FP, Camargo EP, et al. Naturally-acquired humoral immune responses against the N- and C-termini of the Plasmodium vivax, MSP1 protein in endemic regions of Brazil and Papua New Guinea using a multiplex assay. Malar J. 2010;9:29.
Kaslow DC, Hui G, Kumar S. Expression and antigenicity of Plasmodium falciparum major merozoite surface protein (MSP119) variants secreted from Saccharomyces cerevisiae. Mol Biochem Parasitol. 1994;63:283.
Putaporntip C, Hughes AL, Jongwutiwes S. Low level of sequence diversity at merozoite surface protein-1 locus of Plasmodium ovale curtisi and P. ovale wallikeri from Thai isolates. PLoS One. 2013;8:e58962.
ESRI. ArcGIS Desktop: Release 10. Redlands: Environmental Systems Research Institute. 2011.
Nicholas K, Nicholas H. GeneDoc: A tool for editing and annotating multiple sequence alignments. Ver. 2.7.000. 1996. Distributed by the author. 1997. http://iubio.bio.indiana.edu/soft/molbio/ibmpc/genedoc-readme.html.
Burland TG. Dnastar’s lasergene sequence analysis software. Methods Mol Biol. 2000;132:71–91.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–26.
Rozas J, Ferrermata A, Sánchezdelbarrio JC, Guiraorico S, Librado P, Ramosonsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol. 2017;34:3299–302.
Tajima F. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics. 1993;135:599–607.
Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709.
Bharti PK. Genetic diversity in the block 2 region of the merozoite surface protein-1 of Plasmodium falciparum in central India. Malar J. 2012;11:78.
Silva JC, Egan A, Friedman R, Munro JB, Carlton JM, Hughes AL. Genome sequences reveal divergence times of malaria parasite lineages. Parasitology. 2011;138:1737–49.
Chenet SM, Branch OH, Escalante AA, Lucas CM, Bacon DJ. Genetic diversity of vaccine candidate antigens in Plasmodium falciparum isolates from the Amazon basin of Peru. Malar J. 2008;7:93.
Atroosh WM, Almekhlafi HM, Mahdy MA, Saifali R, Almekhlafi AM, Surin J. Genetic diversity of Plasmodium falciparum isolates from Pahang, Malaysia based on MSP-1 and MSP-2 genes. Parasit Vectors. 2011;4:233.
Putaporntip C, Jongwutiwes S, Sakihama N, Ferreira MU, Kho WG, Kaneko A, et al. Mosaic organization and heterogeneity in frequency of allelic recombination of the Plasmodium vivax merozoite surface protein-1 locus. Proc Natl Acad Sci USA. 2002;99:16348–53.
Bannister LH, Hopkins JM, Fowler RE, Krishna S, Mitchell GH. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000;17:1467.
Putaporntip C, Hongsrimuang T, Seethamchai S, Kobasa T, Limkittikul K, Cui L, et al. Differential prevalence of Plasmodium infections and cryptic Plasmodium knowlesi malaria in humans in Thailand. J Infect Dis. 2009;199:1143–50.
Versiani FG, Almeida ME, Mariuba LA, Orlandi PP, Nogueira PA. N-Terminal Plasmodium vivax merozoite surface protein-1, a potential subunit for malaria vivax vaccine. Clin Dev Immunol. 2013;2013:965841.
Tanabe K, Sakihama N, Rooth I, Björkman A, Färnert A. High frequency of recombination-driven allelic diversity and temporal variation of Plasmodium falciparum msp1 in Tanzania. Am J Trop Med Hyg. 2007;76:1037–45.
Acknowledgements
The authors thank all study participants, local health officials and doctors for their participation and support.
Funding
This study was funded by grants from National Natural Science Foundation of China (81601787), by Natural Science Foundation of Jiangsu Province (BK20160192, BK20150001), by the Jiangsu Provincial Key Research and Development Program (BE2016631), by the Fundamental Research Funds for the Central Universities funded by the Ministry of Education of China (JUSRP51710A), by Bill & Melinda Gates Foundation (OPP1161962), by National First-Class Discipline Program of Food Science and Technology (JUFSTR20180101), and by the Jiangsu Provincial Project of Invigorating Health Care through Science, Technology and Education.
Availability of data and materials
The data supporting the conclusions of this article are included within the article and its additional file.
Author information
Authors and Affiliations
Contributions
YC and JC conceived and designed the study. SX, JXT, JC and GDZ collected the samples. RLC, XXZ and LMC performed the acquisition of data and data analysis. RLC, XXZ, YHL and YHX conducted the laboratory work, data handling and analysis and reviewed the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee, Jiangsu Provincial Key Laboratory on Parasite and Vector Control Technology, Jiangsu Institute of Parasitic Diseases (JIPD) (IRB00004221), Wuxi, China. Written informed consent was obtained from all of the participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Neighbor-joining tree of 26 unique alleles of the gene encoding msp1 from 18 Plasmodium parasite species. (TIF 3756 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chu, R., Zhang, X., Xu, S. et al. Limited genetic diversity of N-terminal of merozoite surface protein-1 (MSP-1) in Plasmodium ovale curtisi and P. ovale wallikeri imported from Africa to China. Parasites Vectors 11, 596 (2018). https://doi.org/10.1186/s13071-018-3174-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-018-3174-0