
Abstract
Background
The argasid tick Ornithodoros moubata is the main African vector of the human relapsing fever agent Borrelia duttoni and the African swine fever virus. Together with saliva, the tick midgut forms part of the host-tick-pathogen interface, and numerous midgut proteins play key functions in the blood digestion-related process and the infection and transmission of pathogens. This work explores the composition of the midgut proteome of unfed and fed O. moubata females with the aim to complete the biological information already obtained from the midgut transcriptome and provide a more robust and comprehensive perspective of this biological system.
Methods
Midgut tissues taken from females before feeding and 48 h after feeding were subjected to LC/MS-MS analysis. After functional characterization and classification of the proteins identified, the differences in the proteome between unfed and fed females were analysed and discussed. Additionally, a detailed analysis of particular groups of proteins that are involved in the processes of nutrient digestion and responses to the oxidative stress was carried out.
Results
1491 non-redundant tick proteins were identified: 1132 of them in the midgut of unfed ticks, 1138 in the midgut of fed ticks, and up to 779 shared by both physiological conditions. Overall, the comparative analysis of the midgut proteomes of O. moubata females before and after feeding did not reveal great differences in the number or class of proteins expressed, enzymatic composition or functional classification.
Conclusions
The hemoglobinolytic system in ixodids and argasids is very similar in spite of the fact that they display very different feeding and reproductive strategies. Although the main source of nutrients in ticks are proteins, lipids and carbohydrates also constitute significant nutritional sources and play an important part in the process of blood digestion. The genes and proteins involved in intracellular transport mechanisms, defensive responses, detoxifying responses and stress responses seem to be closely regulated, highlighting the complexity and importance of these processes in tick biology, which in turn assigns them a great interest as targets for therapeutic and immunological interventions.
Similar content being viewed by others

Background
Ticks are haematophagous ectoparasites that classify into two main families, the Ixodidae (hard ticks) and the Argasidae (soft ticks). Both families differentiate in a range of morphological and biological characteristics, for example, their feeding and reproductive strategies. Typically, ixodids are slow feeders that take several days to ingest their blood meal, while argasids are fast feeders that complete their blood meal in minutes to hours. Ixodid females feed only once, lay thousands of eggs and die after oviposition, whereas adult argasids can feed and reproduce up to ten times, laying hundreds of eggs per trophogonic cycle [1,2,3,4,5].
Both tick families represent important threats for human and animal health because they cause direct damage to hosts (including paralysis, toxicosis, irritation and allergy) and transmit a wide range of pathogenic microorganisms to them [4, 6]. This noteworthy vector capacity together with tick abundance has placed ticks as the most important arthropod vectors of diseases affecting livestock, pets and people [7].
The argasid Ornithodoros moubata is distributed throughout southern and eastern African countries [8], where it transmits the African swine fever virus and the East African human relapsing fever causing agent, Borrelia duttoni [9, 10].
Chemical acaricide-based tick control has shown important drawbacks, including a selection of tick resistant strains, environmental pollution and animal product contamination, which have encouraged the search for alternative methods of tick control, such as immunological control with anti-tick vaccines [11].
With this purpose some years ago, our team started to develop an anti-O. moubata vaccine by testing two types of candidate antigens: salivary antigens, which are naturally exposed to the host immune system during tick feeding; and concealed antigens from the intestinal tissue. The studies on salivary antigens identified three protective anti-haemostatic molecules, which when jointly administered to rabbits provided more than 50% vaccine efficacy, making them promising vaccine candidates. Despite this, a fully protective vaccine to O. moubata, based on salivary antigens only, has not been hitherto obtained [12].
On the other hand, the possibility of inducing protective immune responses to intestinal (midgut) antigens of O. moubata seemed reasonably high, since such antigens (namely, the Bm86 glycoprotein from Rhipicephalus microplus) were the basis of the two anti-tick vaccines marketed to date (TickGARD® and GAVAC®) [13].
Together with tick saliva, the tick midgut forms part of the host-tick-pathogen interface, and numerous midgut proteins play key functions in the blood digestion-related process and the infection and transmission of pathogens. Thus, midgut proteins are considered potential targets for the development of new drugs and vaccines aimed at tick control [14,15,16].
In this direction, our former studies with O. moubata, and with the Iberian species O. erraticus, demonstrated that vaccination of animals with extracts of midgut membrane proteins induced protective responses. These responses significantly reduced female feeding and reproduction rates in both species, and the survival rates in O. erraticus nymphs, confirming the potential of the midgut as a source of protective antigens [17, 18].
In ixodid ticks, the midgut molecular machinery for blood digestion, as well as the proteins involved in successful pathogen transmission, have been reasonably well established [19,20,21,22,23,24,25]. In argasids, by contrast, this topic has been little explored, and the information available is scant. Only the identification of several proteolytic enzymes and some proteins associated with defence responses, such as lysozyme and several defensins, have been reported in the midgut of O. moubata [26, 27]. This paucity of information on the argasid midgut, and the potential of proteins involved in physiological processes that take place in the tick midgut as vaccine antigens prompted us to the study the molecular machinery involved in blood digestion in argasids. With this aim, our first step was the characterization of the midgut proteome of O. erraticus females before and after the blood meal. This study described the first midgut proteome in argasids and provided interesting novel information on blood ingestion-induced changes in the expression of midgut proteins in this species, contributing to the understanding of the multi-enzymatic molecular system for blood digestion in argasids [28]. More recently, the midgut transcriptomes of O. moubata females before and after the blood meal have been assembled and analysed, and the genes that were differentially expressed in the midgut after feeding have been identified and functionally annotated. This study has significantly expanded our knowledge of the biochemistry and physiology of blood digestion in argasids [29].
In the current work, our purpose was to investigate the composition of the midgut proteomes of O. moubata females before and after blood meal ingestion. This will complete the biological information already obtained from the midgut transcriptome and provide a more robust and comprehensive perspective of this biological system. To reach this objective, we have applied an experimental approach similar to that previously used by Oleaga et al. [28] for O. erraticus. Thus, midgut extracts from fed and unfed females were obtained and fractionated into soluble and membrane proteins; SDS-PAGE then resolved the fractions, and the gels were sliced into pieces. The proteins in each gel slide were subject to LC-MS/MS followed by searching the NCBInr_metazoa and EST_acari databases for protein identification. Additionally, to maximize the number of protein identifications, a proteomics informed by transcriptome (PIT) analysis was also implemented, which consisted of mining the O. moubata midgut transcriptomic dataset previously obtained by RNAseq [29] as an additional database.
Methods
Ticks and collection of tick midgut samples
The O. moubata specimens were obtained from the laboratory colony of the IRNASA (CSIC), which is regularly fed on rabbits and maintained at 28 °C, 85% relative humidity and a 12/12 h light/dark photoperiod.
Homogeneous batches of O. moubata females taken before feeding (unfed) and at 48 h post-feeding (fed) were dissected in cold phosphate buffered saline (PBS) pH 7.4. The midguts were recovered, rinsed several times with PBS to eliminate host blood and tick hemolymph proteins and inspected to remove contaminant tissues, mostly Malpighian tubules and tracheae [18].
Preparation of midgut protein extracts from unfed and fed females
We followed the procedure previously described by Oleaga et al. [28]. Briefly, batches of 25 midguts were suspended and homogenized in ice-cold PBS with proteinase inhibitors (Roche Diagnostics, Indianapolis, USA), followed by sonication (6 pulses, 30 s/pulse) (Ultrasonic Cell Disrupter Virsonic 300). Tissue homogenates were centrifuged for 20 min at 10,000× g and 4 °C to remove particulate remnants, and the supernatants were re-centrifuged for 1 h at 100,000× g, and 4 °C. Final supernatants were recovered and named as the S fraction (soluble proteins). The pellets were re-suspended in PBS and recentrifuged for 1 h at 100,000 g and 4 °C. These final pellets, enriched in membrane proteins, were recovered and named as the M fraction (membranes). The protein concentrations in these samples were assessed using the BCA Protein Assay Reagent kit (Thermo-Fisher, Rockford, USA). Samples were stored at -20 °C.
Sample fractioning by SDS-PAGE
The four samples of midgut proteins, namely, soluble and membrane-associated proteins from unfed (S-0, M-0) and fed (S-1, M-1) females were resolved by 5–20% gradient SDS-PAGE. Gels were loaded with 20 μg of protein sample per lane and stained with Sypro Ruby (Bio-Rad, Hercules, USA) for protein visualisation and image analysis (ChemiDoc System and Image Lab software, Bio-Rad). Replicated gels were stained with Coomassie Blue. In these gels, each lane (corresponding to a different sample) was sliced into 10 pieces, and the resulting 40 slices were each subjected to protein digestion and mass spectrometry analysis for protein identification.
Enzymatic digestion and liquid chromatography and tandem mass spectrometry analysis (LC-MS/MS)
Trypsin digestion and LC-MS/MS analysis were carried out at the SCSIE_University of Valencia Proteomics Unit, a member of ISCIII ProteoRed Proteomics Platform, following a standardized protocol, described previously [28].
Briefly, gel slices were reduced with DTT, alkylated with iodoacetamide and digested with 20 ng/μl of trypsin (Promega, Madison, USA) overnight at 37 °C. Digestion was stopped with 10% trifluoroacetic acid (TFA) to a final concentration of 0.1%, and the supernatants were filtered through a 0.22 μm filter and dried by centrifugation in a vacuum. Pellets were re-suspended in 6 μl of 5% acetonitrile, 0.1% TFA, and 5 μl of every sample was loaded onto a trap column (Nano LC Column, 3 μ C18-CL, 350 μm × 0.5 mm, Eksigent) and desalted with 0.1% TFA at a flow rate of 3 μl/min for 5 min. The peptides were then loaded onto an analytical column (LC Column, 3 μ C18-CL, 75 μm × 25 cm, Eksigent) and equilibrated in 5% acetonitrile (ACN) and 0.1% formic acid (FA). Elution was carried out with a linear gradient of 5–40% B (B: ACN, 0.1% FA) in A (A: 0.1% FA) for 30 min at a flow rate of 300 nl/min. Eluted peptides were analysed in a nanoESI qQTOF mass spectrometer (5600TripleTOF, ABSCIEX, Ontario, Canada) in IDA mode performing 0.25-s TOF MS scans from 350 to 1250 m/z, followed by 0.05-s product ion scans from 100 to 1500 m/z on the 50 most intense 2–5 charged ions.
Database search and protein identification
Mass spectra were processed with Mascot v.2.2 (Matrix Science, Boston, USA) and the following databases were searched: protein sequences of the National Center for Biotechnology Information (NCBI) with taxonomic restriction to Metazoa (NCBInr_metazoa: 7,616,579 sequences); nucleotide sequences (EST) of the NCBI, restricted to subclass Acari (EST_acari: 2,476,050 sequences); and midgut transcriptome of O. moubata (annotated with restriction to phylum Arthropoda: 6629 sequences; [29]). Searches were performed with tryptic specificity, allowing one missed cleavage and tolerance on the mass measurement of 70 ppm in MS mode and 0.6 Da for MS/MS ions. Carbamidomethylation of Cys was used as a fixed modification and oxidation of Met and deamidation of Asn and Gln as variable modifications. The significance threshold was set at a confidence ≥95% and only proteins with at least two unique significant peptides were selected and shown in the results. Database searching was individually performed for each of the 40 gel slices, and jointly for each protein sample by combining all spectra from the 10 gel slices corresponding to each sample.
After manually inspecting all the proteins identified in the three databases, redundant identifications were removed by preferentially selecting the proteins matched to the O. moubata midgut transcriptome or the protein hit with the highest score. Contaminants, such as keratins and porcine trypsin, were also excluded from the lists of proteins identified.
The relative quantitation of the proteins was carried out using the exponentially modified protein abundance index (emPAI), which is based on protein coverage by the peptide matches in a database search result [30].
Functional characterization and protein classification
The functional characterization and classification of the proteins identified were carried out according to Gene Ontology hierarchy (GO), using the UniProt tools (http://www.uniprot.org) and the PANTHER Classification System (Protein Analysis Through Evolutionary Relationships; http://www.pantherdb.org) [31]. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis was executed using the Blast2GO software (https://www.blast2go.com/).
Results and discussion
Midgut protein extracts
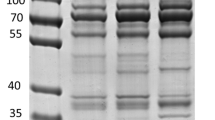
Midgut extracts from fed and unfed O. moubata females were separated by centrifugation into two types of fractions: the fractions enriched in soluble proteins (supernatants S-0, S-1) and the fractions enriched in insoluble membrane-associated proteins (pellets M-0, M-1). Figure 1 shows that all four samples were complex mixtures of proteins with a broad range of molecular sizes, from 10 to 260 kDa. Band patterns were different among samples, particularly in sample S-1, which contained three very intense bands of 10, 52 and 66 kDa. This sample contained the soluble proteins from fed ticks, and these three bands corresponded to host haemoglobin, the heavy chain of immunoglobulins and serum albumin, respectively (Fig. 1).

Sypro Ruby-stained 5–20% polyacrylamide gel showing the protein fractions obtained from midgut homogenates of fed and unfed Ornithodoros moubata ticks. Gel lanes were sliced into the ten pieces indicated at the right, and the resulting gel slices were digested with trypsin and analysed by LC-MS/MS. Lanes S-0 and S-1: supernatants from midgut homogenates from unfed and fed ticks, respectively; Lanes M-0 and M-1: pellets from midgut homogenates from unfed and fed ticks, respectively
Because of their abundance, the presence of these host proteins in the midgut samples from fed ticks could mask the identification of a significant portion of the tick proteins. This problem could also occur in unfed ticks, although to a lesser extent, since host proteins can persist for months after tick feeding and moulting [32]. This problem can be avoided by treating the sample to reduce the amount of haemoglobin, for example, by chloroform precipitation [32]. In the current work, we fractionated the samples into 10 gel slices, which were analysed individually. In this way, the abundant host blood proteins (haemoglobin, immunoglobulin and serum albumin) were concentrated in a few gel slices, avoiding the problem of these proteins hindering the detection of tick proteins present in the remaining gel slices.
Protein identifications
The proteins shown in this section were identified by searching databases with the combined spectra of the 10 gel slices from each fraction (S-0, M-0, S-1 and M-1). To improve the number of identifications, searches were performed in three databases: two of them are publicly accessible (NCBInr_Metazoa and NCBI_EST_Acari), and the third was custom-made from the O. moubata midgut transcriptomic data obtained by Oleaga et al. [29].
Table 1 and Fig. 2 summarize the number of non-redundant proteins identified in each midgut fraction in each database. Since all the samples contained host and tick proteins, the protein origin was assigned to either host or tick by applying the same criteria as Oleaga et al. [28] in their analysis of the O. erraticus midgut proteome. In this way, protein hits to tick and arthropod species, and to non-mammal vertebrates were all considered of tick origin, whereas protein hits to rabbit or any other mammal were considered of host origin.

The number of proteins identified in the fractions of the midgut of unfed and fed O. moubata females, in each database
Regarding proteins of host origin, the highest number of identifications arose from searching the NCBI_Metazoa database, particularly for the samples from fed ticks (59 and 52 proteins in S-1 and M-1; 23 and 9 proteins in S-0 and M-0) (Table 1). Additional file 1: Table S1 shows all the host proteins identified in every sample after searching the NCBI_Metazoa and EST_Acari databases. Since the O. moubata midgut transcriptome only included sequences annotated to arthropods, searching this database did not identify any protein of host origin. All host proteins were excluded from further characterization and analysis in the current work.
Regarding the proteins from O. moubata (Table 1, Fig. 2, Additional file 2: Table S2), the number of identifications significantly varied depending on the searched database. In the NCBI_Metazoa database, we identified between 41 and 113 proteins; in the EST-Acari database, between 257 and 430 proteins; while the highest numbers of identifications, between 307 and 565 proteins, were obtained from mining the transcriptomic sequences of the O. moubata midgut.
Protein identification by mass spectrometry is dependent on the available information in databases, but usually, this information is scant or negligible for non-model organisms. However, the recent development and increasing application of new generation sequencing methods (RNA-seq) provide large amounts of transcriptomic data, which once annotated can be mined for mass spectrometry-based identification of proteins and peptides. This methodological approach is known as proteomics informed by transcriptomics (PIT) analysis, which has demonstrated its utility in proteomic studies on species without a sequenced genome, such as most tick species [33,34,35,36,37]. Our results on O. moubata confirm the usefulness of PIT analysis.
Whole proteins identified for each sample in the three databases were inspected, and redundant identifications were eliminated. As a result, 719 and 516 non-redundant proteins were respectively recorded for the midgut soluble fractions S-0 and S-1, while 813 and 908 non-redundant proteins were, respectively, recorded for the membrane protein-enriched insoluble fractions M-0 and M-1 (Table 1, Additional file 2: Table S2). After that, the proteins identified in each fraction were classified according to their assigned molecular function and biological process in the “Gene Ontology” database. Protein categories including less than five proteins were excluded from further analyses (Figs. 3 and 4).

Classification according to molecular function of the proteins identified in each of the midgut fractions obtained from unfed (S-0, M-0) and fed (S-1, M-1) ticks. Only categories with more than five proteins were considered. The percentage in each category is the ratio between the number of proteins in each category, indicated in parentheses, and total proteins identified in that fraction

Classification according to biological process of the proteins identified in each of the midgut fractions obtained from unfed (S-0, M-0) and fed (S-1, M-1) ticks. Only categories with more than five proteins were considered. The percentage in each category is the ratio between the number of proteins in each category, indicated in parentheses, and the total proteins identified in that fraction
Regarding the classification of molecular function, the more abundant categories in the four samples were the proteins with catalytic (41% in S-0 and S-1; 36% in M-0 and M-1), binding (30 and 29% in S-0 and M-0, respectively; 31% in S-1 and M-1), structural (6 and 10% in S-0 and M-0, respectively; 5 and 9% in S-1 and M-1, respectively) and transporter activities (3 and 5% in S-0 and M-0, respectively; 4 and 6% in S-1 and M-1, respectively). The remaining categories were significantly less represented, except for proteins with unknown function, which contained 15–17% of the proteins identified. Figure 3 also shows that the distribution of the proteins in functional categories was very similar for every fraction, the only difference being a higher ratio of structural proteins in the insoluble fractions M-0 and M-1. This is mainly due to the higher number of ribosomal proteins in the 100,000× g pellets and parallels the distribution observed in the insoluble fractions of the midgut proteome of O. erraticus [28].
The biological processes assigned to the identified proteins were similarly represented in the four samples (Fig. 4). The most abundant categories were metabolic processes (31–33%) and single-organism processes (22–25%), followed by biological regulation (7–9%), localization (6–9%), cellular component organization or biogenesis (5–7%), response to stimulus (4–5%) and signal transduction (3%). Finally, proteins with unknown biological process represented 14–15%.
Comparative analysis of the midgut proteome between unfed and fed O. moubata ticks
All proteins identified in the two samples (soluble and membranes) from each physiological condition (fed or unfed) were combined and compared between both physiological conditions (Additional file 2: Table S2). Overall, we identified 1491 non-redundant proteins: 1132 of them in the midgut of unfed ticks, 1138 in the midgut of fed ticks, and up to 779 shared by both physiological conditions (Fig. 5a, Additional file 2: Table S2).

a Some proteins identified in the midgut of unfed and fed Ornithodoros moubata females. b Classification of proteins using the PANTHER system. Only categories with more than five proteins have been included
To compare the midgut proteomes before and after feeding, we started by classifying the intestinal proteins expressed in each physiological condition into “protein classes” according to the PANTHER system. This system allowed the classification of 543 and 554 intestinal proteins expressed by the unfed and fed ticks, respectively. Figure 5 shows protein classes containing more than five proteins. The most numerous protein classes were oxidoreductases (88 unfed, 83 fed), nucleic acid binding proteins (75 unfed, 81 fed), hydrolases (71 unfed, 75 fed), transferases (56 unfed, 57 fed), cytoskeletal proteins (40 unfed, 34 fed), enzyme modulator (32 unfed, 31 fed) and transporter proteins (23 unfed, 31 fed). Taken together, these paralleled distributions indicate that the activities and functions of the proteins expressed in the O. moubata midgut before and after feeding are very similar, in accordance with what was also observed for the O. erraticus midgut [28].
After this analysis, the proteins identified in each physiological condition as having a functional annotation in the Gene Ontology database were classified by their molecular function and biological process. Figure 6 shows that both midgut proteomes, before and after feeding, presented similar patterns. The molecular functions most represented in both conditions were catalytic (63–64%) and binding (50–53%) activities, followed by structural (12–13%) and transporter activities (7–8%), with the remaining categories being minority ones (Fig. 6a). Regarding the biological process, the most numerous were metabolic (69–71%) and single-organism process (50–52%), followed by process of localization (17–19%), biological regulation (16–18%), cellular component organization or biogenesis (13–14%), response to stimulus (10–11%) and signal transduction (6–7%). There were four additional minority categories, each representing around 2% of the proteins (Fig. 6b).

Classification of proteins identified in the midgut of O. moubata females before feeding (blue bars, unfed) and at 48 h post-feeding (red bars, fed). Molecular function (a) and biological process (b) assigned in the Gene Ontology database
Finally, the active biological pathways in the O. moubata midgut were investigated. Thus, the proteins identified in the midgut were further analysed using the KEGG database. We found that proteins expressed in the midgut from unfed and fed female O. moubata ticks were involved, respectively, in 108 and 111 enzymatic pathways that clustered into 16 different “Classes” (Table 2, Additional file 3: Table S3).
All these proteins -except the enzymes involved in immune response, translation and signal transduction- participate in the different metabolic pathways that occur in the tick midgut related with the processes of blood digestion and nutrient acquisition [38]. The number of sequences involved in the different pathways indicated that the metabolism of carbohydrates (193 unfed, 166 fed), amino acids (131 unfed, 126 fed) and lipids (124 unfed, 111 fed) were the processes with the highest activity. These data suggest that together with haemoglobin, which is considered the main nutrient source from blood digestion in ticks [38], carbohydrates and lipids would also be important nutrient sources for ticks. This notion is also supported by the O. moubata midgut transcriptomic data [29].
The fourth most active enzymatic pathway was the metabolism of cofactors and vitamins, mainly thiamine metabolism, involving 113 enzymes in unfed females and 49 enzymes in fed females (Table 2, Additional file 3: Table S3). The high activity of this pathway in the tick midgut can be somewhat unexpected as these components, cofactors and vitamins, are scarce in the blood of mammals. However, these metabolites can be supplied by the tick intestinal microbiota as has already been demonstrated to occur in the midgut of several haematophagous insects (flies, lice, bugs) and the tick Amblyomma americanum [39, 40].
Another notably active enzymatic pathway is nucleotide metabolism, particularly purine metabolism (110 sequences in unfed and 56 in fed ticks) (Table 2, Additional file 3: Table S3). Purine metabolism is very important to tick physiology as it plays a key role in the disposal of nitrogenized waste in the form of guanine [25]. In addition, newly synthesized purines may participate in other functions, such as ribonucleotide synthesis for transcription in the digestive cells, and, furthermore, particular enzymes might act as modulators of the extracellular levels of ATP and ADP to avoid platelet activation and aggregation, and maintain the fluidity of the ingested blood [25, 41].
Overall, the comparative analysis of the midgut proteomes of O. moubata females before and after feeding did not reveal great differences in the number or class of proteins expressed, enzymatic composition or functional classification. These data are in accordance with what was also observed in the midgut proteomes of O. erraticus and of several ixodid species, which indicates that, at least during the first phases of blood digestion, the proteome remains quite stable and does not experience significant modifications [23, 28, 35]. Conversely, the analysis of tick midgut transcriptomes, including that of O. moubata, shows that the expression levels of numerous genes involved in blood digestion and the associated stress responses varied significantly in response to tick feeding [29].
Indeed, it has repeatedly been observed that the dynamics of the transcriptome and proteome of tick midguts are different. Several causes may account for these differential dynamics, including the different sensitivity of the analytical methods (i.e. mass spectrometry vs RNA-seq), the different half-life of mRNA and proteins, and the post-transcriptional and post-translational regulation levels of gene expression [36, 42].
Proteins involved in the blood ingestion and digestion process
Once the midgut proteomes from unfed and fed O. moubata females had been characterized and compared, we carried out a more detailed analysis of particular groups of proteins that are involved in the processes of nutrient digestion and responses to the oxidative stress associated with blood ingestion and digestion in ticks. Thus, in accordance with the biological processes in the Gene Ontology database, we selected proteins involved in the following processes: metabolism of proteins, carbohydrates and lipids; endocytoses and intracellular transport; and stress responses and detoxification. The tick midgut is the organ where blood digestion and blood nutrient absorption take place, and this requires that the midgut expresses a range of proteins to perform these tasks [43].
Proteolysis
Haemoglobin and serum albumin constitute up to 80% of the blood protein content, and they are the main source of amino acids for ticks, mostly haemoglobin [44]. In ixodids, the process of digesting haemoglobin has been the subject of numerous studies, and it is quite well known [5]. Haemoglobin digestion takes place inside lysosomes or digestive vesicles and is performed by a set of proteolytic enzymes that act sequentially and belong to the following protease families: cysteine peptidases (cathepsin B, L and C, legumain); aspartic endopeptidases (cathepsin D), metallopeptidases (leucine aminopeptidase) and serine peptidases [38]. In Ixodes ricinus, the haemoglobinolysis is started by cathepsins D and L and legumain, which cut haemoglobin into large fragments. These fragments are digested by cathepsin B, and secondarily by cathepsin L, into smaller fragments, which finally are degraded into dipeptides and amino acids by cathepsins C and B, a leucine aminopeptidase and a serine carboxypeptidase [15, 45]. Free amino acids reach the haemolymph via transcitosis and are distributed and incorporated into tick tissues [38]. Regarding albumin, it has been recently demonstrated that it is digested by the same proteolytic machinery as haemoglobin [44]. The other blood proteins seem to remain undigested in the gut lumen [38].
In argasids, the information about the molecular machinery of blood digestion is scanter and is limited to the identification of several proteases, including cathepsins B and C, in gut extracts of O. moubata [27]. This information has recently been enlarged by Oleaga et al. [28], who identified a cysteine peptidase (legumain), several aspartic endopeptidases, metallopeptidases and serine peptidases in the midgut proteome of O. erraticus, all of them likely involved in haemoglobin and albumin digestion.
In the current midgut proteome of O. moubata, we identified up to 32 proteases: 5 in unfed females, 9 in fed females and 18 in both conditions. Up to 28 of these proteins were also found in the midgut transcriptome (Table 3). These proteases belong to the same classes/families as described above for ixodids. Thus we have found homologues to cathepsin B, L and D, longipain (which is the cathepsin B of Haemaphysalis longicornis), legumain, leucine aminopeptidase and ixodid carboxypeptidase (Table 3) [15, 45]. These results suggest that argasids and ixodids use a very similar proteolytic system to digest host haemoglobin.
It is worth mentioning that besides their potential digestive functions, some of the proteases identified in the O. moubata midgut might have additional functions in other biological processes of the tick. For instance, it has been demonstrated that cathepsin D, found in several ixodid species, participates in defence responses by generating antimicrobial peptides [38, 46]. In the current work, we also found a serine protease in the O. moubata midgut (Q6U8A8, Table 3), which contains a sequence motif rich in histidine and aspartic residues similar to that shown by hebraein, an antimicrobial protein from the ixodid tick Amblyomma hebraeum [47]. Further studies are needed to confirm whether these O. moubata proteins have any function in tick antimicrobial defensive responses.
Carbohydrate metabolism and transport
As already indicated by Oleaga et al. [29], information regarding the digestion of carbohydrates and their likely significance as nutrients from blood digestion in ticks is negligible. To date, it has been limited to the identification of several enzymes in the intestinal transcriptomes of four species of ixodids that, owing to their molecular activity, could participate in carbohydrate metabolism [19, 24, 25, 43, 48]. Recently, Oleaga et al. [29] identified up to 113 transcripts in the O. moubata midgut transcriptome coding for enzymes involved in carbohydrate metabolism and transport. In the current proteome, we have identified 24 proteins with several enzymatic activities (hydrolase, isomerase, transferase, phosphatase, oxidoreductase and kinase) and 3 proteins with carbohydrate transporter functions (Table 4). Four of them were found in unfed females only, 4 in fed females and 19 were shared by both physiological conditions. Most of these proteins had also been annotated in the transcriptome (Table 4). Two of these proteins show high emPAI values in unfed females. They are two enzymes that participate in the energy metabolism of glucose: the phosphoenolpyruvate carboxykinase (PEPCK) (emPAI 8.58), which is involved in the transformation of glucose in pyruvate in the cell cytoplasm; and malate dehydrogenase (emPAI 19.85), which replaces oxaloacetate in the Krebs cycle. Another important protein in using glucose as an energy source is the glucose transporter 1 (E2C2M2), also identified in the transcriptome, which would be responsible for transporting blood glucose from the intestinal lumen to the inside of the enterocytes (Table 4).
The detection of these enzymes and transporters in O. moubata, which are involved in obtaining energy from glucose have recently been described in detail in the intestine of Ixodes scapularis [49], suggesting that host blood glucose would also be an important nutrient for both ixodids and argasids.
Lipid metabolism and transport
Up to 117 transcripts likely involved in lipid metabolism and transport have been annotated in the midgut transcriptome of O. moubata females [29]. Here, we have identified 19 proteins: 3 in unfed females only, 4 in fed females only and 12 in both unfed and fed ticks (Table 5). These consisted of enzymes with various activities (lipase, hydrolase, ceramidase, isomerase, ligase and oxidoreductase) and potential transporters of cholesterol and other lipids.
Among the enzymes, there were group XV phospholipase A2, triacylglycerol lipase, inositol-1-phosphate synthetase, acylsphingosine amidohydrolase and sphingolipid desaturase (Table 5). Group XV phospholipase A2 (A0A087U096) is different from the secreted phospholipase A2 identified in O. moubata saliva [50] and from the phospholipase A2 identified in the midgut transcriptome of O. moubata [29]. This group XV phospholipase A2 is known as lysosomal phospholipase because it is found inside lysosomes and it participates in the degradation of endogenous phospholipids [51].
Among lipid transporters, we found four proteins whose function was cholesterol binding and transport (Table 5). Unlike vertebrates, arthropods cannot synthesize cholesterol themselves and must acquire it from their diet, as it is necessary for the biosynthesis of the ecdysteroids or hormones regulating the processes of development and reproduction [25, 52]. One of these proteins is the low-density lipoprotein (LDL) receptor (B7QDC5). LDL is the main plasma cholesterol-carrying lipoproteins. This receptor, which is expressed on the membrane of the intestinal cell, captures LDL molecules and transports them into the enterocytes by a mechanism of endocytosis involving clathrin-coated vesicles. Once inside the cell, free cholesterol binds to endogenous tick proteins, passes to the haemolymph and is distributed throughout the remaining tick tissues [25]. Up to seven transcripts coding for this LDL receptor were identified in the O. moubata midgut transcriptome, lending additional support to the importance of this mechanism in the O. moubata physiology.
Three additional proteins likely engaged in the transport of cholesterol are the Niemann-Pick type C1 (NPC1) proteins (B7Q350, Q6VL7) and the epididymal secretory protein E1 (also known as Niemann-Pick C2 protein - NPC2) (Table 5) [53]. Likewise, erlin-2 is a protein that localizes to areas of lipid accumulation of the endoplasmic reticulum membrane and regulates cholesterol levels inside the cell (Table 5) [54, 55].
Other proteins showing lipid transport function are ankyrin-2 (E2C1X2), three vitellogenins (G9M4I6, B6ZIV7, E1CAX9), a fatty acid transport protein (B7QMW0) and an uncharacterized protein (B7Q381). Ankyrin is a protein that has a domain common to the family of apolipoproteins, which, together with the vitellogenins, belongs to a large protein family of lipid transporters. Vitellogenin, also involved in binding and detoxification of the haem group, is the precursor of vitellin, which is the main component of eggs and provides the necessary energy and amino acids for egg development and oviposition [56, 57] (Table 5).
Endocytosis and intracellular transport
The haemoglobin released after erythrocyte lysis is incorporated into the digestive cells through an endocytic process, mediated by specific receptors present in clathrin-coated vesicles that are internalized to form large endosomes. Albumin is also internalized by endocytosis, but using a non-specific transport mechanism that does not require the involvement of clathrin. The proteolytic enzymes that digest haemoglobin, and likely the albumin, are synthesized as precursors in the endoplasmic reticulum and are transferred to lysosomal vesicles through the Golgi. The lysosomes and the endosomes that contain haemoglobin fuse to form the digestive vesicles where proteolysis takes place [5, 15, 43].
Up to 43 proteins likely involved in the recognition and internalization of haemoglobin inside endosomes, the trafficking and fusion of intracellular vesicles, and the transport of proteins and other molecules inside the digestive cells were identified in the O. moubata midgut proteome: 8 of these proteins were found in unfed females, 14 in fed females and 21 in both physiological conditions (Table 6).
Among the proteins potentially involved in the endocytosis process, and the formation and assembly of clathrin-coated vesicles, we identified several clathrins: three proteins belonging to the adaptor protein (AP) complex (AP-1, AP-2 and AP-3) and a beta-adaptin [58]. In relation to the regulation of these mechanisms, we also identified the phosphatidylinositol-binding clathrin assembly protein, which limits the size of clathrin-coated vesicles and controls the traffic of endocytic membranes by recruiting adapters and other components of the transport machinery [59]. Dynamin-1 plays a key role in the fusion of the endocytic membrane [60] (Table 6).
Among the proteins that may be implicated in vesicle-mediated intracellular transport, we identified, among others, the following: acetylcholine regulator unc-18; alpha-SNAP; synaptobrevin and its homologue VAMP-7; the Rab family of proteins; transport-interaction with t-SNAREs-like protein 1B; surfeit 4; the coatomers COPI and COPII; cargo transport protein EMP24; transmembrane protein Tmp21 and glycoprotein 25 l (Table 6).
The acetylcholine regulator unc-18 protein regulates vesicle transport by preventing the formation of the SNARE complex, which, together with the alpha-SNAP protein, synaptobrevin and surfeit 4, is essential for specific fusion between the membranes of the vesicles [61, 62]. Rab proteins are a family of GTPases that regulate the intracellular transport of vesicles, directing the trafficking and fusion of the vesicle to the target membrane, thus fulfilling a key role in the specificity of the vesicular transport within the cell [63].
Coatomers COPI and COPII (coat complex proteins) are multiprotein complexes that, when assembled, form a coating that covers the vesicles that transport proteins and lipids, and mediates their trafficking between the Golgi apparatus and the endoplasmic reticulum [64, 65] (Table 6). Related to this latter mechanism are the proteins cargo transport protein EMP24, transmembrane protein Tmp21 and glycoprotein 251, which are members of the p24 family. This is a very conserved family of membrane proteins that mediate the assembly of COPI coatomers in the membranes of vesicles [66].
Regarding the intracellular transport of molecules, it has recently been demonstrated that the “ABC transporter” proteins mediate the transport of the haem group from the digestive vesicles to the haemosomes, where it accumulates to form large aggregates [67]. We have identified three of these ABC transporters: G9F9U2, Q3YB2 and B7PUI6. On the other hand, the surfeit 4 protein, besides being involved in the fusion of membranes, is also believed to be involved in the transport of haemoglobin in the cell cytosol [38].
Some of the above-listed proteins were also identified in the intestinal proteomes of Rhipicephalus microplus and O. erraticus, which revealed the importance of mechanisms of intracellular transport within the enterocytes during blood digestion processes [21, 28]. Our results significantly expand the knowledge of the elements that are part of the intracellular transport machinery, highlighting its complexity and the interest for deepening the knowledge of the regulatory mechanisms that mediate these processes.
Oxidative stress response and detoxification
The digestion of haemoglobin inside digestive vesicles of enterocytes releases large amounts of haem group, iron ions, hydrogen peroxide, hydroxyl radicals and other toxic molecules that induce oxidative responses. As other haematophagous organisms, ticks have detoxifying mechanisms that block the oxidative reactions produced by blood digestion, thus protecting themselves from their deleterious effects [68,69,70]. One of these detoxifying mechanisms developed by ticks is the accumulation of excess haem molecules in intracellular organelles called haemosomes [71].
In the midgut transcriptome of O. moubata, Oleaga et al. [29] identified up to 79 genes involved in response to cellular oxidative stress, most of which were upregulated after feeding, in parallel with what was also observed in the mialome of Dermacentor marginatus [19].
In the midgut proteomes analysed in the current work, we identified up to 13 antioxidant proteins with oxidoreductase activity, 12 proteins involved in detoxification processes, up to 40 heat shock proteins (HSPs), also known as stress response proteins, and other chaperones involved in protein folding processes (Table 7).
Related to the antioxidant enzymes responsible for removing hydrogen peroxide, we identified several thioredoxin peroxidases (peroxiredoxins) (Q9GV35, A6N9S1, A6NA14), thioredoxin reductases (B7QN17, B7PTG8), peroxidases (B7Q8W6, B7QGB0), peroxinectin (B7PUM7), glutathione peroxidase (Q2XW17), catalase (T2FDK5) and superoxide dismutase (SOD) (B7PUC4) (Table 7). Some of these proteins have also been identified in the gut of O. erraticus and several ixodid species, where they participate in the defence against cellular oxidative stress [19, 21, 25, 28, 43, 70]. Specifically, catalase is the main enzyme responsible for controlling hydrogen peroxide released in digestive cells after blood ingestion [70]. On the other hand, among the enzymes with oxidoreductase activity, the enzyme SOD stands out due to its abundance, especially in the unfed ticks (emPAI 4.59 in unfed and 2.15 in fed ticks). Besides its antioxidant properties of removing free radicals produced during blood digestion, SOD is likely involved in additional biological processes because it can also regulate the size of the populations of the pathogens transmitted by the tick [72] and participate in transporting the haem group inside the digestive cells [19, 73].
HSPs and other chaperones are a set of highly conserved proteins produced by cells whose concentration increases in stressful situations, such as the increase in temperature after blood ingestion at 37 °C. In such a situation of stress exposure, these proteins stabilize other proteins inside cells, preventing their denaturation, and promoting their folding and assembly to generate the correct tertiary structure [74]. Many upregulated transcripts coding for HSPs and chaperones were identified in the midgut transcriptome of O. moubata after feeding [29].
Table 7 shows the numerous chaperones identified in the O. moubata midgut proteome. Among them, the following proteins are noteworthy because of their abundance: three HSPs of 70 kDa (A0A097A1J8, E4W3Z2, F0J8P3) with emPAI values of 18.61, 8.27 and 7.42; the chaperonin containing t-complex polypeptide (G6DGE9) protein with emPAI values of 4.89 in unfed and 6.06 in fed; and the peptidyl-prolyl cis-after isomerase protein (B7PD56) with an emPAI value in unfed ticks of 4.59. HSP70 not only contributed to cell protection against the stress generated by blood digestion, but it also could participate in removing the clathrin-cover from the endocytic vesicles, thus playing a significant role in the intracellular vesicular transport [75].
Several proteins involved in detoxification processes were also identified in the O. moubata midgut proteome, including members of the family of cytochrome P450 (CYPs) and members of the family of glutathione S-transferases (GSTs) (Table 7). CYPs are involved in phase I of the xenobiotic detoxification system. In O. moubata, we identified up to five components of this family. These enzymes have a haem group in their structure that acts as a prosthetic group. This might suggest some relation of CYPs to the digestion of haemoglobin; however, it has recently been shown that, at least in Ixodes ricinus, their expression is independent of blood ingestion [25].
The GST family comprises several classes of enzymes, some of which participate in phase II of the detoxification system [25]. We have identified up to five GSTs in the midgut of O. moubata, which were also present in the O. erraticus midgut [28] (Table 7).
Conclusions
This work, together with the former analysis of the O. moubata midgut transcriptome [29], have provided a wealth of unprecedented information on the genes and proteins involved in the digestion process in argasids, particularly in the processes of nutrient metabolism and transport and the defensive, detoxifying and antioxidant responses triggered by the ingestion of blood. The identification of a set of proteolytic enzymes belonging to the same classes/families of proteases in the midgut of the argasid tick O. moubata as those described in ixodid ticks, reveals that the haemoglobinolytic system in both tick families is very similar although they display very different feeding and reproductive strategies. Although the main source of nutrients during host blood digestion in ticks is proteins, particularly haemoglobin and albumin, the identification of numerous genes and proteins involved in the metabolism and transport of lipids and carbohydrates reveals that these components also constitute significant nutritional sources and play an important part in the process of blood digestion. The genes and proteins identified in the mialome of O. moubata that are involved in intracellular transport mechanisms, defensive responses, detoxifying responses and stress responses seem to be closely regulated, highlighting the complexity and importance of these processes in tick biology, which in turn assigns them great interest as targets for therapeutic and/or immunological interventions. Our results also confirm the usefulness of the transcriptome as a searchable database for improving identifications in proteomic analyses, especially in those carried out on organisms that do not have their genome sequenced, as is the case for O. moubata.
Abbreviations
- ACN:
-
Acetonitrile
- AP:
-
Adaptor protein
- COP:
-
Coat complex proteins
- CYP:
-
Cytochrome P450
- DTT:
-
Dithiothreitol
- emPAI:
-
Exponentially modified protein abundance index
- EST:
-
Expressed sequence tag
- FA:
-
Formic acid
- GO:
-
Gene Ontology
- GST:
-
Glutathione S-transferases
- HSP:
-
Heat shock protein
- IDA:
-
Information dependent acquisition
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LC/MS-MS:
-
Liquid chromatography tandem mass spectrometry
- LDL:
-
Low-density lipoprotein
- M-0:
-
Membrane-associated proteins from unfed ticks
- M-1:
-
Membrane-associated proteins from fed ticks
- mRNA:
-
Messenger ribonucleic acid
- NCBInr:
-
National Center for Biotechnology Information non-redundant
- PANTHER:
-
Protein Analysis Through Evolutionary Relationships
- PBS:
-
Phosphate buffered saline
- PIT:
-
Proteomics informed by transcriptome
- RNAseq:
-
Ribonucleic acid sequencing
- S-0:
-
Soluble proteins from unfed ticks
- S-1:
-
Soluble proteins from fed ticks
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SOD:
-
Superoxide dismutase
- TFA:
-
Trifluoroacetic acid
- TOF MS:
-
Time of flight mass spectrometry
References
Hoogstraal H. Argasid and nuttalliellid ticks as parasites and vectors. Adv Parasitol. 1985;24:135–238.
Barker SC, Murrel A. Systematics and evolution of ticks with a list of valid genus and species names. In: Bowman AS, Nuttall P, editors. Ticks: biology, disease and control. New York: Cambridge University Press; 2008. p. 1–39.
Latif AA, Putterill JF, de Klerk DG, Pienaar R, Mans BJ. Nuttalliella namaqua (Ixodoidea: Nuttalliellidae): first description of the male, immature stages and re-description of the female. PLoS One. 2012;7:e41651.
Manzano-Román R, Díaz-Martín V, de la Fuente J, Pérez-Sánchez R. Soft ticks as pathogen vectors: distribution, surveillance and control. In: Shah MM, editor. Parasitology. Croatia: Intech, Rijeka; 2012.p.125–162.
Sonenshine DE, Roe RM. Biology of ticks, vol. I. Oxford: Oxford University Press; 2014.
de la Fuente J, Estrada-Pena A, Venzal JM, Kocan KM, Sonenshine DE. Overview: ticks as vectors of pathogens that cause disease in humans and animals. Front Biosci. 2008;13:6938–46.
de la Fuente J, Kopáček P, Lew-Tabor A, Maritz-Olivier C. Strategies for new and improved vaccines against ticks and tick-borne diseases. Parasite Immunol. 2016;38:754–69.
Vial L. Biological and ecological characteristics of soft ticks (Ixodida: Argasidae) and their impact for predicting tick and associated disease distribution. Parasite. 2009;16:191–202.
Cutler SJ. Relapsing fever – a forgotten disease revealed. J Appl Microbiol. 2010;108:1115–22.
Quembo CJ, Jori F, Heath L, Pérez-Sánchez R, Vosloo W. Investigation into the epidemiology of African swine fever virus at the wildlife-domestic interface of the Gorongosa National Park, Central Mozambique. Transbound Emerg Dis. 2016;63:443–51.
Guerrero FD, Miller RJ. Perez de Leon AA. Cattle tick vaccines: many candidate antigens, but will a commercially viable product emerge? Int J Parasitol. 2012;42:421–7.
Díaz-Martín V, Manzano-Román R, Obolo-Mvoulouga P, Oleaga A, Perez-Sanchez R. Development of vaccines against Ornithodoros soft ticks: an update. Ticks Tick-Borne Dis. 2015;6:211–20.
Willadsen P. Anti-tick vaccines. In: Bowman AS, Nuttall P, editors. Ticks: biology, disease and control. New York: Cambridge University Press; 2008. p. 424–46.
Kocan KM, De La Fuente J, Blouin EF, Garcia-Garcia JC. Anaplasma marginale (Rickettsiales: Anaplasmataceae): recent advances in defining host-pathogen adaptations of a tick-borne Rickettsia. Parasitology. 2004;129:S285–300.
Sojka D, Franta Z, Horn M, Caffrey CR, Mareš M, Kopáček P. New insights into the machinery of blood digestion by ticks. Trends Parasitol. 2013;29:276–85.
Richards SA, Stutzer C, Bosman A-M, Maritz-Olivier C. Transmembrane proteins - mining the cattle tick transcriptome. Ticks Tick-Borne Dis. 2015;6:695–710.
García-Varas S. Vacuna anti-Ornithodoros moubata. PhD Thesis. Universidad de Salamanca; 2004.
Manzano-Román R, Encinas-Grandes A, Pérez-Sánchez R. Antigens from the midgut membranes of Ornithodoros erraticus induce lethal anti-tick immune responses in pigs and mice. Vet Parasitol. 2006;135:65–79.
Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics. 2008;9:552.
Rachinsky A, Guerrero FD, Scoles GA. Proteomic profiling of Rhipicephalus (Boophilus) microplus microplus midgut responses to infection with Babesia bovis. Vet Parasitol. 2008;152:294–313.
Kongsuwan K, Josh P, Zhu Y, Pearson R, Gough J, Colgrave ML. Exploring the midgut proteome of partially fed female cattle tick Rhipicephalus (Boophilus) microplus. J Insect Physiol. 2010;56:212–26.
Heekin AM, Guerrero FD, Bendele KG, Saldivar L, Scoles GA, Dowd SE, Gondro C, Nene V, Djikeng A, Brayton KA. Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol Res. 2013;112:3075–90.
Schwarz A, Tenzer S, Hackenberg M, Erhart J, Gerhold-Ay A, Mazur J, et al. A systems level analysis reveals transcriptomic and proteomic complexity in Ixodes ricinus midgut and salivary glands during early attachment and feeding. Mol Cell Proteomics. 2014;13:2725–35.
Kotsyfakis M, Schwarz A, Erhart J, Ribeiro JM. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep. 2015;5:9103.
Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood- and serum-fed Ixodes ricinus ticks. Sci Rep. 2016;6:36695.
Nakajima Y, Van Der Goes van Naters-Yasui A, Taylor D, Yamakawa M. Antibacterial peptide defensin is involved in midgut immunity of the soft tick, Ornithodoros moubata. Insect Mol Biol. 2002;11:611–8.
Grunclová L, Horn M, Vancová M, Sojka D, Franta Z, Mares M, Kopácek P. Two secreted cystatins of the soft tick Ornithodoros moubata: differential expression pattern and inhibitory specificity. Biol Chem. 2006;387:1635–44.
Oleaga A, Obolo-Mvoulouga P, Manzano-Román R, Pérez-Sánchez R. Midgut proteome of an argasid tick, Ornithodoros erraticus: a comparison between unfed and engorged females. Parasit Vectors. 2015;8:525.
Oleaga A, Obolo-Mvoulouga P, Manzano-Román R, Pérez-Sánchez R. Functional annotation and analysis of the Ornithodoros moubata midgut genes differentially expressed after blood feeding. Ticks Tick-Borne Dis. 2017;8:693–708.
Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–72.
Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modelling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–86.
Popara M, Villar M, de la Fuente J. Proteomics characterization of tick-host-pathogen interactions. Methods Mol Biol. 2015;1247:513–27.
Evans VC, Barker G, Heesom KJ, Fan J, Bessant C, Matthews DA. De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nat Methods. 2012;9:1207–11.
Chocu S, Evrard B, Lavigne R, Rolland AD, Aubry F, Jégou B, et al. Forty-four novel protein-coding loci discovered using a proteomics informed by transcriptomics (PIT) approach in rat male germ cells. Biol Reprod. 2014;91:123.
Villar M, Popara M, Ayllón N. Fernández de Mera IG, Mateos-Hernández L, Galindo RC, et al. A systems biology approach to the characterization of stress response in Dermacentor reticulatus tick unfed larvae. PLoS One. 2014;9:e89564.
Kumar D, Bansal G, Narang A, Basak T, Abbas T, Dash D. Integrating transcriptome and proteome profiling: strategies and applications. Proteomics. 2016;16:2533–44.
Luge T, Fischer C, Sauer S. Efficient application of de novo RNA assemblers for proteomics informed by transcriptomics. J Proteome Res. 2016;15:3938–43.
Sonenshine DE, Anderson JM. Mouthparts and digestive system: anatomy and molecular biology of feeding and digestion. In: Sonenshine DE, Roe RM, editors. Biology of ticks. Oxford: Oxford University Press; 2014. p. 122–62.
Smith TA, Driscoll T, Gillespie JJ, Raghavan R. A Coxiella-like endosymbiont is a potential vitamin source for the lone star tick. Genome Biol Evol. 2015;7:831–8.
Narasimhan S, Fikrig E. Tick microbiome: the force within. Trends Parasitol. 2015;31:315–23.
Stutzer C, Mans BJ, Gaspar AR, Neitz AW, Maritz-Olivier C. Ornithodoros savignyi: soft tick apyrase belongs to the 5′-nucleotidase family. Exp Parasitol. 2009;122:318–27.
Maier T, Güell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–73.
Stutzer C, van Zyl WA, Olivier NA, Richards S, Maritz-Olivier C. Gene expression profiling of adult female tissues in feeding Rhipicephalus microplus cattle ticks. Int J Parasitol. 2013;43:541–54. d.
Sojka D, Pytelková J, Perner J, Horn M, Konvičková J, Schrenková JM, et al. Multienzyme degradation of host serum albumin in ticks. Ticks Tick Borne Dis. 2016;7:604–13.
Horn M, Nussbaumerová M, Sanda M, Kovárová Z, Srba J, Franta Z, et al. Hemoglobin digestion in blood-feeding ticks: mapping a multipeptidase pathway by functional proteomics. Chem Biol. 2009;16:1053–63.
Cruz CE, Fogaça AC, Nakayasu ES, Angeli CB, Belmonte R, Almeida IC, et al. Characterization of proteinases from the midgut of Rhipicephalus (Boophilus) microplus involved in the generation of antimicrobial peptides. Parasit Vectors. 2010;3:63.
Lai R, Takeuchi H, Lomas LO, Jonczy J, Rigden DJ, Rees HH, Turner PC. A new type of antimicrobial protein with multiple histidines from the hard tick, Amblyomma hebraeum. FASEB J. 2004;18:1447–9.
Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol. 2016;38:62–72.
Cabezas-Cruz A, Alberdi P, Valdés JJ, VillarM, de la Fuente J. Anaplasma phagocytophilum infection subverts carbohydrate metabolic pathways in the tick vector, Ixodes scapularis. Front Cell Infect Microbiol. 2017;7:23.
Manzano-Román R, Díaz-Martín V, González-González M, Matarraz S, Álvarez-Prado AF, LaBaer J, et al. Self-assembled protein arrays from an Ornithodoros moubata salivary gland expression library. J Proteome Res. 2012;11:5972–82.
Shayman JA, Kelly R, Kollmeyer J, He Y, Abe A. Group XV phospholipase a2, a lysosomal phospholipase a2. Prog Lipid Res. 2011;50:1–13.
Ress HH. Hormonal control of tick development and reproduction. Parasitology. 2004;129:127–43.
King G, Sharom FJ. Proteins that bind and move lipids: MsbA and NPC1. Crit Rev Biochem Mol Biol. 2012;47:75–95.
Browman DT, Resek ME, Zajchowski LD, Robbins SM. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J Cell Sci. 2006;119:3149–60.
Huber MD, Vesely PW, Datta K, Gerace L. Erlins restrict SREBP activation in the ER and regulate cellular cholesterol homoeostasis. J Cell Biol. 2013;203:427–36.
Smolenaars MM, Madsen O, Rodenburg KW, Van der Horst DJ. Molecular diversity and evolution of the large lipid transfer protein superfamily. J Lipid Res. 2007;48:489–502.
Taheri M, Nabian S, Ranjbar M, Mazaheri Nezhad R, Gerami Sadeghian A, Sazmand A. Study of vitellogenin in Boophilus annulatus tick larvae and its immunological aspects. Trop Biomed. 2014;31:398–405.
Owen DJ, Collins BM, Evans PR. Adaptors for clathrin coats: structure and function. Annu Rev Cell Dev Biol. 2004;20:153–91.
Haucke V. Phosphoinositide regulation of clathrin-mediated endocytosis. Biochem Soc Trans. 2005;33:1285–9.
Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. 2012;13:75–88.
Bracher A, Perrakis A, Dresbach T, Betz H, Weissenhorn W. The X-ray crystal structure of neuronal Sec1 from squid sheds new light on the role of this protein in exocytosis. Structure. 2000;8:685–94.
May AP, Whiteheart SW, Weis WI. Unraveling the mechanism of the vesicle transport ATPase NSF, the N-ethylmaleimide-sensitive factor. J Biol Chem. 2001;276:21991–4.
Bhuin T, Roy JK. Rab proteins: the key regulators of intracellular vesicle transport. Exp Cell Res. 2014;328:1–19.
Watson PJ, Frigerio G, Collins BM, Duden R, Owen DJ. Gamma-COP appendage domain - structure and function. Traffic. 2004;5:79–88.
Beck R, Rawet M, Wieland FT, Cassel D. The COPI system: molecular mechanisms and function. FEBS Lett. 2009;583:2701–9.
Carney GE, Bowen NJ. p24 proteins, intracellular trafficking, and behavior: Drosophila melanogaster provides insights and opportunities. Biol Cell. 2004;96:271–8.
Lara FA, Pohl PC, Gandara AC, Ferreira Jda S, Nascimento-Silva MC, Bechara GH, et al. ATP binding cassette transporter mediates both heme and pesticide detoxification in tick midgut cells. PLoS One. 2015;10:e0134779.
Paes MC, Oliveira MB, Oliveira PL. Hydrogen peroxide detoxification in the midgut of the blood-sucking insect, Rhodnius prolixus. Arch Insect Biochem Physiol. 2001;48:63–71.
Graça-Souza AV, Maya-Monteiro C, Paiva-Silva GO, Braz GR, Paes MC, et al. Adaptations against heme toxicity in blood-feeding arthropods. Insect Biochem Mol Biol. 2006;36:322–35.
Citelli M, Lara FA, da Silva Vaz I Jr, Oliveira PL. Oxidative stress impairs heme detoxification in the midgut of the cattle tick, Rhipicephalus (Boophilus) microplus. Mol Biochem Parasitol. 2007;151:81–8.
Lara FA, Lins U, Paiva-Silva G, Almeida IC, Braga CM, Miguens FC, et al. A new intracellular pathway of haem detoxification in the midgut of the cattle tick Boophilus microplus: aggregation inside a specialized organelle, the hemosome. J Exp Biol. 2003;206:1707–15.
Crispell G, Budachetri K, Karim S. Rickettsia parkeri colonization in Amblyomma maculatum: the role of superoxide dismutases. Parasit Vectors. 2016;9:291.
Pacello F, Langford PR, Kroll JS, Indiani C, Smulevich G, Desideri A, et al. A novel heme protein, the cu, Zn-superoxide dismutase from Haemophilus ducreyi. J Biol Chem. 2001;276:30326–34.
Carmelo E, Zurita AI, González AC, Martínez E, Valladares B. Proteínas de choque térmico en parásitos: la Hsp70 y el sistema inmune. Rev Iber Parasitol. 2006;66:39–46.
Sousa R, Lafer EM. The role of molecular chaperones in clathrin mediated vesicular trafficking. Front Mol Biosci. 2015;2:26.
Acknowledgements
The authors are grateful to Rocío Vizcaíno Marín, María González Sánchez and Diana Ramos Pintado, from the Instituto de Recursos Naturales y Agrobiología de Salamanca (IRNASA, CSIC) (Spain), and Luz Valero from the SCSIE_University of Valencia Proteomics Unit for their skilful technical assistance. We acknowledge the support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Funding
This research was funded by project AGL2013–42745-P granted by the Spanish Ministry of Economy and Competitiveness.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request. The midgut transcriptome data used during this study were deposited in DDBJ/ENA/GenBank under accession number GFJQ00000000 as a Transcriptome Shotgun Assembly project (BioProject: PRJNA377416).
Author information
Authors and Affiliations
Contributions
AO, RPS conceived the study and designed the experiments. POM maintained the tick colony and prepared the midgut protein extracts. AO, RPS, RMR and POM collaborated in the data analysis. AO, RPS and RMR collaborated in writing and editing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All the procedures involving animal experimentation were performed according to Spanish rules and regulations and the obtained approval from the Board for Animal Wellness (OEBA) of IRNASA (CSIC).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Non-redundant host proteins identified in Soluble fraction, S-0, from midgut of unfed Ornithodoros moubata female ticks (XLSX 26 kb)
Additional file 2:
Table S2. Non-redundant tick proteins identified in the midgut of unfed ticks (unfed group) and engorged ticks at 48 h post-feeding (fed group) (XLSX 295 kb)
Additional file 3:
Table S3. Biological pathways represented in the midgut unfed ticks, number of sequences and enzymes involved in each pathway. The analysis of the midgut proteome was done in the KEEG pathway database using the Blast2GO software (XLSX 44 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Oleaga, A., Obolo-Mvoulouga, P., Manzano-Román, R. et al. A proteomic insight into the midgut proteome of Ornithodoros moubata females reveals novel information on blood digestion in argasid ticks. Parasites Vectors 10, 366 (2017). https://doi.org/10.1186/s13071-017-2300-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2300-8