
Abstract
Background
The blood fluke, Schistosoma japonicum still causes severe disease in China, the Philippines and Indonesia. Although there have been some studies the molecular epidemiology of this persistent and harmful parasite, few have explored the possibility and implications of selection in S. japonicum populations.
Methods
We analyzed diversity and looked for evidence of selection at three nuclear genes (SjIpp2, SjFabp and SjT22.6) in 13 S. japonicum populations.
Results
SjT22.6 was found to exhibit high nucleotide diversity and was under positive selection in the mountainous region of mainland China. As a tegumental protein, its secondary and tertiary structure differed between S. japonicum strains from the mountainous and lakes regions. In contrast, SjIpp2 and SjFabp had relatively low levels of nucleotide diversity and did not show significant departure from neutrality.
Conclusions
As a tegument-associated antigen-encoding gene of S. japonicum, SjT22.6 has high nucleotide diversity and appears to be under positive selection in the mountainous region of mainland China.
Similar content being viewed by others

Background
Approximately 207 million people suffer from schistosomiasis [1]. Schistosoma japonicum is endemic in the People’s Republic of China [2], the Philippines [3] and parts of Indonesia [4]. In mainland China, this parasite is particularly prevalent in the lake/marshland regions around the River Yangtze and some mountainous regions in southwest China [5]. Strenuous control efforts during the last five decades have greatly reduced the infection levels and sizes of endemic areas [6]. However, ecosystem changes caused by environmental deterioration and the construction of new infrastructure projects, such as the Three Gorges Dam, contributed to the resurgence of schistosomiasis in the early 21st century [7]. Given the great need for prevention and control of this disease, a thorough understanding of the evolutionary history and population genetic structure of S. japonicum is urgently required [8, 9].
Different types of molecular markers have been applied to investigate the genetic variability of S. japonicum populations, such as restriction fragment length polymorphism [10], isoenzymes [11], random amplified polymorphic DNA [12], mitochondrial DNA sequences [13, 14] and microsatellites [8, 15]. Recent phylogenetic analyses showed that S. japonicum populations in the middle and lower reaches of the River Yangtze are well differentiated from those in the mountainous areas of western China [16–18]. However, so far no study has used specific nuclear genes as molecular markers to assess the effect of selection among Chinese S. japonicum populations.
Nuclear genes can be sensitive for addressing questions about genetic variation and in tracing genetic bottlenecks and identifying selection [19]. Liu et al. [20] proposed that three nuclear genes in S. japonicum might be under positive selection, including those which encode a protein phosphatase inhibitor 2 (SjIpp2), a fatty acid-binding protein (SjFabp) and a tegument-associated antigen (SjT22.6). The first of these, SjIpp2, likely stops, prevents or reduces the activity of a protein phosphatase [21]. SjFabp belongs to a family of lipid-binding proteins [22] and SjT22.6 is a tegumental surface membrane-anchored calcium-binding antigen, which belongs to a family consisting of platyhelminth tegument-specific proteins [23].
In the present study, we analyzed gene sequences in individual S. japonicum worms for SjIpp2, SjFabp and SjT22.6 from 13 populations, including nine locations across mainland China (covering the lakes region and mountainous region) and four locations elsewhere in Asia (Taiwan, Indonesia, Japan and the Philippines). First, we analyzed the diversity of these genes in S. japonicum populations. Then we looked for evidence of positive selective pressure acting on these genes as predicted by our previous study [20]. Finally, we used bioinformatics tools to predict whether the protein structure changed under positive selection.
Methods
Sample collection
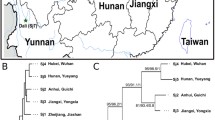
Adult individuals of S. japonicum were obtained from 13 locations, including nine from mainland China and four from elsewhere in Asia (Table 1). In mainland China, the sampling was carried out in the lakes region (Guichi and Tongling City in Anhui Province, Shashi City in Hubei Province, Yueyang and Changde City in Hunan Province, Duchang and Nanchang City in Jiangxi Province) and the mountainous region (Eryuan County in Yunnan Province and Xichang City in Sichuan Province). Infected snails (Oncomelania hupensis) were collected from each locality in mainland China then transported to the laboratory of NIPD, China CDC, Shanghai. Collection information concerning the infected snails was provided in [24]. In brief, numbers of infected snails from seven localities were as follows: Guichi, 7; Tongling, 10; Shashi, 30; Yueyang, 29; Changde, 21; Duchang, 11; Xichang, 15. Numbers of infected snails from Nanchang and Eryuan are not available. Cercariae were released from pooled infected snails from each locality and used to infect laboratory-raised rabbits. Forty-five days after infection, the adult schistosomes were perfused from the mesenteric veins of infected rabbits and washed in saline, then preserved in 95% (v/v) ethanol at 4 °C. Four other locations from which samples were obtained were Chinese Taiwan (Changhua), Indonesia (Lake Lindu, Sulawesi), Japan (Yamanashi) and the Philippines (Leyte). The lyophilized adult worms from Indonesia and Taiwan were provided by Dr. John Cross, Uniformed Services University of the Health Sciences, Bethesda, USA. The adult worms from Japan (Kofu) were gifted by Dr. Hiroshi Yamasaki, National Institute of Infectious Diseases, Tokyo, Japan. The S. japonicum individuals from the Philippines were taken into culture originally in 1969 by Dr. Scholice. The material sent to us consisted of lyophilized adult worms, which was provided by Dr. John Bruce, Centre for Tropical Diseases, University of Lowell, USA.
Genomic DNA was extracted from each adult worm using the DNeasy Blood & Tissue Kit and Animal Tissues (Spin-Column) protocol from Qiagen (Hilden, Germany), and stored at -20 °C [15, 24]. For most locations, 2–5 individual worms were randomly selected for genetic analysis. However, following initial analyses, 28 additional individual worms were sampled from the mountainous region to examine genetic variation and natural selection in SjT22.6.
Gene amplification and sequencing
To obtain the sequences of the complete gene, we first extracted mRNA sequences of the three targeted gene fragments from NCBI (as only mRNA sequences of these target genes were deposited there): SjIpp2 (AY815218.1), SjFabp (EZ000092.2) and SjT22.6 (AY813797.1). Using these, we then obtained the complete gene sequences by local Blast searches against the S. japonicum genome sequence database [25], enabling us to design primers to amplify and sequence the entire region coding for the mRNA.
The complete mRNA sequence of SjIpp2 was obtained from NCBI (AY815218.1, 992 bp). The coding regions for this gene are composed of two exons, which were amplified separately from the S. japonicum genome DNA using two PCR reactions (Additional file 1: Figure S1a). We analyzed the full length of the gene (sites 1–992) and the coding region (sites 23–616) separately. The complete mRNA sequence of SjFabp was obtained from NCBI (EZ000092.2, 699 bp). Only a portion of the coding region and non-coding region of this gene (sites 210–675) could be successfully amplified (Additional file 1: Figure S1b). We analyzed the genome sequence (sites 210 to 675) and the partial coding region (sites 210–513) of this fragment. The complete mRNA sequence of SjT22.6 was obtained from NCBI (AY813797.1, 887 bp). Again, only a portion of the coding region and non-coding region of this fragment (sites 283–815) was successfully amplified using PCR (Additional file 1: Figure S1c). We analyzed the genome sequence (sites 283–728) and the partial coding region (sites 283–624) of this fragment. The primers used in the present study were shown in Additional file 2: Table S1.
Each DNA fragment was amplified in a 20 μl PCR reaction, containing 1 μl of each primer at a final concentration of 10 μM, 2 μl genomic DNA from an individual worm and 10 μl 2× Taq PCR Master Mix (TaKaRa, Japan). For PCR amplification, templates were denatured at 94 °C for 5 min, followed by 35 cycles with denaturation at 94 °C for 30 s, annealing for 45 s at 51 °C for SjIpp2-1, 54 °C for SjIpp2-2, 55 °C for SjFabp, 46 °C for SjT22.6 and extension at 72 °C (40s for SjIpp2-1, 60 s for SjIpp2-2, 100 s for SjFabp and 40 s for SjT22.6), ending with a final extension at 72 °C for 10 min. All the PCR products were examined using agarose gel electrophoresis (1% w/v) to verify amplification efficiency. The PCR products were sequenced by an ABI 3730 DNA Analyzer (BGI, Shanghai, China) using the PCR primers from both directions. All the sequences were submitted to GenBank under accession numbers KY494243–KY494287 for SjIpp2, KY494288–KY494346 for SjFabp and KY494347–KY494416 for SjT22.6.
Genetic diversity
The sequences were assembled manually using Vector NTI [26] and aligned using the ClustalW algorithm [27] in MEGA v. 5.10 [28]. The numbers of haplotypes (H), nucleotide diversity (π) and the Watterson estimator (θw) were obtained using DnaSP Version 5 [29]. The Watterson estimator is a method for describing the genetic diversity in a population, which is estimated by counting the number of polymorphic sites. It is a measure of the “population mutation rate” (the product of the effective population size and the neutral mutation rate) from the observed nucleotide diversity of a population [30].
Haplotype network and selection
The haplotype networks were constructed using HapView [31] to visualize phylogenies. Rates of nonsynonymous substitutions (dN) and synonymous substitutions (dS) were obtained and Tajima’s D test and Fu’s Fs test were implemented using DnaSP Version 5. A ratio of nonsynonymous to synonymous divergence (dN/dS) higher than 1 was considered as evidence of positive selection [32]. Furthermore, Tajima’s D test and Fu’s Fs test were applied to test for departure from neutral evolution. Tajima’s D test is calculated according to the difference between segregating sites and the average of nucleotide differences [33], and Fu’s Fs test compares the number of haplotypes observed with the expected number of haplotypes in a random sample [34].
Protein structure
Inferred amino acid sequences were aligned using the ClustalW algorithm in MEGA 5.10. Protein models were predicted by Phyre2 [35], and the energy minimization of the model was performed using YAMASA [36]. Secondary structures were extracted from the predicted model by ProFunc [37]. Protein-ligand binding sites were predicted using COFACTOR [38–40]. Structural visualization and editing were done using PyMol software (http://www.pymol.org).
Results
Genetic diversity
In total, 45 individual S. japonicum adult worms were successfully sequenced for SjIpp2, 59 for SjFabp and 70 for SjT22.6. Among these, 21 unique haplotypes were detected for SjIpp2, 7 for SjFabp and 17 for SjT22.6; 10, 4 and 10 unique haplotypes were obtained for the coding regions of the three genes, respectively (Table 1 and Additional file 3: Table S2).
Overall, the level of nucleotide diversity within S. japonicum populations was low for SjIpp2 (π = 0.006) and SjFabp (π = 0.002). In contrast, the nucleotide diversity for SjT22.6 (π = 0.022) was approximately 4-fold higher than that for SjIpp2 and 10-fold higher than that for SjFabp. Furthermore, the Watterson estimator for this gene (θw = 0.015) was 2 times higher than SjIpp2 (θw = 0.007) and 5 times higher than SjFabp (θw = 0.003). Interestingly, for SjT22.6, the nucleotide diversity of S. japonicum populations from the mountainous region (π = 0.030) was approximately six times greater than those from the lakes region (π = 0.005). Outside mainland China, the SjT22.6 sequences from Chinese Taiwan or Indonesia worms were identical and this gene could not be amplified for individuals from Japan and the Philippines.
Haplotype network
The most abundant haplotype of the SjIpp2 coding region (IH1; 23 out of 45) was shared by the 11 locations in the lakes region of mainland China and elsewhere (Fig. 1a), but was not found in mountainous regions of the mainland. For the SjFabp coding region, almost all of the S. japonicum individuals shared a single haplotype (FH1; 56 out of 59, Fig. 1b), regardless of geographical origin. For the SjT22.6 coding region, the most abundant haplotype (TH1; 18 out of 70) was shared by seven locations in mainland China, including Sichuan; the second most abundant haplotype (MHap; 14 out of 70) was obviously very distinct from all others (Fig. 1c), and found only in worms from mountainous regions (Sichuan and Yunnan).

Haplotype networks for Schistosoma japonicum based on the coding region of SjIpp2 (a), SjFabp (b) and SjT22.6 (c). Each color represents a locality. The distance between two haplotypes corresponds to the numbers of substitutions. Abbreviations of the geographical localities are shown in Table 1
The network of the SjIpp2 coding region sequences showed that a single haplotype (IH4, Fig. 1a), was common only at the two mountainous localities in China. Similarly, one of the haplotypes for the SjT22.6 coding region (MHap, Fig. 1c) was only found among worms from the mountainous regions, where it was common. Otherwise there was no obvious geographical distinction between mainland China and elsewhere. However, in the network of the SjT22.6 coding region, all the Chinese Taiwan individuals exhibited a single haplotype (CTW, Fig. 1c), distinct from others. The networks generated from genome sequences (coding + non-coding) (Additional file 4: Figure S2) were consistent with those inferred from coding region sequences only (Fig. 1).
Selection
The dN/dS values were less than one among all the populations for SjIpp2 or SjFabp, suggesting both of them were under negative or purifying selection. However, The Tajima’s D and Fu’s Fs tests did not show significant departure from neutrality. This was also observed in S. japonicum populations from the lakes region for SjT22.6. However, for this gene, the dN/dS radio was two for the alignment of S. japonicum individuals from mountainous regions, suggesting positive selection. This was further supported by the Tajima’s test (D = 3.227, P < 0.001) and Fu’s test (Fs = 7.259, P < 0.01) (Table 1).
Protein structure prediction
Protein sequence analysis of SjT22.6 showed that the variations mainly occurred between MHap and other haplotypes (Additional file 5: Figure S3), so further comparison between MHap and the reference sequence (from lake region, GenBank ID: AAW25529.1) was conducted. The secondary structure of the two sequences differed at 13 amino acid sites. Three α-helixes (α1, α2, α3) and four β-shifts (β1, β2, β3, β4) were detected in both of the sequences (Fig. 2a). The length of α1 inferred from the reference sequence was shorter than in MHap, leading to differences in tertiary structures (Fig. 2b).

Protein structure prediction based on the coding region of SjT22.6 for the main mountainous region haplotype and the reference sequence. a Secondary structure. b Predicted tertiary structure. c The binding affinity of SjT22.6 with the potential peptide (ICp) for MHap and the reference sequence
Functional predictions showed that SjT22.6 was homologous to dynein light chain 1, a member of the dynein superfamily. The major functional identification region encompassed the first 20 amino acids (Additional file 6: Table S3) where α1 existed, with one amino acid substitution (K8ref vs E8MHap). The binding site in MHap and the reference sequence, characterized as the conserved motif KXTQT [41], differed in binding strength with dynein intermediate chain (ICp) [42]. Obviously, ICp interacted more powerfully with the reference protein than with MHap (Fig. 2c), indicating functional divergence between these two proteins.
Discussion
In this study, we found that the nuclear gene SjT22.6 of S. japonicum was under positive selection in the mountainous area of mainland China. Furthermore, the secondary and tertiary structures of the protein encoded by SjT22.6 differed in some S. japonicum individuals from the mountainous region, when compared with those from the lakes region and elsewhere in Asia.
A high level of nucleotide diversity was detected in SjT22.6, a gene encoding a tegument-associated antigen [43]. Such high diversity has also been found in other membrane proteins of S. japonicum [44, 45]. In general, high levels of polymorphism in proteins are due to recombination events and favored by positive selection in which selective forces, such as immune responses and drugs, drive the accumulation of mutations and maintain diversity in the population [46]. Many previous studies have indicated that genes involved in infection or maintaining important functions can exhibit particularly high mutation rates [47–49]. Thus, SjT22.6, as an immune response target [43], has tended to evolve more rapidly. The accumulated mutations may alter the protein sequence and help the worms to evade host immune attack. Interestingly, in the present study, SjT22.6 could not be amplified in S. japonicum strains from Japan and the Philippines. This was unexpected because the blood flukes in these two countries both have ancestors from mainland China [16]. This lack of amplification might be due to mutations occurring in the primer regions following the arrival of the parasites in Japan and the Philippines [50].
In this study, positive selection of SjT22.6 has occurred in the mountainous region of mainland China, but not in other regions. Several research groups have shown strong differentiation between S. japonicum populations in mountainous regions and those in the lakes area of the middle and lower Yangtze [16–18]. Among the selection pressures that might specifically impinge upon the blood flukes in the mountainous regions are the differences in intermediate and final hosts. While only speculation at this stage, adaptations to new hosts, and especially to a different subspecies of snail host, may have left the signature of selection in the population. Subspecies of Oncomelania hupensis have substantial genetic and morphological differences among different endemic areas of schistosomiasis japonica [51–53]. Therefore, substantial genetic change might accompany a geographical and host shift. The major definitive hosts of S. japonicum in the lakes region of mainland China are bovines and humans [54], while in the mountainous region, in addition to bovines and humans, there is a wide range of additional hosts, such as rodents and dogs [55].
As a platyhelminth-specific calcium-binding antigen, SjT22.6 is believed to have an N-terminal calcium-binding EF-hand domain and a C-terminal dynein light chain-like domain (DLC) [56]. The DLC domains of tegumental proteins are important for normal cellular homeostasis [57]. In S. mansoni, they might participate in the transport of vesicles within the tegumental cytoplasm [58] and the shuttling of vesicles into the tegument surface [59], probably within dynein motor complexes. In this study, the first α-helix of the main haplotype of SjT22.6 in the mountainous region (MHap) was longer and had less binding power than that of the reference sequence. These changes might have potential effects on the cellular homeostasis of S. japonicum, which might further influence the host-parasite interaction.
The other two gene fragments, SjFabp and SjIpp2, had low levels of nucleotide diversity. For SjFabp, most individuals shared a single haplotype and the dN/dS ratio was less than one. Thus, this gene fragment might be under purifying selection. Furthermore, the networks of these three genes indicated that the Sichuan and Yunnan populations from mountainous areas often shared haplotypes. This might reflect common ancestry [16] rather than gene flow between the two populations, which is rare because of the geographical isolation.
Conclusions
In conclusion, as a tegument-associated antigen-encoding gene of S. japonicum, SjT22.6 has high nucleotide diversity, appearing to be under positive selection in the mountainous region of mainland China. The protein structure and binding power of the most common SjT22.6 haplotype in the mountainous region differed from those of the reference sequence and of many haplotypes from elsewhere. In contrast, SjIpp2 and SjFabp had relatively lower diversity and did not show signatures of positive selection. Future research should aim to obtain a comprehensive understanding of the specific function of SjT22.6, such as its effect on the interaction between the worm and its hosts. Overall, by using three nuclear genes as genetic markers, the current findings provide valuable insights and fundamental genetic and evolutionary information concerning S. japonicum. Further research on these, and particularly on SjT22.6, is clearly required to confirm whether these can contribute to new interventions in the fight to eliminate schistosomiasis.
References
Utzinger J, Becker SL, Knopp S, Blum J, Neumayr AL, Keiser J, Hatz CF. Neglected tropical diseases: diagnosis, clinical management, treatment and control. Swiss Med Wkly. 2012;142:24.
Wu W, Feng AC, Huang YX. Research and control of advanced schistosomiasis japonica in China. Parasitol Res. 2015;114(1):17–27.
Magalhaes RJS, Salamat MS, Leonardo L, Gray DJ, Carabin H, Halton K, et al. Geographical distribution of human Schistosoma japonicum infection in The Philippines: tools to support disease control and further elimination. Int J Parasitol. 2014;44(13):977–84.
Satrija F, Ridwan Y, Jastal, Samarang, Rauf A. Current status of schistosomiasis in Indonesia. Acta Trop. 2015;141:349–53.
Zhao QP, Sen Jiang M, Dong HF, Nie P. Diversification of Schistosoma japonicum in mainland China revealed by mitochondrial DNA. Plos Neglect Trop D. 2012;6(2):11.
Lei ZL, Zheng H, Zhang LJ, Zhu R, Xu ZM, Xu J, et al. Endemic status of schistosomiasis in People’s Republic of China in 2013. Chin J Schistosom Control. 2014;26(6):591–7.
Li SZ, Zheng H, Abe EM, Yang K, Bergquist R, Qian YJ, et al. Reduction patterns of acute schistosomiasis in the People’s Republic of China. Plos Neglect Trop D. 2014;8(5):e2849.
Shrivastava J, Qian BZ, McVean G, Webster JP. An insight into the genetic variation of Schistosoma japonicum in mainland China using DNA microsatellite markers. Mol Ecol. 2005;14(3):839–49.
Zarowlecki MZ, Huyse T, Littlewood DTJ. Making the most of mitochondrial genomes - Markers for phylogeny, molecular ecology and barcodes in Schistosoma (Platyhelminthes : Digenea). Int J Parasitol. 2007;37(12):1401–18.
Bowles J, Hope M, Tiu WU, Liu XS, McManus DP. Nuclear and mitochondrial genetic markers highly conserved between Chinese and Philippine Schistosoma japonicum. Acta Trop. 1993;55(4):217–29.
Hope M, Foley DH, McManus DP. Electrophoretically-detected allozyme variation reveals only moderate differentiation between Chinese and Philippine Schistosoma japonicum. Acta Trop. 1995;60(2):101–8.
Gasser RB, BaoZhen Q, Nansen P, Johansen MV, Bogh H. Use of RAPD for the detection of genetic variation in the human blood fluke, Schistosoma japonicum, from mainland China. Mol Cell Probes. 1996;10(5):353–8.
Sorensen E, Drew AC, Brindley PJ, Bogh HO, Gasser RB, Qian BZ, et al. Variation in the sequence of a mitochondrial NADH dehydrogenase I gene fragment among six natural populations of Schistosoma japonicum from China. Int J Parasitol. 1998;28(12):1931–4.
Zhao GH, Mo XH, Zou FC, Li J, Weng YB, Lin RQ, Xia CM, Zhu XQ. Genetic variability among Schistosoma japonicum isolates from different endemic regions in China revealed by sequences of three mitochondrial DNA genes. Vet Parasitol. 2009;162(1-2):67–74.
Yin M, Li H, McManus DP, Blair D, Su J, Yang Z, et al. Geographical genetic structure of Schistosoma japonicum revealed by analysis of mitochondrial DNA and microsatellite markers. Parasit Vector. 2015;8(1):757.
Yin MB, Zheng HX, Su J, Feng Z, McManus DP, Zhou XN, et al. Co-dispersal of the blood fluke Schistosoma japonicum and Homo sapiens in the Neolithic Age. Sci Rep. 2015;5:8.
Young ND, Chan KG, Korhonen PK, Chong TM, Ee R, Mohandas N, et al. Exploring molecular variation in Schistosoma japonicum in China. Sci Rep. 2015;5:12.
Attwood SW, Ibaraki M, Saitoh Y, Nihei N, Janies DA. Comparative phylogenetic studies on Schistosoma japonicum and its snail intermediate host Oncomelania hupensis: Origins, dispersal and coevolution. Plos Neglect Trop D. 2015;9(7):28.
Vignal A, Milan D, SanCristobal M, Eggen A. A review on SNP and other types of molecular markers and their use in animal genetics. Genet Sel Evol. 2002;34(3):275–305.
Liu F, Lu J, Hu W, Wang SY, Cui SJ, Chi M, et al. New perspectives on host-parasite interplay by comparative transcriptomic and proteomic analyses of Schistosoma japonicum. Plos Pathog. 2006;2(4):268–81.
Huang FL, Glinsmann WH. Separation and characterization of two phosphorylase phosphatase inhibitors from rabbit skeletal muscle. Eur J Biochem. 1976;70(2):419–26.
Furlong ST, Caulfield JP. Schistosoma mansoni: synthesis and release of phospholipids, lysophospholipids, and neutral lipids by schistosomula. Exp Parasitol. 1989;69(1):65–77.
Zhang ZP, Xu HX, Gan WJ, Zeng SX, Hu XC. Schistosoma japonicum calcium-binding tegumental protein SjTP22.4 immunization confers praziquantel schistosomulumicide and antifecundity effect in mice. Vaccine. 2012;30(34):5141–50.
Yin MB, Hu W, Mo XJ, Wang SY, Brindley PJ, McManus DP, et al. Multiple near-identical genotypes of Schistosoma japonicum can occur in snails and have implications for population-genetic analyses. Int J Parasitol. 2008;38(14):1681–91.
Zhou Y, Zheng HJ, Chen YY, Zhang L, Wang K, Guo J, et al. The Schistosoma japonicum genome reveals features of host-parasite interplay. Nature. 2009;460(7253):345–U356.
Lu G, Moriyama EN. Vector NTI, a balanced all-in-one sequence analysis suite. Briefings in Bioinformatics. 2004;5(4):378–88.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2.
Watterson GA. Number of segregating sites in genetic models without recombination. Theor Popul Biol. 1975;7(2):256–76.
Salzburger W, Ewing GB, Von Haeseler A. The performance of phylogenetic algorithms in estimating haplotype genealogies with migration. Mol Ecol. 2011;20(9):1952–63.
Biswas S, Akey JM. Genomic insights into positive selection. Trends Genet. 2006;22(8):437–46.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–25.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–58.
Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77:114–22.
Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res. 2005;33:W89–93.
Roy A, Yang JY, Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012;40(W1):W471–7.
Roy A, Zhang Y. Recognizing protein-ligand binding sites by global structural alignment and local geometry refinement. Structure. 2012;20(6):987–97.
Yang JY, Roy A, Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013;41(D1):D1096–103.
Benison G, Karplus PA, Barbar E. The interplay of ligand binding and quaternary structure in the diverse interactions of dynein light chain LC8. J Mol Biol. 2008;384(4):954–66.
Hall J, Karplus PA, Barbar E. Multivalency in the assembly of intrinsically disordered dynein intermediate chain. J Biol Chem. 2009;284(48):33115–21.
Santiago ML, Hafalla JCR, Kurtis JD, Aligui GL, Wiest PM, Olveda RM, et al. Identification of the Schistosoma japonicum 22.6-kDa antigen as a major target of the human IgE response: Similarity of IgE-binding epitopes to allergen peptides. Int Arch of Allergy Immunol. 1998;11(2):94–104.
Zhang W, Li J, Duke M, Jones MK, Kuang L, Zhang J, et al. Inconsistent protective efficacy and marked polymorphism limits the value of Schistosoma japonicum tetraspanin-2 as a vaccine target. Plos Neglect Trop D. 2011;5(5):e1166.
Cai PF, Bu LY, Wang JA, Wang ZS, Zhong XA, Wang H. Molecular characterization of Schistosoma japonicum tegument protein tetraspanin-2: Sequence variation and possible implications for immune evasion. Biochem Biophys Res Commun. 2008;372(1):197–202.
Escalante AA, Cornejo OE, Rojas A, Udhayakumar V, Lal AA. Assessing the effect of natural selection in malaria parasites. Trends Parasitol. 2004;20(8):388–95.
Barrett LG, Thrall PH, Dodds PN, van der Merwe M, Linde CC, Lawrence GJ, Burdon JJ. Diversity and evolution of effector loci in natural populations of the plant pathogen Melampsora lini. Mol Biol Evol. 2009;26(11):2499–513.
Blanc G, Ngwamidiba M, Ogata H, Fournier PE, Claverie JM, Raoult D. Molecular evolution of Rickettsia surface antigens: evidence of positive selection. Mol Biol Evol. 2005;22(10):2073–83.
Obbard DJ, Jiggins FM, Halligan DL, Little TJ. Natural selection drives extremely rapid evolution in antiviral RNAi genes. Curr Biol. 2006;16(6):580–5.
Dlugosch KM, Parker IM. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol. 2008;17(1):431–49.
Li SZ, Wang YX, Yang K, Liu Q, Wang Q, Zhang Y, et al. Landscape genetics: the correlation of spatial and genetic distances of Oncomelania hupensis, the intermediate host snail of Schistosoma japonicum in mainland China. Geospatial Health. 2009;3(2):221–31.
Saijuntha W, Jarilla B, Leonardo AK, Sunico LS, Leonardo LR, Andrews RH, et al. Genetic structure inferred from mitochondrial 12S ribosomal RNA sequence of Oncomelania quadrasi, the intermediate snail host of Schistosoma japonicum in the Philippines. Am J Trop Med Hyg. 2014;90(6):1140–5.
Zhou XN, Kristensen TK. Genetic and morphological variations in populations of Oncomelania spp. in China. Southeast Asian J Trop Med Public Health. 1999;30(1):166–76.
Gray DJ, Williams GM, Li YS, Chen HG, Forsyth SJ, Li RS, et al. A cluster-randomised intervention trial against Schistosoma japonicum in the Peoples’ Republic of China: Bovine and human transmission. Plos ONE. 2009;4(6):7.
Lu DB, Rudge JW, Wang TP, Donnelly CA, Fang GR, Webster JP. Transmission of Schistosoma japonicum in marshland and hilly regions of China: Parasite population genetic and sibship structure. Plos Neglect Trop D. 2010;4(8):10.
Thomas CM, Fitzsimmons CM, Dunne DW, Timson DJ. Comparative biochemical analysis of three members of the Schistosoma mansoni TAL family: Differences in ion and drug binding properties. Biochimie. 2015;108:40–7.
Hoffmann KF, Strand M. Molecular characterization of a 20.8-kDa Schistosoma mansoni antigen - Sequence similarity to tegumental associated antigens and dynein light chains. J Biol Chem. 1997;272(23):14509–15.
Skelly PJ, Shoemaker CB. The Schistosoma mansoni host-interactive tegument forms from vesicle eruptions of a cyton network. Parasitol. 2001;122:67–73.
Hockley DJ, McLaren DJ. Schistosoma mansoni: changes in the outer membrane of the tegument during development from cercaria to adult worm. Int J Parasitol. 1973;3(1):13–25.
Acknowledgements
Not applicable.
Funding
This research was funded by the National Natural Science Foundation of China (No. 91431104) and the Foundation of National Science and Technology Major Program (Grant Nos. 2012ZX10004–220 and 2008ZX10004–011).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. The datasets generated and/or analysed during the current study are available in the GenBank (accession numbers KY494243–KY494287 for SjIpp2, KY494288–KY494346 for SjFabp and KY494347–KY494416 for SjT22.6).
Authors’ contributions
MY and WH designed the study, YL, QW, BX, XM and HL carried out the molecular work. MY, DB and YL contributed to data analyses. MY, DM, DB, ZF and WH wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
All animal experiments in this study were conducted strictly according to the rules of the Association for Assessment and Accreditation of Laboratory Animal Care International. This study was approved by the ethics committee of the National Institute of Parasitic Diseases, Chinese Center for Disease Control and Prevention (NIPD, China CDC).
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Figure S1.
The locations of the three genes in S. japonicum genome for SjIpp2 (a), SjFabp (b) and SjT22.6 (c). The red rectangles represent the coding regions of the genes. The blue rectangles represent the non-coding regions of these genes. (PDF 225 kb)
Additional file 2: Table S1.
Primers sequences for the three genomic fragments. (DOCX 13 kb)
Additional file 3: Table S2.
Numbers of observed haplotypes of S. japonicum isolates from different locations based on three genomic fragments. (DOCX 15 kb)
Additional file 4: Figure S2.
Networks for haplotypes of S. japonicum based on three genome fragments with (a) SjIpp2, (b) SjFabp and (c) SjT22.6. Each color represents a locality. The distance between two haplotypes corresponds to the number of substitutions. Abbreviations of the geographical localities are shown in Table 1. (PDF 199 kb)
Additional file 5: Figure S3.
Haplotype network for S. japonicum based on the amino acid sequence of SjT22.6. Each color represents a locality. The distance between two haplotypes corresponded to the number of substitutions. The number represents each mutation site of MHap compared with the reference sequence. Abbreviations of the geographical localities are shown in Table 1. (PDF 195 kb)
Additional file 6: Table S3.
The structural alignment of the reference sequence (ref, AAW25529.1) and MHap. (DOCX 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, Y., Yin, M., Wu, Q. et al. Genetic diversity and selection of three nuclear genes in Schistosoma japonicum populations. Parasites Vectors 10, 87 (2017). https://doi.org/10.1186/s13071-017-2033-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2033-8