
Abstract
Background
The DNA barcoding system using the cytochrome c oxidase subunit 1 mitochondrial gene (cox1 or COI) is highly efficient for discriminating vertebrate and invertebrate species. In the present study, we examined the suitability of cox1 as a marker for Trypanosoma cruzi identification from other closely related species. Additionally, we combined the sequences of cox1 and the nuclear gene glucose-6-phosphate isomerase (GPI) to evaluate the occurrence of mitochondrial introgression and the presence of hybrid genotypes.
Methods
Sixty-two isolates of Trypanosoma spp. obtained from five of the six Brazilian biomes (Amazon Forest, Atlantic Forest, Caatinga, Cerrado and Pantanal) were sequenced for cox1 and GPI gene fragments. Phylogenetic trees were reconstructed using neighbor-joining, maximum likelihood, parsimony and Bayesian inference methods. Molecular species delimitation was evaluated through pairwise intraspecific and interspecific distances, Automatic Barcode Gap Discovery, single-rate Poisson Tree Processes and multi-rate Poisson Tree Processes.
Results
Both cox1 and GPI genes recognized and differentiated T. cruzi, Trypanosoma cruzi marinkellei, Trypanosoma dionisii and Trypanosoma rangeli. Cox1 discriminated Tcbat, TcI, TcII, TcIII and TcIV. Additionally, TcV and TcVI were identified as a single group. Cox1 also demonstrated diversity in the discrete typing units (DTUs) TcI, TcII and TcIII and in T. c. marinkellei and T. rangeli. Cox1 and GPI demonstrated TcI and TcII as the most genetically distant branches, and the position of the other T. cruzi DTUs differed according to the molecular marker. The tree reconstructed with concatenated cox1 and GPI sequences confirmed the separation of the subgenus Trypanosoma (Schizotrypanum) sp. and the T. cruzi DTUs TcI, TcII, TcIII and TcIV. The evaluation of single nucleotide polymorphisms (SNPs) was informative for DTU differentiation using both genes. In the cox1 analysis, one SNP differentiated heterozygous hybrids from TcIV sequences. In the GPI analysis one SNP discriminated Tcbat from TcI, while another SNP distinguished TcI from TcIII.
Conclusions
DNA barcoding using the cox1 gene is a reliable tool to distinguish T. cruzi from T. c. marinkellei, T. dionisii and T. rangeli and identify the main T. cruzi genotypes.
Similar content being viewed by others


Background
How many species are there on Earth? Estimations suggest that approximately 90% of species remain undescribed [1]. The identification and classification of biodiversity is a practice that has always fascinated humankind. The Greek philosopher Aristotle (4th century BC) developed the first classification system, which was used for nearly 2000 years [2]. In the 1700s, Carl Linnaeus [3] developed the concept of binomial nomenclature using Latin, which was the language of educated men at his time. Binomial nomenclature is a standard method for scientists that speak different languages to classify living things to clearly communicate their discoveries. Recently, DNA sequences have been applied in the classification of life forms. However, different methods and DNA regions have been used to compare the same taxonomic groups, frequently leading to conflicting results.
In the search for a simple method to identify and compare species, Hebert et al. [4] proposed DNA barcoding, a new system of species identification using the cytochrome c oxidase subunit 1 mitochondrial gene (cox1 or COI) as a standardized single molecular marker for the classification of animal species. One of the requirements of the DNA barcoding approach is that species identification is associated with a voucher belonging to a curated biological collection, enabling follow up and a strategy for corroborating species identification [5]. Until recently, national barcode networks have been established in 11 countries, including Brazil, which uses the Brazilian Barcode of Life (BrBOL) [6].
In addition to the identification of known and new species, barcoding with the cox1 gene is suggested as a standard for cryptic taxa discovery, association of different life stages of the same species and wildlife conservation genetics [5]. Cox1 appears to have a better phylogenetic signal than the other mitochondrial genes [7]. Some authors argue that the evolution of the cox1 gene is sufficiently rapid to discriminate between closely related species and investigate intraspecific diversity [4, 8]. The cox1 region is highly efficient for discriminating vertebrate and invertebrate species [5, 9, 10], but is not suited for plants and some fungal species [4, 11, 12]. The use of cox1 for identifying protozoa and other unicellular organisms is still in its initial stage but has been demonstrated as a promising barcode marker for dinoflagellates, Paramecium sp., Nemertea [13,14,15]. Moreover, joint analyses of nuclear and mitochondrial markers may be necessary to distinguish species [1].
Therefore, the small subunit ribosomal DNA (SSU rDNA) has been proposed as a first step for a protist barcode, followed by the use of a second marker specific for each group [6]. To confirm a gene as a barcode marker, it is essential to calculate the “barcoding gap”, that is, the gap between the maximum intraspecific and minimum interspecific genetic distances that establish the limits between species [16]. Several molecular targets for trypanosomatid identification have previously been used: internal transcribed spacer (ITS) [17], mini-exon [18], glucose-6-phosphate isomerase (GPI) [19,20,21], cytochrome b (cytb) [22, 23], multilocus sequencing typing (MLST) [24], the variable regions V7 V8 of SSU rDNA, and glyceraldehyde-3-phosphate dehydrogenase (gGAPDH) genes [22, 25, 26]. However, there is no consistency concerning which DNA region to use as a trypanosomatid barcode, and none of these markers were evaluated as a universal marker for biodiversity analysis.
With the emergence of molecular techniques with higher analytical power, an increasing number of new species of Trypanosoma have been reported [26, 27] and it is becoming increasingly clear that there is a high diversity of Trypanosoma spp. within the subgenus Schizotrypanum of Trypanosoma. The extreme morphological similarity and phylogenetic proximity of these species, and the use of tools with lower analytical power, resulted in the isolation of numerous species of T. (Schizotrypanum) from the wild transmission cycle remaining only at the generic level or wrongly diagnosed as Trypanosoma cruzi, highlighting the importance of a universal method to characterize T. cruzi, its subpopulations and representatives of the subgenus Schizotrypanum.
Trypanosoma cruzi (Trypanosomatida: Trypanosomatidae) is a successful parasite that is capable of establishing an extracellular cycle in dozens of triatomine species (Hemiptera: Reduvidae) and infecting almost every cellular type from more than 100 mammal species distributed from the south of the USA to the south of Latin America [28, 29]. Although described as one taxon, T. cruzi has a remarkable genetic heterogeneity that was already recognized by the very first authors who researched it [30,31,32]. The T. cruzi population structure has been classified as clonal or, at least, primarily clonal [32, 33] and hybridization events have also been observed, suggesting that genetic recombination occurs in this group [33, 34]. Moreover, the extent to which these hybridization events are responsible for the enormous heterogeneity of this parasite remains a controversial issue [19, 35,36,37]. Several techniques, (biological, biochemical and molecular) applied to define T. cruzi subpopulations have led to different designations (Fig. 1) [25, 30, 35, 38,39,40,41,42,43,44,45,46,47,48,49,50,51]. Presently, T. cruzi subpopulations are assembled in six discrete typing units (DTUs) - named TcI, TcII, TcIII, TcIV, TcV, TcVI [29] - and a genotype called Tcbat, initially described as being associated with Chiroptera [25], although it was previously isolated from a child [52] and from mummified human tissue [53]. However, Barnabé et al. [51] recently proposed the subdivision of T. cruzi into three mitochondrial clades (mtTcI, mtTcII and mtTcIII) based on the analysis of the nucleotide sequences available in the GenBank database, showing that the classification of T. cruzi subpopulations remains a debatable issue.

Designations of T. cruzi subpopulations through time and according to the method employed. The numbers represent dates of publication. The red star indicates current consensus of T. cruzi nomenclature and subdivisions
In the present study, we tested cox1 as a DNA barcode to identify T. cruzi from other closely related species belonging to the subgenus Schizotrypanum and to examine the genetic diversity within T. cruzi and its DTUs to further understand the ecology of the species of T. (Schizotrypanum). Additionally, we evaluated cox1 as a target to also identify Trypanosoma (Tejeraia) rangeli, as this trypanosome shares the geographical distribution, vectors and mammal hosts of T. cruzi and is also included in the T. cruzi clade [54]. To evaluate the occurrence of mitochondrial introgression events and the presence of hybrid genotypes we tested a combination of cox1 (uniparental inheritance) and the nuclear gene glucose-6-phosphate isomerase (GPI) (biparental inheritance). GPI is one of the genes sequenced for an extensive number of T. cruzi isolates distributed over several geographic regions [51], enabling a comparison of the sequences generated in the present study. Thus, the precise identification of these Trypanosoma species is of the utmost importance. In addition, the present study will enhance the amount of nucleotide sequences available for comparison, since the GenBank database still lacks a broader deposit on trypanosomatid sequences for the cox1 gene.
Methods
Samples
The present study included 62 isolates of Trypanosoma spp. obtained from 16 different genera of free-ranging wild mammals and from triatomines of the genera Rhodnius and Triatoma, distributed in five of the six Brazilian biomes (Amazon Forest, Atlantic Forest, Caatinga, Cerrado and Pantanal) (Table 1 Fig. 2b). Isolates were cryopreserved and deposited in the Coleção de Trypanosoma de Mamíferos Silvestres, Domésticos e Vetores, Fiocruz - COLTRYP (Oswaldo Cruz Foundation, Rio de Janeiro - RJ/Brazil) in previous studies. Details on animal capture and parasite culture methods are described elsewhere [55,56,57]. After thawing, the cells were sown in axenic culture media (NNN and LIT). When the cultures reached the exponential growth phase, DNA was extracted using the phenol-chloroform method, as described elsewhere [58]. The T. dionisii samples used in the present study were molecularly identified using 18S (SSU) and gGAPDH sequences and subsequently deposited in COLTRYP.

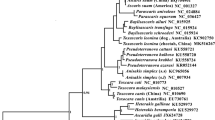
Phylogenetic tree based on cox1 and the geographical origin of the isolates under study. a The cox1 gene differentiates T. cruzi DTUs TcI, TcII, TcIII and TcIV, Tcbat, T. c. marinkellei, T. dionisii and T. rangeli. The heterozygous hybrids TcV and TcVI cannot be differentiated and were placed into the same cluster as TcIV. The numbers at the nodes correspond to NJ, ML, MP and BI support values, respectively (only values >60 are shown). The scale-bar shows the number of nucleotide substitutions per site. The different haplotypes correspond to the diversity observed in cox1 sequences and are represented by colors in the tree. b The map represents the distribution of the characterized isolates among Brazilian biomes. Each colored circle indicates a different haplotype. Circle size represents the number of haplotypes. Abbreviations: Brazilian states: AC, Acre; BA, Bahia; CE, Ceara; ES, Espírito Santo; GO, Goiás; MS, Mato Grosso do Sul; PA, Pará; PI, Piauí; RJ, Rio de Janeiro; SC, Santa Catarina; TO, Tocantins
Nine T. cruzi references were also genotyped and used as DTU standards. Colombiana (COLPROT 004), Dm28c (COLPROT 010), Y (COLPROT 106), 3663 (COLPROT 608), 4167 (COLPROT 607) DNA samples obtained from the Protozoa Collection - COLPROT (FIOCRUZ, Rio de Janeiro, Brazil); Sc43cl1, Bug2148cl1, CLBrener and Tulacl2 DNA samples were a kind gift from Dr. Michael Miles from the London School of Hygiene & Tropical Medicine (London, UK). The T. rangeli lineage R1625 DNA sample (COLPROT 002) was obtained from COLPROT (Table 1). In addition, cox1 and GPI sequences were retrieved from the GenBank database and used as references (Table 1).
PCR and sequencing
A fragment of the mitochondrial gene cox1 was amplified using a set of forward (5′-CCA WAC AAC AAA CAT ATG ATG CTG C-3′) and reverse (5′-TCC HGA TAT GGT WTT KCC YCG-3′) primers. Polymerase chain reaction (PCR) was conducted in a 50 μl final reaction volume containing 2.5 mM MgCl2 (Invitrogen, Carlsbad, California, USA), 0.25 mM of each nucleotide (Thermo Scientific, Waltham, Massachusetts, USA), 0.25 mM of each primer (IDT, Coralville, Iowa, USA), 1.0 U of Platinum Taq DNA polymerase (Invitrogen), and 50 ng of DNA template. The amplification was performed using a Veriti 96-Well Thermal Cycler (Applied Biosystems, Foster City, California, USA) with the following cycle conditions: denaturation at 95 °C for 3 min; followed by 35 cycles at 95 °C for 1 min, 54 °C for 1 min, and 72 °C for 1 min; and a final elongation step at 72 °C for 10 min. Nucleotide sequences were also determined using a fragment of the nuclear gene GPI. The primers and cycling conditions are described elsewhere [20]. The PCR products were separated on 1.5% agarose gels and stained with GelRed (Biotium Inc., Fremont, California, USA). The fragments were purified using the Wizard Genomic DNA Purification Kit, according to manufacturer’s instructions (Promega, Madison, Wisconsin, USA), and direct sequencing of both strands of DNA was performed with BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) using an ABI 3730 DNA sequencer available at the RPT01A/FIOCRUZ sequencing facilities.
Data analysis
The sequences were manually edited using Geneious software version 8.1.6. (Biomatters, Auckland, New Zealand) and aligned using the CLUSTAL X version 2.1 multiple alignment program [59]. All sequences were translated to confirm the absence of premature stop codons. All sequences generated were deposited in the GenBank database (Table 1).
The heterozygous hybrid lineages (TcV and TcVI) amplified for GPI were subjected to haplotype reconstruction using the PHASE algorithm implemented in DnaSP v5.10.01 [60].
The neighbor-joining (NJ) method and Kimura 2-parameters (K2P) model were applied for both cox1 and GPI genes according to the barcode approach [4]. NJ analyses were performed with MEGA version 6 [61]. For each node, bootstrap percentages (BP) were computed after 1000 resamplings.
The maximum likelihood (ML) method was also applied to each topology. The model of nucleotide substitution that best fitted the cox1 data was the Hasegawa-Kishino-Yano’s model (HKY), with a gamma-distributed rate (Γ). For GPI, the best-fit model was the Tamura-Nei model, with a gamma-distributed rate. These models were selected using the Akaike Information Criterion corrected for small samples (AICc) approach implemented in the program jModelTest [62]. ML analyses were performed using PhyML 3.0 [63]. For each node, BP were computed after 1000 resamplings.
Maximum Parsimony (MP) analyses were performed using PAUP* 4.0b10 [64]. For the tree search and bootstrap we used a heuristic search with 100 random sequence addition replicates through tree bisection and reconnection (TBR) branch-swapping algorithm. Bayesian inference (BI) was run in MrBayes v3.2.6 [65] with a general time reversible model with gamma-distributed rate variation across sites and a proportion of invariable sites (GTR + Γ + I). The runs converged after 1,000,000 generations, by sampling every 100th generation and discarding the first 25% of the trees as ‘burn-in’. Cox1 and GPI sequences were concatenated in SequenceMatrix 1.8 [66] and submitted to NJ, ML, MP and BI analysis as described above.
The number of haplotypes, nucleotide diversity (π) and haplotype diversity (Hd) were calculated for both genes, except for Tcbat, which had a single sequence available in GenBank. The analyses were run in DnaSP v5.10.01 [60].
Molecular species delimitation was evaluated using distance-based methods and coalescent-based models. Distance-based analyses included the pairwise intraspecific and interspecific distances calculated using MEGA version 6 [61] and the Automatic Barcode Gap Discovery (ABGD) method, which detects a gap in the distribution of pairwise distances and uses this information to partition the sequences into groups of hypothetical species [67]. ABGD analysis was conducted in the web version: Jukes Cantor, K2P and p distances were calculated, and the remaining parameters were used as default [67]. Coalescent-based analysis included the single-rate Poisson Tree Processes (PTP) model [68], which considers that every species evolved at the same rate, and the multi-rate Poisson Tree Processes (mPTP) [69], which assumes a different evolution rate for each species. PTP and mPTP analyses were conducted using the web version of this software [69].
Results
Cox1 and GPI gene fragments were successfully amplified for a panel of 62 Trypanosoma spp. isolates and ten reference strains (Table 1). All sequences were translated to amino acids and compared to cox1 and GPI proteins. No indels (insertions/deletions) or stop codons were detected. No pseudo genes or contaminants were observed.
Phylogenetic tree reconstruction using cox1 as barcode
The clusters observed in the cox1 trees were the same for all methods tested, indicating that these groups are robust and do not depend on the evolutionary methods selected (Fig. 2a). Cox1 discriminated species belonging to the subgenus Schizotrypanum and T. cruzi DTUs. TcI and Tcbat were closely related but clearly constitute two different DTUs with a statistical support of 99, 70, 92 and 100 in NJ, ML, MP and BI analysis, respectively (Fig. 2a). TcIII and TcIV sequences were separated into two different clusters with bootstrap values of 99, 87, 93 and 100 in NJ, ML, MP and BI analysis, respectively. For the heterozygous hybrid lineages, TcV and TcVI formed an indistinguishable group in the same cluster as TcIV.
Phylogenetic tree reconstruction using GPI
Both GPI and cox1 helped recognize Schizotrypanum species, but not T. cruzi DTUs. Depending on the method used for the phylogenetic tree reconstruction there was a slightly different topology. Tcbat and TcI clustered together and could not be differentiated in the tree (Fig. 3). In NJ analysis TcIII constituted a separate DTU close to TcI (Fig. 3a). However, with ML, MP and BI methods TcI, Tcbat and TcIII clustered together (bootstrap of 80, 77 and 82, respectively) (Fig. 3b). GPI sequences generated for TcV and TcVI presented electropherograms with double peaks (i.e. with two bases at the same position) and were submitted to haplotype reconstruction prior to use in the final alignments and tree reconstructions. This analysis resulted in two sequences for each hybrid sample corresponding to alleles. One allele was closer to TcII, and the other allele was closer to TcIII (Fig. 3).

Phylogenetic tree based on nuclear gene GPI. a Tree inferred with neighbor-joining method and Kimura-2-parameter model. GPI recognizes and differentiates T. cruzi DTUs TcI, TcII, TcIII and TcIV, T. c. marinkellei, T. dionisii and T. rangeli. One allele of TcV and TcVI sequences cluster with TcII, and the other allele clusters with TcIII. Tcbat was placed in the same cluster as TcI. b The tree inferred from maximum likelihood, parsimony and Bayesian inference. T. cruzi, T. c. marinkellei, T. dionisii and T. rangeli are clearly separated from each other. DTUs TcI and TcII are the most genetically divergent. Tcbat, TcI and TcIII fall in the same cluster. One allele of TcV and TcVI clustered with TcII, and the other allele clustered with the group comprising TcI, TcIII and Tcbat. The numbers at the nodes correspond to ML, MP and BI support values, respectively (only values >60 are shown). The scale-bar shows the number of nucleotide substitutions per site
The geographical distribution of the trypanosomatid isolates under study is represented in Fig. 2b. Both cox1 and GPI sequences demonstrated the differences between T. cruzi, T. c. marinkellei, T. dionisii and T. rangeli and also, to some degree, T. cruzi lineages. Cox1 and GPI phylogenies equally demonstrated that TcI and TcII are the most genetically distant branches, but showed differences concerning the positions of the DTUs TcIII, TcIV, TcV, TcVI and Tcbat in the phylogenetic trees. The mitochondrial gene cox1 may be a better discriminator of T. cruzi lineages, identifying five DTUs and TcV/TcVI as a single group (Fig. 2a). Additionally, these differences between mitochondrial and nuclear tree topologies, no incongruence was observed in DTU assignment (Table 1), and mitochondrial introgression events were absent in the present sample set.
Identification of T. cruzi DTUs through single nucleotide polymorphisms (SNPs)
Some T. cruzi sequences were not clearly assigned to a DTU based solely on information from the trees. In the cox1 tree, TcIV and TcV/TcVI sequences were arranged in the same cluster (Fig. 2a), whereas in GPI analyses, TcI, TcIII and Tcbat separation was blurred (Fig. 3). Therefore, the multiple sequence alignment of cox1 and GPI data was considered for the evaluation of single nucleotide polymorphisms (SNPs) using T. cruzi sequences only. These polymorphisms were informative to DTU differentiation for both genes.
In the cox1 gene fragment analysis, we identified 84 polymorphic sites. We observed a single nucleotide polymorphism (SNP) that differentiates the heterozygous hybrids from TcIV sequences. A T (thymine) was present at position 1264 of the cox1 gene in all ten TcIV sequences analyzed, whereas TcV and TcVI sequences display a C (cytosine) at the same site. No polymorphism differentiating TcV from TcVI was observed (Additional file 1: Figure S1).
In the GPI analysis we identified 20 polymorphic sites concerning all T. cruzi sequences. A thymine at position 315 separates Tcbat from TcI (cytosine) and one guanine to adenine change separates TcI from TcIII at position 396 of the gene (Additional file 2: Figure S2). No polymorphism discriminating TcV from TcVI sequences was observed.
Phylogenetic tree reconstruction using concatenated data
The concatenation of cox1 and GPI gene fragments confirmed, with robust statistical support values, the separation of species belonging to the subgenus Schizotrypanum and the T. cruzi DTUs.
TcI, TcII, TcIII and TcIV sequences constituted clearly separated clades. In addition, concatenated data supported Tcbat as a sister clade to TcI. The heterozygous hybrids TcV and TcVI could not be differentiated and formed a cluster separate from TcIV. The topologies observed in the trees were the same for the four methods tested (NJ, ML, MP and BI) and were supported by values above 80 in the main branches (Fig. 4).

Phylogenetic tree based on the concatenation of cox1 and GPI sequences. The concatenated data show a clear separation between T. cruzi DTUs TcI, TcII, TcIII and TcIV, Tcbat, T. c. marinkellei, T. dionisii and T. rangeli. The heterozygous hybrids TcV and TcVI were not differentiated from each other. The numbers at the nodes correspond to NJ, ML, MP and BI support values, respectively (only values >60 are shown). The scale-bar shows the number of nucleotide substitutions per site
Genetic diversity evaluated with cox1 and GPI genes
Although the cox1 gene did not distinguish TcV from TcVI, this gene demonstrated diversity in T. cruzi DTUs TcI, TcII and TcIII and T. c. marinkellei and T. rangeli (Additional file 3: Table S1, Fig. 2b). GPI sequences also displayed distinct haplotypes in T. c. marinkellei and T. rangeli, but lower intra-DTU diversity. The correlation between haplotype and geographical area or host species was not evident.
In the cox1 analysis, TcI was demonstrated as the most diverse DTU with the highest nucleotide diversity and haplotype diversity of all DTUs, followed by TcIII and TcII. The TcI isolates and reference strains in the present study were distributed in eight haplotypes throughout five Brazilian biomes (Fig. 2b) and six different host orders (Carnivora, Chiroptera, Didelphimorphia, Hemiptera, Primates and Rodentia) (Table 1). In the Amazon, we observed the highest number of different TcI haplotypes in the Para state (hap 2, 3 and 5). Haplotypes 2 and 5 were observed only in the Para State, while haplotype 3 was widely distributed and detected in four different biomes (Amazon, Atlantic Forest, Caatinga and Pantanal). Additionally, in Para, we observed two different haplotypes in three TcI isolates derived from Didelphis marsupialis (Table 1). Thus, establishing a correlation between the TcI haplotype and location or host species would be premature. Substantial genetic diversity was also observed in TcIII sequences. In four TcIII isolates derived from Monodelphis domestica (Cerrado biome), Galictis vittata (Atlantic Forest), Rhodnius pictipes (Amazon biome) and a reference strain from Panstrongylus geniculatus (Amazon biome), we identified three different haplotypes (Table 1, Fig. 2b). These TcII isolates were divided into three haplotypes and originated from primates, a marsupial and triatomines captured in the Atlantic Forest and from a rodent captured in the Caatinga biome. The reference strains were isolated from humans in the Atlantic Forest. The three TcII haplotypes were distributed in the state of Rio de Janeiro, isolated from a Philander frenatus and two Leontopithecus rosalia (Table 1). Genetic diversity was not detected among the TcIV, TcV and TcVI isolates. Diversity could not be evaluated for Tcbat, reflecting the unique sequence available for this DTU.
The seven T. c. marinkellei isolates displayed three haplotypes (Additional file 3: Table S1, Fig. 2b). One haplotype was formed by isolates originated from P. discolor from the Goiás State (Cerrado) and the Bahia state (Atlantic Forest); another haplotype comprised isolates originated from P. hastatus from the Acre State (Amazon biome); and a third group was formed by the reference strain TCC 344, isolated from C. perspicillata (Amazon) (Table 1, Fig. 2a). Despite the low number of isolates, an apparent correlation between haplotype and host species was observed.
We compared two T. rangeli isolates previously identified as lineages C and D [54, 70, 71]. Sequences generated with cox1 exhibited two different haplotypes (Table 1, Additional file 3: Table S1). Trypanosoma dionisii sequences showed no diversity. Isolates derived from two different species of phyllostomid bats from the same geographical area and collected during the same field expedition (Table 1, Additional file 3: Table S1).
In the GPI analysis, intra-DTU diversity was lower than observed with cox1. TcI, TcII, TcIII and TcIV sequences displayed two different haplotypes each. In TcIV, one haplotype was formed by the nine isolates identified herein and the reference strain 4167, while the other haplotype only comprised the reference strain CANIII, which was not available for cox1 analysis. No diversity within TcV/TcVI sequences was detected.
The highest diversity in GPI sequences was observed in T. c. marinkellei with five haplotypes. One haplotype comprised three isolates from the Goiás state (Cerrado) and the other haplotypes corresponded to the other four isolates (Additional file 3: Table S1, Fig. 3). The two T. rangeli sequences generated with GPI were identified elsewhere as lineage C [71]. However, these sequences displayed distinct haplotypes (Additional file 3: Table S1, Fig. 3). No diversity within T. dionisii sequences was observed. No correlation between haplotype, host or geographical location could be established.
Barcoding gap and species delimitation with cox1 and GPI sequences
Based on the analysis of the barcode gaps we assessed and compared the efficiency of cox1 and GPI for the identification of trypanosomatids. In cox1 the mean genetic distance between T. cruzi sequences and T. c. marinkellei was 13%. The mean intraspecific divergence for T. cruzi was 7.6%, a value lower than the mean interspecific value. However, comparing the minimum and maximum values, we observed an overlap of the genetic distances between T. cruzi and T. c. marinkellei (10.1–15.3%) and the intraspecific divergence for T. cruzi (0–12.7%), indicating the absence of a limit that separates these two subspecies (i.e. absence of a barcoding gap). The divergence between T. cruzi and T. dionisii limits was 13.0–16.0%, while the divergence between T. cruzi and T. rangeli was 14.7–21.3%, indicating that the genetic distance separating T. cruzi, T. dionisii and T. rangeli as different species is 0.3 and 2.0%, respectively (Additional file 4: Table S2).
GPI was not as discriminative as cox1. The mean interspecific divergence between T. cruzi sequences and T. c. marinkellei, T. dionisii and T. rangeli was 3.6, 7.8 and 13.7%, respectively.
We observed differences between T. c. marinkellei sequences in the trees (Fig. 3) confirmed by an intraspecific distance ranging from 0 to 1.0% (Additional file 5: Table S3). Distance within T. rangeli sequences was 0.7% and there was no genetic difference between T. dionisii sequences. For T. cruzi, the intraspecific genetic distance ranged from 0 to 3.0%, i.e. lower than the mean interspecies values (Table 2). Similar to cox1, we observed an overlap of the genetic distances between T. cruzi and T. c. marinkellei with GPI, indicating the absence of a “barcoding gap”.
Trypanosoma cruzi and T. c. marinkellei were separated into distinct groups according to ABGD, PTP and mPTP analysis using cox1 and GPI data. Here, we report the results for the three substitution models and both initial and recursive partitions in the output of ABGD. The results varied from 6 to 21 for cox1 depending on the substitution model used, confirming diversity in T. cruzi sequences and the separation of T. cruzi, T. c. marinkellei, T. rangeli and T. dionisii (Table 2). Trypanosoma cruzi sequences were divided into a minimum of three groups in the three models tested. One group corresponded to Tcbat and TcI; another group corresponded only to TcII sequences only; and a third group comprised TcIII, TcIV, TcV and TcVI sequences. A maximum of 13 different groups were observed, with TcI sequences divided into eight different groups, showing the higher intra-DTU diversity of TcI compared to the other DTUs (Additional file 6: Table S4). In all models and partitions, T. dionisii sequences were arranged in one group.
The number of ABGD groups for GPI data varied from 2 to 14 depending on the model applied (Table 2). Trypanosoma cruzi sequences were separated into different groups. TcI sequences were grouped together, showing less variability with GPI. TcII sequences were divided into two groups: one group with only TcII sequences and another group with TcII, TcV (allele 1) and TcVI (allele 1). TcIII sequences were grouped together with sequences representing the other TcV and TcVI alleles. TcIV sequences were combined in one group, except for the reference strain CANIII, which was placed in a separated group. Trypanosoma cruzi marinkellei and T. rangeli sequences were divided into groups, reaffirming their diversity (Additional file 7: Table S5), while T. dionisii sequences formed one group in all tests (Table 2, Additional file 7: Table S5).
The number of groups recovered by ABGD was higher than the number of species studied. However, this finding confirms the genetic diversity within T. cruzi DTUs, T. c. marinkellei and T. rangeli observed in the phylogenetic trees (Figs. 2a and 3).
The PTP and mPTP models identified, respectively, a total of 10 and 7 putative species in the cox1 dataset (Table 3). Four of these putative species were subdivisions of T. cruzi, indicating the heterogeneity of this taxon. The PTP model also recognized diversity within T. c. marinkellei and T. rangeli sequences.
PTP and mPTP provided a similar number of putative species for GPI sequences (Table 3). Trypanosoma cruzi was divided into three groups, and T. c. marinkellei sequences were allocated into one group. The difference between models was observed in the T. rangeli sequences, separated into two groups or placed into one group.
In cox1 and GPI analysis using both models, T. cruzi, T. c. marinkellei, T. rangeli and T. dionisii were recognized as different species. The diversity of T. cruzi was confirmed, and no diversity was observed in T. dionisii sequences.
Discussion
In the present study, the DNA barcoding approach using the cox1 gene has been demonstrated to be efficient at recognizing Trypanosoma species and their major subpopulations. With cox1, we distinguished T. cruzi from T. c. marinkellei, T. donisii and T. (Tejeraia) rangeli, fulfilling the main DNA barcode demands of a short gene fragment that can be sequenced in diverse sample sets and generating comparable sequences that enable the distinction of species from each other [4]. We also generated a library of trypanosome sequences for cox1 and GPI genes. Each specimen analyzed is linked to an identification number, collection date, country, region and host of origin, geographical coordinates and other information that enable the tracking of the origin of the specimen and ensure the reproducibility of subsequent experiments.
Trypanosoma cruzi is currently divided into seven DTUs [29]. Using cox1, we identified five T. cruzi groups (TcI, TcII, TcIII, TcIV and Tcbat). The DTUs TcI and TcII are consistently shown as the most genetically distant groups, well separated by cox1 in all four methods tested (Fig. 2a). This structure has been observed by other authors in trees with high bootstrap support values, sustaining TcI and TcII as the two discernible DTUs, independently of gene or method used [25, 51, 72, 73]. Furthermore, this system showed the potential for separating genetically closer DTUs. We observed Tcbat as a separated cluster within T. cruzi and its proximity to TcI, consistent with Marcili et al. [25]. According to other studies using cytb, V7 V8 rRNA and gGAPDH genes this relationship is unanimous [26, 51, 74, 75]. However, the data on Tcbat are still limited, as only one sequence was generated with cox1 available in GenBank. Our cox1 sequences also showed the homozygous hybrids TcIII and TcIV forming distinct groups in all methods tested (Fig. 2a). These DTUs are proposed to have been originated from the genetic exchange between TcI and TcII and evolved separately giving origin to TcIII and TcIV [76, 77]. The genetic proximity between TcIII and TcIV is undeniable, and their separation and position in the phylogenetic trees is altered by the gene and method of inference selected. Based on cytb (inferred by MP), V7 V8, gGAPDH, GPI genes and MLST approaches, TcIII and TcIV were identified as two separate DTUs [24,25,26, 28, 51]. However, in other studies using the cytb gene (inferred by neighbor-joining and maximum likelihood), TcIII and TcIV were identified as a single group [34, 51, 77]. TcV and TcVI sequences were indistinguishable and clustered with TcIV (Fig. 2a), consistent with previous cytb mitochondrial gene results [25]. In some studies, independently of the molecular marker and phylogenetic method applied, TcV and TcVI were also indistinguishable from each other. However, these hybrids clustered together with TcII or TcIII when analyzed using nuclear markers [25, 26, 34, 78]. In contrast, in studies using 4 to 10 gene fragments and neighbor-joining trees, TcV and TcVI appeared as two distinct DTUs [24, 79]. In the cox1 analysis, we observed one SNP that differentiates TcIV sequences from TcV/TcVI (Additional file 1: Figure S1). This nucleotide polymorphism, combined with the phylogenetic tree, was demonstrated as relevant to DTU assignment. However, TcV and TcVI are the less conspicuous lineages, and their separation remains an issue.
Cox1 was also suitable to determine diversity within DTUs TcI, TcII and TcIII (Additional file 3: Table S1). The number of sequences classified as TcI and the number of different haplotypes in these DTU sequences were the highest, compared to the other DTUs. The diversity within TcI is consistent with previous studies and may be explained by TcI being a multi-host lineage widely distributed throughout Brazilian biomes, representing the DTU with the largest set of samples analyzed, and consequently, the DTU with the most published studies compared to the other DTUs [20, 51, 52, 80, 81]. The nucleotide and haplotype diversity of the TcII sequences generated using cox1 were lower. However, this effect may not reflect the reality, but rather may show subsampling. In the Rio de Janeiro State, we observed one TcII haplotype circulating in a specimen of L. rosalia and a different haplotype in another specimen of L. rosalia. This observed diversity may reflect primates captured in different years and the changes in TcII haplotype circulation in that area. Nevertheless, the same host can harbor different haplotypes from the same parasite, and one haplotype can prevail over another in different moments of isolation. Diversity within TcII has previously been demonstrated through the sequencing of the glycoprotein 72 gene (gp72) and showed that this DTU has a higher distribution range than previously considered [56]. The high haplotype diversity observed in TcIII could result from overestimation, since we identified three different haplotypes in the four sequences analyzed, belonging to isolates from different Brazilian regions. However, this scenario shows TcIII distributed in a wide geographical range, infecting marsupials, carnivores and triatomines (Table 1). This finding clearly indicates that the richness within TcIII, and its dispersion is yet to be explored. Diversity in TcIII has previously been observed based on V7 V8, cytb, GPI, MLST approaches, but no correlation with geographical area or host species was evident [24, 25, 28]. In the present study, TcIV samples were isolated from triatomines and rodents from the same geographical area (Table 1). This aspect might explain the observation of only one haplotype in TcIV sequences. However, isolates from rodents were collected 11 years before the parasites isolated from the intestinal content of triatomines. Thus, we propose that TcIV haplotype circulation in the Pantanal area was at least equally predominant throughout more than a decade. However, the TcIV reference was isolated from a triatomine in the Amazon region (Table 1) and had the same haplotype as the Pantanal isolates. This finding could reflect the conservation of the cox1 region in TcIV. A correlation between TcIV diversity and the geographical region has been suggested by other studies based on mitochondrial genes cytb and cytochrome c oxidase subunit 2-NADH dehydrogenase subunit 1 (cox2-nad1) [28, 34, 77].
The cox1 tree topologies, independently of the method applied, showed T. c. marinkellei as a sister clade to the monophyletic clade formed by all T. cruzi DTUs (Fig. 2a); we also observed genetic diversity within T. c. marinkellei (Additional file 3: Table S1). Even with the characterization of a low number of isolates, the samples were separated into two groups, and a sequence retrieved from GenBank was positioned in a third group (Fig. 2a). Heterogeneity within T. c. marinkellei has previously been reported [75, 82, 83]. Subdivision into two major groups (T.c.m.I and II) and a potential third group (lineage Z) was proposed using multilocus enzyme electrophoresis (MLEE) and random amplified polymorphic DNA (RAPD). No association with a host or geographical distribution was confirmed [82]. This lack of evidence for an association and the different markers used prevented the comparison of these data.
We also observed genetic differences between T. rangeli reference sequences R1625 and SC58 classified, respectively, as lineage C and lineage D [54, 70]. Previous studies have proposed the subdivision of T. rangeli in five lineages (A-E), based on spliced leader and SSU rDNA [54, 70, 71]. Even with only two sequences, we suggest that cox1 can distinguish different T. rangeli lineages and is a promising tool for use in species identification.
In the present study, we showed the first T. dionisii sequences for the cox1 gene. The nucleotide sequences were deposited in GenBank, contributing to the enhancement of the barcode public library for Trypanosoma species. We did not observe diversity among these sequences (Fig. 2a, Additional file 3: Table S1), likely because the samples were collected from bats of the same area. No subdivisions in groups or subpopulations have been proposed for T. dionisii until recently. Although potential diversity within this species can be observed in cytb and 18S (SSU) phylogenetic trees, these data were not reported [84].
The concomitant analysis of the mitochondrial gene cox1 and the nuclear gene GPI enable the confirmation of the absence of mitochondrial introgression events in the sample set. The frequency of this genetic phenomenon is unknown and has primarily been observed in heterozygous hybrid DTUs TcV and TcVI, where uniparental inheritance of maxicircle kDNA is the rule [28, 36, 56, 85]. Cox1 has limitations and does not work as a single barcode in all situations [1], and since T. cruzi possesses heterozygous hybrid lineages and mitochondrial introgression events have previously been reported, we proposed this cox1-GPI barcoding system. Additionally, the concatenated analysis of cox1 and GPI confirmed T. cruzi DTU separation (Fig. 4).
Consistent with the cox1 results, GPI distinguished T. cruzi from T. c. marinkellei, T. donisii and T. (Tejeraia) rangeli. Additionally, with GPI, TcI and TcII were separated into two conspicuous groups. This nuclear gene recognizes a lower number of T. cruzi groups (Fig. 3) and considerably lower intra-DTU diversity compared to cox1 (Additional files 4 and 5: Tables S2 and S3). In contrast to the cox1 results, Tcbat and TcI were clustered together. The lower power of resolution to discriminate DTUs and intra-DTU diversity might reflect the fact that GPI is a housekeeping nuclear gene, which shows a lower evolution rate than mitochondrial genes [28, 85]. Furthermore, we compared the GPI sequences with the single Tcbat sequence available in GenBank and the results may change depending on the number of sequences available. It is likely that a larger set of Tcbat sequences would resolve the incongruence between the mitochondrial and nuclear gene trees. In addition, we also observed differences in DTU placement in the trees according to the method of inference in the GPI analyses. TcIII sequences formed a cluster separate from TcI when we applied the neighbor-joining method and clustered together with TcI when maximum likelihood, parsimony and Bayesian inference were used. This effect may reflect the fact that the neighbor-joining method was based on the genetic distance matrix, where a pairwise distance matrix is produced, and the tree is inferred from this matrix; maximum likelihood, parsimony and Bayesian inference were character-based methods of inference where each position of the alignment is analyzed [86]. However, in the GPI analysis, we detected SNPs that enable the differentiation of TcI, TcIII and Tcbat (Additional file 2: Figure S2). We observed diversity within T. c. marinkellei and generated the first five T. dionisii sequences for GPI. We could not compare T. rangeli sequences generated with GPI, as both sequences analyzed belonged to lineage C [54, 70].
Barcoding gap in trypanosomatids is still an unresolved issue. Therefore, there are no parameters or cut-off values available to compare with these results. As expected, we did not observe a barcoding gap between T. cruzi and T. c. marinkellei because T. c. marinkellei is considered a subspecies of T. cruzi [87] (Additional files 4 and 5: Tables S2 and S3).
The debate concerning the definition of species will always exist since species are not discrete units, but rather continuous entities.
Conclusions
The use of partial sequences of cox1 and GPI genes can clearly identify and separate T. cruzi samples from T. c. marinkellei, T. dionisii and T. rangeli. The two-locus barcoding system using cox1 and the nuclear gene GPI revealed that mitochondrial introgression was absent from the sample set. Additionally, the resolution of cox1 at the intraspecific level shows great potential for DTU characterization, separating five DTUs and recognizing the heterozygous hybrids TcV and TcVI as one group different from all the other DTUs; the resolution of cox1 at the intraspecific level also demonstrates intra-DTU genetic diversity. Moreover, with cox1, we evaluated the diversity within T. c. marinkellei sequences and identified two T. rangeli lineages. Therefore, the cox1 gene is a promising DNA barcode for studying the genus Trypanosoma and represents a simple, fast and reliable marker.
Abbreviations
- ABGD:
-
Automatic Barcode Gap Discovery
- BI:
-
Bayesian inference
- BrBOL:
-
Brazilian Barcode of Life
- COLPROT:
-
Protozoa Collection
- COLTRYP:
-
Trypanosomatid collection from wild and domestic mammals and vectors
- cox1:
-
cytochrome c oxidase subunit 1 gene
- cox2-nad1:
-
cytochrome c oxidase subunit 2-NADH dehydrogenase subunit 1 gene region
- cytb :
-
cytochrome b
- DTU:
-
Discrete typing unit
- gGAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- GPI :
-
Glucose-6-phosphate isomerase
- ITS:
-
Internal transcribed spacer
- K2P:
-
Kimura 2-parameter
- LIT:
-
Liver infusion tryptose
- ML:
-
Maximum likelihood
- MLST:
-
Multilocus sequencing typing
- MP:
-
Maximum parsimony
- mPTP:
-
multi-rate Poisson Tree Processes
- NJ:
-
Neighbor-joining
- NNN:
-
Novy-McNeal-Nicole medium
- PTP:
-
single-rate Poisson Tree Processes
- SSU rDNA:
-
Small subunit ribosomal DNA
- V7 V8:
-
Variable regions 7 and 8
References
Blaxter M. Imagining Sisyphus happy: DNA barcoding and the unnamed majority. Philos Trans R Soc Lond Ser B Biol Sci. 2016;371(1702):1–5.
The Internet Classics Archive. The History of Animals by Aristotle. Translated by D’Arcy Wentworth Thompson. http://classics.mit.edu//Aristotle/history_anim.html. Accessed 20 Feb 2017.
Linnaeus C. Systema naturae per regna tria naturae, secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis. Tomus I. 1758. http://www.biodiversitylibrary.org/item/10277#page/3/mode/1up. Accessed 20 Feb 2017.
Hebert PDN, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc Biol Sci. 2003;270:313–21.
Trivedi S, Aloufi AA, Ansari AA, Ghosh SK. Role of DNA barcoding in marine biodiversity assessment and conservation: an update. Saudi J Biol Sci. 2016;23:161–71.
Hebert PDN, Hollingsworth PM, Hajibabaei M, Hebert PDN. From writing to reading the encyclopedia of life. Philos Trans R Soc Lond B Biol Sci. 2016;371:1–9.
Strüder-Kypke MC, Lynn DH. Comparative analysis of the mitochondrial cytochrome c oxidase subunit I (COI) gene in ciliates (Alveolata, Ciliophora) and evaluation of its suitability as a biodiversity marker. Syst Biodivers. 2010;8:131–48.
Lin X, Stur E, Ekrem T. Exploring genetic divergence in a species-rich insect genus using 2790 DNA barcodes. PLoS One. 2015;10:e0138993.
Tavares ES, Baker AJ. Single mitochondrial gene barcodes reliably identify sister-species in diverse clades of birds. BMC Evol Biol. 2008;8:81.
Oba Y, Ôhira H, Murase Y, Moriyama A, Kumazawa YDNA. Barcoding of Japanese click beetles (Coleoptera, Elateridae). PLoS One. 2015;10:e0116612.
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, et al. From the cover: nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc Natl Acad Sci USA. 2012;109:6241–6.
de Boer HJ, Ouarghidi A, Martin G, Abbad A, Kool ADNA. Barcoding reveals limited accuracy of identifications based on folk taxonomy. PLoS One. 2014;9(1):e84291.
Stern RF, Horak A, Andrew RL, Coffroth MA, Andersen RA, Küpper FC, et al. Environmental barcoding reveals massive dinoflagellate diversity in marine environments. PLoS One. 2010;5(11):e13991.
Zhao Y, Gentekaki E, Yi Z, Lin X. Genetic differentiation of the mitochondrial cytochrome c oxidase subunit I gene in genus Paramecium (Protista, Ciliophora). PLoS One. 2013;8:e77044.
Sundberg P, Kvist S, Strand M. Evaluating the utility of single-locus DNA barcoding for the identification of ribbon worms (phylum Nemertea). PLoS One. 2016;11:1–17.
Meyer CP, Paulay G. DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 2005;3:e422.
Cupolilo E, Grimaldi Junior G, Momen H, Beverly SM. Intergenic region typing (IRT): a rapid molecular approach to the characterization and evolution of Leishmania. Mol Biochem Parasitol. 1995;3(1-2):145–55.
Fernandes O, Sturm NR, Derré R, Campbell DA. The mini-exon gene: a genetic marker for zymodeme III of Trypanosoma cruzi. Mol Biochem Parasitol. 1998;95:129–33.
Lewis MD, Ma J, Yeo M, Carrasco HJ, Llewellyn MS, Miles MA. Genotyping of Trypanosoma cruzi: systematic selection of assays allowing rapid and accurate discrimination of all known lineages. Am J Trop Med Hyg. 2009;81:1041–9.
Brenière SF, Aliaga C, Waleckx E, Buitrago R, Salas R, Barnabé C, et al. Genetic characterization of Trypanosoma cruzi DTUs in wild Triatoma infestans from Bolivia: predominance of TcI. PLoS Negl Trop Dis. 2012;6:e1650.
Perez E, Monje M, Chang B, Buitrago R, Parrado R, Barnabé C, et al. Predominance of hybrid discrete typing units of Trypanosoma cruzi in domestic Triatoma infestans from the Bolivian Gran Chaco region. Infect Genet Evol. 2013;13:116–23.
Cavazzana M, Marcili A, Lima L, da Silva FM, Junqueira ACV, Veludo HH, et al. Phylogeographical, ecological and biological patterns shown by nuclear (ssrRNA and gGAPDH) and mitochondrial (Cyt b) genes of trypanosomes of the subgenus Schizotrypanum parasitic in Brazilian bats. Int J Parasitol. 2010;40:345–55.
Ramírez JD, Duque MC, Montilla M, Cucunubá Z, Guhl F. Natural and emergent Trypanosoma cruzi I genotypes revealed by mitochondrial (Cytb) and nuclear (SSU rDNA) genetic markers. Exp Parasitol. 2012;132:487–94.
Diosque P, Tomasini N, Lauthier JJ, Messenger LA, Monje Rumi MM, Ragone PG, et al. Optimized multilocus sequence typing (MLST) scheme for Trypanosoma cruzi. PLoS Negl Trop Dis. 2014;8(8):e3117.
Marcili A, Lima L, Cavazzana M, Junqueira ACV, Veludo HH, Maia Da Silva F, et al. A new genotype of Trypanosoma cruzi associated with bats evidenced by phylogenetic analyses using SSU rDNA, cytochrome b and Histone H2B genes and genotyping based on ITS1 rDNA. Parasitology. 2009;136:641–55.
Lima L, Espinosa-Álvarez O, Ortiz PA, Trejo-Varón JA, Carranza JC, Pinto CM, et al. Genetic diversity of Trypanosoma cruzi in bats, and multilocus phylogenetic and phylogeographical analyses supporting Tcbat as an independent DTU (discrete typing unit). Acta Trop. 2015;151:166–77.
Botero A, Cooper C, Thompson CK, Clode PL, Rose K, Thompson RA. Morphological and phylogenetic description of Trypanosoma noyesi sp. nov.: an Australian wildlife trypanosome within the T. cruzi clade. Protist. 2016;167:425–39.
Lewis MD, Llewellyn MS, Yeo M, Acosta N, Gaunt MW, Miles MA. Recent, independent and anthropogenic origins of Trypanosoma cruzi hybrids. PLoS Negl Trop Dis. 2011;5:e1363.
Zingales B, Miles MA, Campbell DA, Tibayrenc M, Macedo AM, Teixeira MMG, et al. The revised Trypanosoma cruzi subspecific nomenclature: rationale, epidemiological relevance and research applications. Infect Genet Evol. 2012;12:240–53.
Chagas C. Nova tripanozomiaze humana: estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. Mem Inst Oswaldo Cruz. 1909;1:159–218.
Miles MA, Toye PJ, Oswald SC, Godfrey DG. The identification by isoenzyme patterns of two distinct strain-groups of Trypanosoma cruzi, circulating independently in a rural area of Brazil. Trans R Soc Trop Med Hyg. 1977;71(3):217–25.
Tibayrenc M, Ward P, Moya A, Ayala FJ. Natural populations of Trypanosoma cruzi, the agent of Chagas disease, have a complex multiclonal structure. Proc Natl Acad Sci USA. 1986;83:115–9.
Tibayrenc M, Ayala FJ. The population genetics of Trypanosoma cruzi revisited in the light of the predominant clonal evolution model. Acta Trop. 2015;151:156–65.
Brisse S, Henriksson J, Barnabé C, Douzery EJP, Berkvens D, Serrano M, et al. Evidence for genetic exchange and hybridization in Trypanosoma cruzi based on nucleotide sequences and molecular karyotype. Infect Genet Evol. 2003;2:173–83.
Machado CA, Ayala FJ. Nucleotide sequences provide evidence of genetic exchange among distantly related lineages of Trypanosoma cruzi. Proc Natl Acad Sci USA. 2001;98(13):7396–401.
Barnabé C, Brenière SF. Scarce events of mitochondrial introgression in Trypanosoma cruzi: new case with a Bolivian strain. Infect Genet Evol. 2012;12:1879–83.
Messenger LA, Miles MA. Evidence and importance of genetic exchange among field populations of Trypanosoma cruzi. Acta Trop. 2015;151:150–5.
Andrade SG, Magalhães JB. Biodemes and zymodemes of Trypanosoma cruzi strains: correlations with clinical data and experimental pathology. Rev Soc Bras Med Trop. 1997;30:27–35.
Barrett T, Hoff R, Mott K, Miles M, Godfrey D, Teixeira R, et al. Epidemiological aspects of three Trypanosoma cruzi zymodemes in Bahia state, Brazil. Trans R Soc Trop Med Hyg. 1980;74:84–90.
Morel C, Chiari E, Camargo EP, Mattei DM, Romanha AJ, Simpson L. Strains and clones of Trypanosoma cruzi can be characterized by pattern of restriction endonuclease products of kinetoplast DNA minicircles. Proc Natl Acad Sci USA. 1980;77:6810–4.
Zingales B, Andrade SG, Briones MRS, Campbell DA, Chiari E, Fernandes O, et al. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz. 2009;104:1051–4.
Tibayrenc M, Ayala FJ. Isozyme variability in Trypanosoma cruzi, the agent of Chagas disease: genetical, taxonomical, and epidemiological significance. Evolution. 1988;42:277–92.
Tibayrenc M, Ayala FJ. Towards a population genetics of microorganisms: the clonal theory of parasitic protozoa. Parasitol Today. 1991;7:228–32.
Tibayrenc M. Population genetics of parasitic protozoa and other microorganisms. Adv Parasitol. 1995;36:48–115.
Souto RP, Fernandes O, Macedo AM, Campbell DA, Zingales B. DNA markers define two major phylogenetic lineages of Trypanosoma cruzi. Mol Biochem Parasitol. 1996;83:141–52.
Tibayrenc M. Genetic epidemiology of parasitic protozoa and other infectious agents: the need for an integrated approach. Int J Parasitol. 1998;28:85–104.
Brisse S, Barnabé C, Tibayrenc M. Identification of six Trypanosoma cruzi phylogenetic lineages by random amplified polymorphic DNA and multilocus enzyme electrophoresis. Int J Parasitol. 2000;30:35–44.
Kawashita SY, Sanson GF, Fernandes O, Zingales B, Briones MR. Maximum-likelihood divergence date estimates based on rRNA gene sequences suggest two scenarios of Trypanosoma cruzi intraspecific evolution. Mol Biol Evol. 2001;18:2250–9.
Mendonça MBA, Nehme NS, Santos SS, Cupolillo E, Vargas N, Junqueira A, et al. Two main clusters within Trypanosoma cruzi zymodeme 3 are defined by distinct regions of the ribosomal RNA cistron. Parasitology. 2002;124:177–84.
De Freitas JM, Augusto-Pinto L, Pimenta JR, Bastos-Rodrigues L, Gonçalves VF, Teixeira SMR, et al. Ancestral genomes, sex, and the population structure of Trypanosoma cruzi. PLoS Pathog. 2006;e24:2.
Barnabé C, Mobarec HI, Jurado MR, Cortez JA, Brenière SF. Reconsideration of the seven discrete typing units within the species Trypanosoma cruzi, a new proposal of three reliable mitochondrial clades. Infect Genet Evol. 2016;39:176–86.
Ramirez JD, Llewellyn MS. Reproductive clonality in protozoan pathogens - truth or artefact? Mol Ecol. 2014;23:4195–202.
Guhl F, Auderheide A, Ramírez JD. From ancient to contemporary molecular eco-epidemiology of Chagas disease in the Americas. Int J Parasitol. 2014;44(9):605–12.
Maia da Silva F, Marcili A, Lima L, Cavazzana M, Ortiz PA, Campaner M, et al. Trypanosoma rangeli isolates of bats from Central Brazil: genotyping and phylogenetic analysis enable description of a new lineage using spliced-leader gene sequences. Acta Trop. 2009;109:199–207.
Rocha FL, Roque ALR, Arrais RC, Santos JP, Lima VDS, Xavier SCDC, et al. Trypanosoma cruzi TcI and TcII transmission among wild carnivores, small mammals and dogs in a conservation unit and surrounding areas, Brazil. Parasitology. 2013;140:160–70.
Lima VDS, Xavier SCDC, Maldonado IFR, Roque ALR, Vicente ACP, Jansen AM. Expanding the knowledge of the geographic distribution of Trypanosoma cruzi TcII and TcV/TcVI genotypes in the Brazilian Amazon. PLoS One. 2014;9(12):e116137.
COLTRYP: Coleção de Trypanosoma de Mamíferos Silvestres, Domésticos e Vetores. http://coltryp.fiocruz.br/. Accessed 10 Jan 2017.
Vallejo GA, Guhl F, Chiari E, Macedo AM. Species specific detection of Trypanosoma cruzi and Trypanosoma rangeli in vector and mammalian hosts by polymerase chain reaction amplification of kinetoplast minicircle DNA. Acta Trop. 1999;72:203–12.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Darriba D, Taboada GL, Posada D. jModelTest2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772.
Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704.
Swofford DL, Waddell PJ, Huelsenbeck JP, Foster PG, Lewis PO, Rogers JS. Bias in phylogenetic estimation and its relevance to the choice between parsimony and likelihood methods. Syst Biol. 2001;50(4):525–39.
Ronquist F, Teslenko M, vander Mark P, Ayres D, Darling A, Höhna S, et al. MrBayes 3.2: eficient Bayesian phylogenetics inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Vaidya G, Lohman DJ, Meier R. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics. 2011;27:171–80.
Puillandre N, Lambert A, Brouillet S, Achaz G. ABGD, automatic barcode gap discovery for primary species delimitation. Mol Ecol. 2012;21:1864–77. web-interface: http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html. Accessed 10 Jan 2017
Zhang J, Kapli P, Pavlidis P, Stamatakis A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics. 2013;29:2869–76.
Kapli P, Lutteropp S, Zhang J, Kobert K, Pavlidis P, Stamatakis A, et al. Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics.btx025. 2017. web-interface: http://mptp.h-its.org/#/tree. Accessed 5 May 2017.
Maia Da Silva F, Noyes H, Campaner M, Junqueira ACV, Coura JR, Añez N, et al. Phylogeny, taxonomy and grouping of Trypanosoma rangeli isolates from man, triatomines and sylvatic mammals from widespread geographical origin based on SSU and ITS ribosomal sequences. Parasitology. 2004;129:549–61.
Maia Da Silva F, Junqueira ACV, Campaner M, Rodrigues AC, Crisante G, Ramirez LE, et al. Comparative phylogeography of Trypanosoma rangeli and Rhodnius (Hemiptera: Reduviidae) supports a long coexistence of parasite lineages and their sympatric vectors. Mol Ecol. 2007;16:3361–73.
Briones MR, Souto RP, Stolf BS, Zingales B. The evolution of two Trypanosoma cruzi subgroups inferred from rRNA genes can be correlated with the interchange of American mammalian faunas in the Cenozoic and has implications to pathogenicity and host specificity. Mol Biochem Parasitol. 1999;104:219–32.
Flores-López CA, Machado CA. Analyses of 32 loci clarify phylogenetic relationships among Trypanosoma cruzi lineages and support a single hybridization prior to human contact. PLoS Negl Trop Dis. 2011;5(8):e1272.
Hamilton PB, Lewis MD, Cruickshank C, Gaunt MW, Yeo M, Llewellyn MS, et al. Identification and lineage genotyping of south American trypanosomes using fluorescent fragment length barcoding. Infect Genet Evol. 2011;11:44–51.
García L, Ortiz S, Osorio G, Torrico MC, Torrico F, Solari A. Phylogenetic analysis of Bolivian bat trypanosomes of the subgenus Schizotrypanum based on cytochrome b sequence and minicircle analyses. PLoS One. 2012;7:1–7.
Westenberger SJ, Barnabé C, Campbell DA, Sturm NR. Two hybridization events define the population structure of Trypanosoma cruzi. Genetics. 2005;171:527–43.
Tomasini N, Diosque P. Evolution of Trypanosoma cruzi: clarifying hybridisations, mitochondrial introgressions and phylogenetic relationships between major lineages. Mem Inst Oswaldo Cruz. 2015;110(3):403–13.
da Costa AP, Nunes PH, Leite BHS, Ferreira JIGS, Tonhosolo R, da Rosa AR, et al. Diversity of bats trypanosomes in hydroeletric area of Belo Monte in Brazilian Amazonia. Acta Trop. 2016;164:185–93.
Lauthier JJ, Tomasini N, Barnabé C, Rumi MMM, D’Amato AMA, Ragone PG, et al. Candidate targets for multilocus sequence typing of Trypanosoma cruzi: validation using parasite stocks from the Chaco region and a set of reference strains. Infect Genet Evol. 2012;12:350–8.
Jansen AM, Xavier SCC, Roque ALR. The multiple and complex and changeable scenarios of the Trypanosoma cruzi transmission cycle in the sylvatic environment. Acta Trop. 2015;151:1–15.
Zumaya-Estrada FA, Messenger LA, Lopez-Ordonez T, Lewis MD, Flores-Lopez CA, Martínez-Ibarra AJ, et al. North American import? Charting the origins of an enigmatic Trypanosoma cruzi domestic genotype. Parasit Vectors. 2012;5:226.
Barnabé C, Brisse S, Tibayrenc M. Phylogenetic diversity of bat trypanosomes of subgenus Schizotrypanum based on multilocus enzyme electrophoresis, random amplified polymorphic DNA, and cytochrome b nucleotide sequence analyses. Infect Genet Evol. 2003;2:201–8.
Pinto CM, Ocaña-Mayorga S, Tapia EE, Lobos SE, Zurita AP, Aguirre-Villacís F, et al. Bats, trypanosomes, and triatomines in Ecuador: new insights into the diversity, transmission, and origins of Trypanosoma cruzi and Chagas disease. PLoS One. 2015;10:e0139999.
Marcili A, da Costa AP, Soares HS, Acosta IDCL, de Lima JTR, Minervino AHH, et al. Isolation and phylogenetic relationships of bat trypanosomes from different biomes in Mato Grosso, Brazil. J Parasitol. 2013;99:1071–6.
Messenger LA, Llewellyn MS, Bhattacharyya T, Franzén O, Lewis MD, Ramírez JD, et al. Multiple mitochondrial introgression events and heteroplasmy in Trypanosoma cruzi revealed by maxicircle MLST and next generation sequencing. PLoS Negl Trop Dis. 2012;e1584:6.
Lemey P, Salemi M, Vandamme AM, editors. The Phylogenetic handbook. A practical approach to Phylogenetic analysis and hypothesis testing. 2nd ed. USA: Cambridge University Press; 2009.
Hamilton PB, Teixeira MMG, Stevens JR. The evolution of Trypanosoma cruzi: the “bat seeding” hypothesis. Trends Parasitol. 2012;28:136–41.
Acknowledgments
The authors would like to thank Dr. Vera Bongertz for English corrections and Carlos Ardé and Marcos Antônio dos Santos Lima for technical support. The authors would also like to thank Dr. Michael Miles from the London School of Hygiene & Tropical Medicine (London, UK) and the Protozoa Collection - COLPROT from FIOCRUZ, Brazil for providing DNA samples for the present study and the RPT01A/FIOCRUZ sequencing facilities for sequencing the samples.
Funding
The present study was funded through a grant from the Oswaldo Cruz Institute - FIOCRUZ; PAPES VI; CNPq and FAPERJ. A doctoral grant was provided by the Oswaldo Cruz Institute to MSR. AMJ is a “Cientista do Nosso Estado”, provided by FAPERJ and is financially supported through CNPq (“Bolsista de Produtividade, nível 1”, CNPq). The funders played no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
All sequences analyzed are available in GenBank under the accession numbers within the article.
Author information
Authors and Affiliations
Contributions
MSR, KAM and AMJ conceived and designed the experiments. MSR performed and analyzed the molecular characterization. KAM provided additional suggestions on content. MSR and AMJ drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. No special permission was required for the present study. We used DNA extracted from the cultures obtained from animals collected during previous field expeditions conducted by our group. The field expeditions were endorsed by the Ethics Committee of FIOCRUZ (Oswaldo Cruz Foundation, Brazil) (CEUA L-015/04; CEUA P-292-06).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Figure S1.
Comparison between TcIV and TcV/TcVI nucleotide sequences generated with the cox1 barcode. a Alignment of TcIV sequences with TcV and TcVI shows one single nucleotide polymorphism differentiating TcIV samples from the hybrids. b Electropherogram confirms the presence of a T (thymine) in TcIV in the same position, showing a C (cytosine) in TcV and TcVI sequences. (TIFF 724 kb)
Additional file 2: Figure S2.
Comparison between TcI, Tcbat and TcIII nucleotide sequences generated with GPI. a Sequence alignment shows one single nucleotide polymorphism differentiating TcI from Tcbat and one polymorphism separating TcI from TcIII sequences. b Electropherogram confirms the presence of A (adenine) in TcIII sequences in the same position, showing a G (guanine) in TcI sequences. The Tcbat sequence was retrieved from GenBank and the electropherogram is not publicly available. (TIFF 527 kb)
Additional file 3: Table S1.
Number of haplotypes, nucleotide diversity and haplotype diversity of sequences generated with cox1 and GPI. (DOCX 12 kb)
Additional file 4: Table S2.
Inter- and intraspecific genetic distance based on cox1 sequences. (DOCX 14 kb)
Additional file 5: Table S3.
Inter- and intraspecific genetic distance based on GPI sequences. (DOCX 15 kb)
Additional file 6: Table S4.
Cox1 sequences partition into groups inferred with ABGD, based on Kimura 2-parameters. (DOCX 14 kb)
Additional file 7: Table S5.
GPI sequences partition into groups inferred with ABGD, based on Kimura 2-parameters. (DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rodrigues, M.S., Morelli, K.A. & Jansen, A.M. Cytochrome c oxidase subunit 1 gene as a DNA barcode for discriminating Trypanosoma cruzi DTUs and closely related species. Parasites Vectors 10, 488 (2017). https://doi.org/10.1186/s13071-017-2457-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2457-1