
Abstract
Background
Culicoides spp. biting midges transmit bluetongue virus (BTV), the aetiological agent of bluetongue (BT), an economically important disease of ruminants. In southern India, hyperendemic outbreaks of BT exert high cost to subsistence farmers in the region, impacting on sheep production. Effective Culicoides spp. monitoring methods coupled with accurate species identification can accelerate responses for minimising BT outbreaks. Here, we assessed the utility of sampling methods and DNA barcoding for detection and identification of Culicoides spp. in southern India, in order to provide an informed basis for future monitoring of their populations in the region.
Methods
Culicoides spp. collected from Tamil Nadu and Karnataka were used to construct a framework for future morphological identification in surveillance, based on sequence comparison of the DNA barcode region of the mitochondrial cytochrome c oxidase I (COI) gene and achieving quality standards defined by the Barcode of Life initiative. Pairwise catches of Culicoides spp. were compared in diversity and abundance between green (570 nm) and ultraviolet (UV) (390 nm) light emitting diode (LED) suction traps at a single site in Chennai, Tamil Nadu over 20 nights of sampling in November 2013.
Results
DNA barcode sequences of Culicoides spp. were mostly congruent both with existing DNA barcode data from other countries and with morphological identification of major vector species. However, sequence differences symptomatic of cryptic species diversity were present in some groups which require further investigation. While the diversity of species collected by the UV LED Center for Disease Control (CDC) trap did not significantly vary from that collected by the green LED CDC trap, the UV CDC significantly outperformed the green LED CDC trap with regard to the number of Culicoides individuals collected.
Conclusions
Morphological identification of the majority of potential vector species of Culicoides spp. samples within southern India appears relatively robust; however, potential cryptic species diversity was present in some groups requiring further investigation. The UV LED CDC trap is recommended for surveillance of Culicoides in southern India.
Similar content being viewed by others

Background
Bluetongue (BT) is an economically important disease of sheep in the southern Indian states of Tamil Nadu, Karnataka, Telangana and Andhra [1]. Outbreaks of BT have a major impact on sheep rearing in southern India due to the high proportion of subsistence level sheep farmers in the region, who have limited access to vaccines and palliative care for their livestock [2]. The aetiological agent of BT, bluetongue virus (BTV), is biologically transmitted between ruminant hosts by competent vectors of the genus Culicoides (Diptera: Ceratopogonidae) [3]. In India, the epidemiology of BTV is highly complex, potentially involving multiple vector species and with at least 21 BTV serotypes identified by serology [1], some of which may have been introduced during efforts to improve ruminant production [4].
Seven putative BTV vector species are known to occur in India (Culicoides actoni Smith, 1929; C. brevitarsis Kieffer, 1917; C. dumdumi Sen & Das Gupta, 1959; C. fulvus Sen & Das Gupta, 1959; C. imicola Kieffer, 1913; C. oxystoma Kieffer 1910 and C. peregrinus Kieffer, 1910) [1, 5–7], although this implication is derived primarily from vector competence data collected in other countries. Culicoides imicola and C. oxystoma have been reported to extend across the Afrotropical, Saharo-Arabian and Oriental regions [8, 9] (geographic regions defined as per Holt et al. [10]). In contrast, C. actoni, C. brevitarsis, C. dumdumi, C. fulvus and C. peregrinus have been recorded in the Australian, Oceanian and Oriental region [11–15], but not in the Saharo-Arabian and Afrotropical regions. The combination of multiple potential vector species and a huge diversity of BTV strains [16, 17] makes India one of the most challenging areas in which to dissect transmission cycles and highlights the importance of this region due to it sharing features of the Afrotropical, Saharo-Arabian, Oriental and Australasian ecozones [6]. While broad relationships between Culicoides spp. abundance and transmission have been suggested [1], these remain very poorly defined and hence unpredictable.
The Culicoides fauna of the Oriental region has been the focus of an authoritative taxonomic review based on morphology [18]. Wirth & Hubert’s review [18], however, did not extend to a comprehensive review of the Culicoides fauna of the Indian subcontinent and the Culicoides fauna of India has only been subject to sporadic morphological studies, e.g. Das Gupta [19, 20]. Checklists of Indian species of Culicoides have been produced [21–23]; however, many contain misidentifications and synonymous species [22, 23] and/or propose new species with no supporting taxonomic data [23], rendering them of limited use with regard to compiling biodiversity inventories or investigating Culicoides-borne arbovirus epidemiology. In addition, molecular DNA analyses of the Indian Culicoides fauna are limited to a single DNA barcode [24] report focussed on five species sampled from a single location, with little comment regarding the specificity of the DNA barcodes relative to other Culicoides species or populations [25]. Further DNA barcode and molecular studies are required to underpin morphological studies of the Culicoides fauna of India, as has been accomplished elsewhere to clarify species-level taxonomic descriptions [26, 27].
Creating a fundamental base for Culicoides species diagnostics in India is a prerequisite for dissecting BTV epidemiology accurately in this country. Wider questions also exist regarding the phylogenetic and taxonomic relationships of Culicoides populations in southern India with those from other regions including the degree of haplotype connectivity between global populations of vector species. Attempts to resolve these questions may be achieved through the development of morphological and genetic datasets of Culicoides spp. from India that are comparable with those being produced elsewhere (for review, see Harrup et al. [28]).
In addition to accurate species identification, a second fundamental requirement for accurate surveillance of Culicoides populations in southern India is the selection of appropriate monitoring tools. Systematic sampling of Culicoides populations using light-suction trapping next to livestock has been used to demarcate geographic and temporal risk of BTV transmission in Europe, Africa and Australia [29–33]. In both Europe and Africa, Culicoides spp. surveillance programmes are reliant upon the use of ultraviolet (UV) (~390 nm peak wavelength) light-suction traps, which have previously been shown to be highly effective at collecting C. imicola [33], the principle vector of BTV in Africa and the Mediterranean Basin. In Australia, however, green light emitting diode (LED) (~520 nm peak wavelength) light-suction traps are used to collect Culicoides spp. as part of the National Arbovirus Monitoring Program [34]. The preference for using green light for the collection of Culicoides spp. in Australia is the result of field studies demonstrating that the primary BTV vector species in this region, C. brevitarsis, has a greater sensitivity to this wavelength than to other colours including UV [31]. As both C. imicola and C. brevitarsis occur in India [18], it is important to define prior to the initiation of wide-scale surveillance project which light wavelength is most effective for surveillance of these and other Culicoides spp. in southern India. In addition, the logistical challenge of establishing field surveys and specifically the limited access to reliable mains electricity for recharging of batteries used to power light-suction trap surveys has limited adult Culicoides surveillance in India to single state studies involving one or a few sites. These challenges must be assessed prior to the deployment of large-scale surveillance programmes in order to confirm the selected equipment is both effective at the collection of Culicoides spp. and robust enough to withstand local conditions.
In this study we DNA barcoded Culicoides spp. collected across southern India and assessed the phylogenetic utility of these DNA barcodes to provide species identifications in agreement with morphology-based taxonomic identifications. We included publically available DNA barcodes from global replicates of targeted species in our analyses to determine if the populations in India contained unique and/or cosmopolitan genetic diversity. In addition, we also assess the use of two commercially available LED Center for Disease Control (CDC) light-suction traps [35, 36] as a precursor to wide-scale surveillance of Culicoides spp. in southern India. In the absence of logistically feasible trapping methods that are reflective of biting rates on ruminants, the key considerations for such a surveillance scheme are that the selected light-suction traps collect a wide-diversity of Culicoides spp. and at a sufficient abundance to consistently discern seasonal patterns in abundance.
Methods
Specimen selection and morphological identification
Seventy-three Culicoides specimens from seven sites and representing 12 morphologically identified species (Additional file 1: Table S1) and one currently unknown species were selected for genetic characterisation (Fig. 1) [TN01 (n = 16); TN02 (n = 16); TN08 (n = 10); TN10 (n = 14); TN11 (n = 9); TN12 (n = 3); KA01 (n = 5)]. Sites TN01 and TN12 were located near Chennai, Tamil Nadu, TN02 near Kattupakkam, Tamil Nadu and TN11 near Erode, Tamil Nadu (Fig. 1), and are all located in areas characterised as having tropical wet and dry climates [37], being particularly effected by the north east monsoon. Site TN08 is located near Pudukottai, Tamil Nadu, a semi-arid area with high temperatures throughout the year and relatively low rainfall. Site TN10 is near Ooty in Tamil Nadu, an area with a subtropical highland climate [37]. Site KA01 is located near Bangalore, Karnataka in an area with a tropical savannah climate [37] where the southwest monsoon has a greater influence than the northeast monsoon on climate conditions. Sites TN01, TN02, TN08, TN10, TN11 and KA01 were all located in areas principally utilised for subsistence and/or semi-intensive sheep, goat, cattle and buffalo rearing (Fig. 2). Site TN12 is located in an area of coastal inter-tidal marsh with low-level subsistence sheep, goat, cattle and buffalo farming present in the area (Fig. 3).

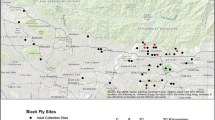
Geographical location of sites from which Culicoides spp. specimens selected for genetic analysis were collected: TN01 (n = 16); TN02 (n = 16); TN08 (n = 10); TN10 (n = 14); TN11 (n = 9); TN12 (n = 3); KA01 (n = 5)

Collection sites, examples of typical semi-intensive farming habitat found in southern India. a TN01 (Chennai, Tamil Nadu, India); b TN02 (Kattupakkam, Tamil Nadu India)

Collection Site TN12 (Chennai, Tamil Nadu) example of a coastal inter-tidal habitat with b cattle grazing, an example of typical subsistence farming habitat found in coastal regions of southern India
Specimens selected for genetic analysis were collected either using an UV LED CDC light-suction trap (Model 2770, 390 nm peak wavelength: BioQuip Products, Rancho Dominguez, CA, USA) or a sweep net and stored in 70 % ethanol prior to identification. Culicoides specimens were selected for genetic characterisation following preliminary identification using an SMZ-140 stereomicroscope (Motic, Barcelona, Spain) and the keys of Boorman [38], Gangopadhyay & Das Gupta [22] and Wirth & Hubert [18], and the descriptions of Majumdar et al. [39] and Nandi & Mazumdah [40, 41].
Molecular identification
DNA extraction
Total DNA was extracted from individual Culicoides specimens using a non-destructive technique [42]. Specimens were individually incubated in 200 μl of DXT Tissue Digest Reagent (QIAGEN, Crawley, UK) with 1 % Proteinase K (QIAGEN) for 16 h at 40 °C. Culicoides were then stored at 4 °C in 70 % ethanol prior to slide mounting. The remaining tissue digest solution was incubated at 70 °C for 15 min to inactivate the proteinase K, and then ethanol-precipitated to remove PCR inhibitors using Pellet Paint® Co-Precipitant (Merck Millipore, Darmstadt, Germany) to improve DNA yield. The purified DNA extractions were re-suspended in 100 μl of 10 mM Tris HCl, pH 8.0 (Buffer EB: QIAGEN) and stored at 4 °C.
COI DNA barcode assay
Amplification of a 658 bp fragment of the mitochondrial COI gene barcoding region [24] was achieved by polymerase chain reaction (PCR) using an Eppendorf® Mastercycler® Pro (Eppendorf®, Chennai, India) thermal cycler. Reactions were performed in a total volume of 25 μl consisting of 2.5 μl nuclease-free water (QIAGEN), 12.5 μl QIAGEN TopTaq Master Mix, 2.5 μl CoralLoad Concentrate (QIAGEN), 1.25 μl of the 20 μM forward primer LCO1490 (5′-GGT CAA CAA ATC ATA AAG ATA TTG G-3′ [43]), 1.25 μl of the 20 μM reverse primer HCO2198 (5′-TAA ACT TCA GGG TGA CCA AAA AAT CA-3′ [43]) and 5.0 μl of template DNA (approximately 5–25 ng DNA) for each reaction. Positive and negative controls for the amplification reactions were carried out at every PCR round. The PCR cycling conditions were as follows: an initial denaturation step at 94 °C for 3 min followed by 35 cycles of 94 °C for 30 s, 46 °C for 30 s, 72 °C for 1 min, and a final extension step at 72 °C for 10 min. Reactions were stored at 4 °C until further processing. PCR products were visualised through electrophoresis on 2 % (w/v) pre-cast agarose gels containing ethidium bromide (E-Gel™ 48 gels: ThermoFisher Scientific, UK) run for 8 min. Successful amplification of the COI DNA barcode region was indicated by the presence of a band at approximately 720 bp, identified by comparison with E-Gel® Low Range Quantitative DNA Ladder (100–2000 bp: ThermoFisher Scientific).
PCR purification and COI sequencing
Dimer formation from the primers was not observed and purification of the remaining PCR product was performed using the MinElute® PCR purification kit (QIAGEN) following manufacturers recommended guidelines (v. 03/2008). The resulting products were sent for bi-directional sequencing using primers HCO2198 (reverse) and LCO1490 (forward) at a commercial facility (Eurofins, Bangalore, India). The resulting electropherograms were edited and forward and reverse sequences assembled and trimmed to remove primer sequence using CodonCode Aligner v. 5.1.5 (CodonCode Aligner, Centerville, MA, USA). Corresponding specimen collection data and DNA sequences including electropherograms have been made publically available via the Barcode of Life Data System (BOLD) [44] as dataset DS-CULIN (dx.doi.org/10.5883/DS-CULIN) and DNA sequences are also available in the GenBank database under accession numbers KT307786–KT307856.
Phylogenetic analysis
Consensus sequences were compared to previously published sequences in the GenBank database using the standard nucleotide BLAST tool [45], in addition to comparison to as yet unreleased sequence data in the BOLD database [44] using the Barcode Identification Engine in BOLD v3. Sequences from GenBank included in the phylogenetic analysis (n = 196) are listed in Additional file 1: Table S1. GenBank sequences were included in the analysis to assess if the morphological identifications made within this study were conspecific with those made from other geographical regions and were not confounded with species morphologically similar enough to result in misidentification, e.g. C. brevitarsis and C. bolitinos Meiswinkel, 1989 [26]. All sequences were aligned using MUSCLE [46] and quality checked using GUIDANCE [47] (100 bootstraps). All included sequences were aligned with a high degree of confidence (GUIDANCE alignment score > 0.999). The general time reversible model with gamma-distribution rates plus invariant Sites (GTR + Γ + I) was identified using ModelTest2 [48, 49] v 2.1.4 as the optimal model of nucleotide substitution in the alignment (outgroup excluded), based on the lowest Bayesian Information Criterion (BIC) and Akaike Information Criterion (AIC) scores.
The phylogenetic relationships among taxa were resolved using a Bayesian Inference (BI) approach [50, 51], with the topology rooted on the partial COI sequence of Anopheles gambiae Giles, 1902 (NC002084 [52]). The BI tree was constructed using MrBayes v.3.2.2 [50, 51] and twenty million tree generations in four chains were run, sampling every 1000th and discarding the first 25 %, before constructing a 50 % majority rule consensus tree reporting Bayesian posterior probabilities. The absence of indicators of a lack of convergence in the final consensus BI topology was confirmed via the examination of the sampled Markov chain Monte Carlo tree topologies using AWTY [53].
Relationships between the observed haplotypes within the C. brevitarsis - C. asiana Bellis, 2014 (nomen novum for C. asiatica Bellis [54] preoccupied by C. asiaticus Gutsevich & Smatov 1966; specimens redescribed by Bellis et al. [26]), C. imicola and C. oxystoma clades were assessed by constructing Median-Joining networks. Roehl haplotype data files (RDF) were created with DnaSP v.5.10 [55] and imported into Network v.4.6.1.2 [56] and networks were calculated with the Median-Joining algorithm [57] with equal weights for all characters, using maximum parsimony [58] post-processing. Uncorrected intra- and inter-specific percentage sequence distances were generated using the packages Spider v 1.3-0 [59] and Ape v.3.2 [60], implemented in R v.3.1.2 [61]. Missing nucleotides were treated in all sequence comparisons using a pairwise deletion option.
Morphological voucher specimens
Following DNA extraction specimens were then individually dissected and slide mounted in Euparal following the techniques of Nevill & Dyce [62]. Mounted specimens were re-examined following mounting and identifications confirmed using the keys of Boorman [38], Gangopadhyay & Das Gupta [22] and Wirth & Hubert [18], and the descriptions of Majumdar et al. [39] and Nandi & Mazumdah [40, 41].
Light-suction trap comparison
Commercially produced, modified CDC design light-suction traps fitted with LEDs were compared (Model 2770: BioQuip Products, Rancho Dominguez, CA, USA). The LED platforms emitted peak light wavelengths of either 390 nm (UV) or 570 nm (Green) and were powered by 6 V batteries. An inline photo-switch (BioQuip) was also used on each trap to standardise collection periods, reduce battery consumption during the trial and replicate their planned use in the surveillance programme. Two locations over 50 m apart at site TN01 (Fig. 1) were chosen for this study (trap height approximately 1.5 m), both of which were in close proximity to ruminant livestock (1–5 m from cattle; 40–50 m from sheep). Trapping was conducted during November 2013, during the predicted peak BTV transmission period, for 20 nights, with the positions of the green and UV LED CDC traps rotated between the two sites on alternate nights. Overnight collections (approximately three hours before sunset to three hours after sunrise) were made into water containing a drop of detergent and then transferred the following morning to 70 % ethanol for storage prior to identification.
Following collection, specimens of Culicoides were identified morphologically under a stereomicroscope using keys [18, 63] and comparison to reference specimens from the local area produced as part of the phylogenetic analysis conducted within this study. Specimens were sexed and females were further separated based on their abdominal pigmentation status (unpigmented/ nulliparous, blood-fed, gravid, or pigmented/ parous) [64, 65]. In large collections (estimated by the investigator to contain more than 1,000 Culicoides spp. specimens), a standardised process of subsampling was used to estimate the abundance and diversity of Culicoides spp. present [66]. In summary, for samples which were subsampled, insect collections were washed with water through a series of stainless steel test sieves (3.35 mm, 2.00 mm, 1.00 mm and 300 μm mesh diameter). The contents of the 300 μm sieve were then transferred to a weigh boat and weighed. Successive 1 g portions of the sample in the weigh boat were then sorted and identified. Successive 1 g portions were taken and completely sorted and identified until at least 650 individuals of Culicoides (any species) had been identified. The total number of specimens of a particular species of Culicoides in the original sample was then estimated as equal to the [(Total weight of the sieved sample/ number of grams of sample identified) × number of Culicoides of the species of interest identified in the subsample].
Statistical analysis
Relationships between female Culicoides abundance and five fixed effects were examined using generalised linear mixed models with a negative binomial error distribution (fitted using the glmmadmb package v 0.7.5 [67, 68] in R [61]), three main effects of light type, position and species (including the five most abundant species) and two possible interactions (species × light type, to reflect that light types may be more attractive to some species than others and species × position, to reflect that a trap position may have been closer or further from a larval development site which may vary by Culicoides species). In all models, a random effect of trapping day was used. All possible combinations of the fixed effects were examined (including an intercept only model) and the model with the lowest AIC [69] was selected. The same procedure was followed for males, with the exception that a zero-inflated negative binomial error distribution was used to account for the higher proportion of zero catches for males which resulted in significantly overdispersed residuals in ordinary negative binomial models. The diversity of Culicoides species collected by the two trap types, i.e. the species richness, was further compared using the Margalef’s index, such that Margalef’s index = (S - 1)/ln N, where S is the total number of species collected in a sample, i.e. one trap collection, N is the total number of individuals in the sample and ln is the natural logarithm [70].
Results
Phylogenetic analysis
Full length primer truncated DNA barcode sequences of 658 bp were recovered from 71 of the 73 specimens sampled from India, representing 12 morphologically identified species: C. actoni; C. anophelis Edwards, 1922; C. brevitarsis; C. huffi Causey, 1938; C. imicola; C. innoxius Sen & Das Gupta, 1959; C. kepongensis Lee, 1988; C. mesghalii Navai, 1973; C. oxystoma; C. peliliouensis Tokunaga, 1936; C. peregrinus; and C. similis Carter, Ingram & Macfie, 1920 (Additional file 1: Table S2), and one currently unidentified species. Amino acid frame shifts and stop codons were not evident among sequence translations, indicating pseudogenes were not likely to be included in the alignments. The COI sequences obtained from the GenBank (Additional file 1: Table S1) overlapped the alignment of the sequences generated in this study by between 434 and 658 bp.
Thirteen species from the collections in this study and a further six potentially morphologically confounding species (19 in total) were represented within the phylogenetic study (Figs. 4, 5, 6, 7, 8 and 9). Species clades represented in the Bayesian Inference (BI) phylogeny were concordant with morphological identifications with the exception of one specimen, TPI:ENT:IBVNET-CULI-TN-65, which could not be assigned to a species based on the morphological descriptions currently available in the literature, and is therefore recorded as ‘Unknown Species (I)’ (Fig. 4, Additional file 1: Table S2 and Additional file 2: Table S3). No discordant Barcode Index Numbers (BINs) [71] were observed for the species identifications of specimens collected within this study, with the exception of C. huffi, which resulted in two BINs being assigned thereby indicating the potential presence of two distinct taxa (see Additional file 1: Table S2). In addition, C. actoni, C. brevitarsis, C. imicola and C. peregrinus currently all have multiple BINs per species name currently assigned within BOLD [44], indicating the presence of either misidentified specimens or unresolved cryptic diversity within the publically avaliable data.

Bayesian Inference phylogenetic tree inferred from COI DNA barcode sequences with species, and subgeneric (thick black line, italic font) or species group (thick black line, Roman font) indicated. Bayesian posterior probability node support values greater than 0.7 shown. Coloured diamonds indicate specimens from the IBVNet project coloured by collection site (KA1: red; TN01: purple; TN02: green; TN08: blue; TN10: orange; TN11: yellow; TN12: black) with specimen number followed by GenBank accession number in brackets. Culicoides imicola and C. oxystoma clade summarised, see Figs. 5 and 6 for further details

Bayesian Inference phylogenetic tree of the C. imicola clade inferred from COI DNA barcode sequences. Bayesian posterior probability node support values greater than 0.7 shown. Coloured diamonds indicates specimens from the IBVNet project coloured by collection site (KA01: red; TN01: purple; TN02: green; TN08: blue; TN10: orange; TN11: yellow; TN12: black) with specimen number followed by GenBank accession number in brackets. See Fig. 4 for the relative placement of the C. imicola clade with respect to other specimens analysed within this study

Bayesian Inference phylogenetic tree of the C. oxystoma clade inferred from COI DNA barcode sequences. Bayesian posterior probability node support values greater than 0.7 shown. Coloured diamonds indicates specimens from the IBVNet project coloured by collection site (KA1: red; TN01: purple; TN02: green; TN08: blue; TN10: orange; TN11: yellow; TN12: black) with specimen number followed by GenBank accession number in brackets. See Fig. 4 for the relative placement of the C. oxystoma clade with respect to other specimens analysed within this study

Most parsimonious Median-Joining Network (ε = 0) depicting the phylogenetic relationships among, and geographical assignment, of C. imicola COI haplotypes. The size of each circle is proportional to the corresponding haplotype frequency. Branch lengths are proportional to the number of nucleotide changes between haplotypes. Black circles indicate median vectors (mv) that represent hypothetical missing or unsampled ancestral haplotypes. Number of nucleotide changes indicated on longer branches (Sequences AF080528, AF080529 and AF080536 excluded from analysis)

Most parsimonious Median-Joining Network (ε = 0) depicting the phylogenetic relationships among, and geographical assignment, of C. oxystoma COI haplotypes. The size of each circle is proportional to the corresponding haplotype frequency. Branch lengths are proportional to the number of nucleotide changes between haplotypes. Black circles indicate median vectors (mv) that represent hypothetical missing or unsampled ancestral haplotypes. Number of nucleotide changes indicated on longer branches

Most parsimonious Median-Joining Network (ε = 0) depicting the phylogenetic relationships among, and geographical assignment, of C. brevitarsis and C. asiana COI haplotypes. The size of each circle is proportional to the corresponding haplotype frequency. Branch lengths are proportional to the number of nucleotide changes between haplotypes. Black circles indicate median vectors (mv) that represent hypothetical missing or unsampled ancestral haplotypes. Number of nucleotide changes indicated on longer branches
Deep interspecific differences within the COI DNA barcode region were present between the majority of Culicoides assessed within this study; however, there was no clear barcoding gap [72, 73] across all current species assignments (Fig. 10). The greatest intraspecific sequence differences were reported from within specimens morphologically identified as C. actoni (mean: 5.8 %; range: 1.5–8.1 %), followed by C. brevitarsis (mean: 3.2 %; range: 0–5.6 %), C. huffi (mean: 2.5 %; range: 0–4.9 %), C. imicola (mean: 1.2 %; range: 0–9.0 %) and C. oxystoma (mean: 2.9 %; range: 0–5.8 %) (Additional file 2: Table S3; Fig. 11). These levels of sequence variation is more akin to interspecific values (Additional file 2, Table S3), indicating cryptic taxa in the samples sequenced within these taxa, or morphological misidentifications in our dataset. All other pairwise intraspecific sequence differences were less than or equal to 2.0 %, i.e. within published ranges of intraspecific variation [74] (Additional file 2: Table S3; Fig. 11). The least mean sequence difference was between C. dubius Arnaud, 1956 and C. peliliouensis (mean: 5.3 %; range: 5.2–5.6 %); C. asiana and C. brevitarsis (mean: 7.0 %; range: 6.3–7.9 %), and C. oxystoma and C. subschultzei Cornet & Brunhes, 1994 (mean: 9.1 %; range: 7.8–10.4 %) and C. brevitarsis and C. imicola (mean: 10.7 %; range: 9.0–15.0 %) (Additional file 2: Table S3). All other pairwise interspecific sequence difference were greater than 10.0 % (Additional file 2: Table S3). No misidentifications occurred between species identified within the study and those considered isomorphic or morphologically similar for which COI DNA barcode sequence data was available.

Box-and-whisker plots (left) and frequency distribution plot (right) of interspecific, i.e. closest non-conspecific (yellow) and intraspecific, i.e. the furthest intraspecific distance among its own species (red) pairwise genetic distances (uncorrected percentage sequence distances) across all species in this study. Areas where the intra- and inter-specific distances overlap shown in orange

Box-and-whisker plots of the interspecific, i.e. closest non-conspecific (yellow) and intraspecific, i.e. the furthest intraspecific distance among its own species (red) pairwise genetic distances (uncorrected percentage sequence distances) by species. Areas where the intra- and inter-specific distances overlap shown in orange
No geographic clustering was observed in the C. imicola haplotypes (Figs. 5 and 7). Three specimens collected in previous studies from Spain (GenBank: AF080528, AF080529 and AF080536) were, however, found to have between 2.7 and 4.6 %, 3.5–5.4 % and 6.0–9.0 % sequence difference, respectively, to other specimens identified as C. imicola. The range of intraspecific sequence differences within C. imicola when these three sequences were excluded was significantly reduced from mean of 1.2 % (range: 0–9.0 %) to mean of 1.0 % (range: 0–3.9 %). Sequences AF080528, AF080529 and AF080536 are likely to be either misidentifications or poor quality sequences and were excluded from subsequent investigations of haplotype relationships within C. imicola.
A significant degree of geographic clustering was observed in the C. oxystoma haplotypes (Figs. 6 and 8), with haplotypes previously recorded from Senegal [9] clustering separately from specimens collected in the current study and from those in Australia, China, India, Israel and Japan [75, 76] which formed an Oriental-Australasian clade with between 3.7 and 5.8 % (mean: 4.3 %) sequence difference between the Oriental-Australasian and Senegalese clades (Fig. 8).
Specimens morphologically identified as C. brevitarsis collected in southern India were strongly supported by BI as a monophyletic clade (100 % posterior probability) with sequence difference between 5.0–5.6 % (mean: 5.3 %) (Fig. 9) to specimens collected from Australia, China, the Solomon Islands and Timor-Leste [26]. They also differed from C. asiana specimens from Japan (= C. brevitarsis [75], misidentification [26]) (mean sequence difference: 6.9 %; range: 6.5–7.3 %) (Fig. 9).
The three C. actoni sequences demonstrated 8.1 and 1.5 % sequence difference between the specimen collected in this study from southern India and the sequences previous published from Australia [26] and Japan [75], respectively. In comparison, Japanese and Australian C. actoni specimens showed 7.9 % sequence difference.
Green versus UV wavelength comparison trial
A total of 7284 Culicoides were collected using the green LED CDC trap, while an estimated total of 120,460 individuals were collected using the UV LED CDC trap. Both trap types collected Culicoides on each day of the trial, with a maximum estimated single night catch of 13,022 individuals (trap night seven) in the UV LED CDC trap, and 650 individuals (trap night 14) in the green LED CDC trap. Trap catches were heavily biased towards female specimens in both the UV LED CDC trap (90.9 %) and the green LED CDC trap (89.0 %) collections (Table 1). Non-pigmented females dominated the collections in both trap types (51.9 % UV; 48.3 % green), followed by blood-fed (20.4 % UV; 21.8 % green) and pigmented individuals (18.0 % UV; 18.5 % green), with only a few gravid females collected (0.6 % UV; 0.4 % green) (Table 1). The relative proportion of the trap catches made up of the different parity states were not significantly different between the UV and green LED CDC traps; however, the UV LED CDC trap collected significantly more male and female Culicoides than the green LED CDC trap (Paired t-test: t = 6.464, df = 19, P < 0.001). Over 16 times more females were collected using the UV LED CDC trap than in the green LED CDC trap.
Eight species (morphologically identified) were represented within the LED CDC trap collections (in order of abundance): C. oxystoma, C. peregrinus, C. imicola, C. brevitarsis, C. anophelis, C. innoxius, C. peliliouensis and C. huffi. All species were present in both UV and green LED CDC trap collections, with the exception of C. huffi, which was absent from green LED CDC trap collections (Table 1). Trap catches from both trap types were dominated by C. oxystoma and C. peregrinus which together represented 65.6 and 32.7 % of Culicoides specimens in the UV, and 82.7 and 13.6 % of Culicoides specimens in the green trap collections respectively, with the other species collected making up just 3.7 and 1.8 % of the trap catch collectively for the green and the UV trap, respectively (Table 1, Fig. 12).

Log10 median abundance of the specimens collected per trap night for the five most abundant Culicoides spp. collected by green light emitting diode (LED) Center for Disease Control (CDC) traps as compared to ultraviolet (UV) LED CDC traps, stratified by species and sex
The relative abundance of the most abundant species, C. oxystoma and C. peregrinus, significantly varied between the green and UV traps (Fig. 12). Overall diversity (i.e. the number of different Culicoides spp. present within a trap catch) did not vary significantly between the green and UV LED CDC traps, as measured by the Margalef’s index (mean: 0.50; range: 0.32–0.73 and mean: 0.57; range: 0.33–0.91 for the UV and green LED CDC traps, respectively) (Fig. 13).

Box-and-whisker plots of Margalef’s Index illustrating the relative variability in species richness in relation to collection by the ultraviolet (UV) compared to green light emitting diode (LED) Center for Disease Control (CDC) trap
Due to the low numbers collected, C. innoxius, C. huffi and C. peliliouensis were excluded from the following statistical analysis. Numbers of female Culicoides collected by the LED CDC traps were significantly affected by the variables species and light type and the by the interaction variable species × light type (Table 2). The UV light trap collected significantly more female C. peregrinus than the green LED CDC trap. There were, however, no significant differences in the numbers of female C. anophelis, C. brevitarsis, C. imicola or C. oxystoma collected by the UV compared to green LED CDC trap (Table 2; Fig. 12). Species and light type were also key determinants of the numbers of male Culicoides collected (Table 2). The UV LED CDC traps collected a significantly larger number of male C. imicola, C. oxystoma and C. peregrinus than the green LED CDC traps (Table 2; Fig. 12). There were, however, no significant differences in the numbers of male C. anophelis and C. brevitarsis, collected by the UV compared to green LED CDC trap (Table 2; Fig. 12).
Discussion
This study presents the first detailed multi-site genetic analysis of Culicoides in southern India with new COI DNA barcode sequence data presented for 12 previously recognised species, and potentially up to four cryptic or unidentified taxa. To the best of our knowledge, Culicoides mesghalii and C. kepongensis are recorded for the first time in India and C. peliliouensis and C. similis are recorded for the first time in southern India. The study also reveals the potential for deep sequences differences symptomatic of cryptic species diversity within C. actoni, C. brevitarsis and C. huffi, which require further investigation. In addition, further evidence of geographic clustering and potential cryptic species diversity within C. oxystoma is presented, in support of that identified by Bakhoum et al. [9]. With these caveats, morphological identification of the species considered the most likely vectors of BTV in India was demonstrated to be robust, with no misidentifications of the main putative vector species. In addition, commercially produced UV LED light-suction traps were shown to outperform traps fitted with green LED’s for Culicoides spp. collection, based on both the total number and diversity of specimens of Culicoides collected. These findings collectively address many of the logistical requirements for effective Culicoides spp. sampling in southern India.
The provision of DNA barcode data for putative BTV vector species in southern India fills an important gap in our knowledge of the phylogeny of these species. An east–west split in haplotypes of C. imicola specimens from the Mediterranean basin was previously identified using COI sequencing by Calvo et al. [77], Dallas et al. [78] and Nolan et al. [79]. The present study has shown that four C. imicola COI haplotypes are present in southern India; two are unique to India, one is identical to specimens collected in Israel and South Africa and the other is identical to specimens collected in China, Israel, South Africa and Portugal. The status of the latter sample from Portugal (GenBank: AF079975) requires re-evaluation as this sample does not fit with previously proposed eastern-western haplotype demarcation in the Mediterranean basin. In addition, three publically available sequences (GenBank: AF080528, AF080529 and AF080536) labelled as C. imicola but with sequences differences of 2.9, 3.7 and 7.5 % respectively to the nearest other C. imicola sequence have been highlighted as likely to be the result of misidentification, cross-contamination or poor sequence quality. Genetic divergence and population structure within C. imicola, the principal Afrotropical vector of BTV, are subject to on-going investigations [80, 81] that will assist in clarifying the status of this pan-continental species.
The status of the subgenus Remmia Glukhova (= Schutzei group) of Culicoides in Asia has historically been fraught with confusion [18]. This is particularly the case in India with numerous publications citing the presence of C. schultzei [12–14, 81–86], despite the fact that C. schultzei is an Afrotropical species while the type-locality of C. oxystoma is Kolkata in India. These two species are, however, both morphologically [18, 87] and genetically [9, 76, 88] distinct, and current evidence agrees with the earlier proposal by Cornet [89] that C. oxystoma is the species present in India. Furthermore, Indian C. oxystoma specimens form part of the Saharo-Arabian-Oriental-Australian clade, but further investigation is needed to see if the two genetic groupings of Bakhoum et al. [9] correlate with the morphological variation observed by Wirth & Hubert [18], and whether they are supported by data from additional genetic markers.
Recent re-evaluations of the Imicola Complex by Bellis et al. [26] provide strong evidence that C. brevitarsis specimens identified in Japan are in fact the novel species C. asiana [26] (= C. asiatica Bellis, 2014 [54]). Further cryptic diversity between Australian and Chinese C. brevitarsis was suggested by analysis of the COI region, but this was not supported by analysis of the fused carbamoyl phosphate synthetase, aspartate transcarbamylase and dihydroorotase (CAD) nuclear genes [26]. Similar investigations are required to explore the potential cryptic diversity identified between the Indian and Australian specimens of C. brevitarsis. There is no evidence that the morphologically indistinguishable C. bolitinos [26, 90] is present in southern India as this species is genetically distinct from the Indian C. brevitarsis (mean sequence difference compared to South African C. bolitinos: 12.2 %; range: 11.6–12.6 %). The Indian C. brevitarsis is also genetically distinct (mean sequence difference: 11.1 %; range: 10.6–11.5 %) from C. bolitinos specimens recently collected in Reunion Island (GenBank: KF186129 and KF186130) [91], which have been provisionally identified as C. bolitinos, but which have been shown to be genetically distinct from South African C. bolitinos (mean sequence difference: 6.4 %; range 5.6–6.9 %). The latter relationship and the implications of this potential cryptic diversity within C. bolitinos are yet to be resolved.
The single C. actoni COI sequence from India is consistent with the Asian clade of this species, and is consequently different to the species in Australia [92] upon which vector competence studies for this species are based [93]. As such, there is no data on the vector potential of Indian C. actoni. Nevertheless, the Indian species, which almost certainly belongs to C. actoni (senso stricto) [92], is closely related to the proven vector in Australia and warrants investigation into its potential as a vector of BTV.
The sequence difference between C. peregrinus specimens from China, Japan, India and Thailand was 1.2–1.7 %. However, Australian specimens have been the basis of C. peregrinus BTV vector incrimination studies [93] and the south Indian specimens of C. peregrinus assessed within this study show only limited sequence divergence (mean sequence difference: 2.0 % (1.8–2.3 %) from specimens recently collected in Australia (GenBank: KR075719–KR075721) [94], indicating these associations are intraspecific and are still valid.
Culicoides anophelis has previously been reported from southern India [11, 12, 95] and is widespread throughout southern Asia [18] possibly ranging as far east as New Guinea [96]. The sequence identity of C. anophelis specimens collected in this study were concordant with the only available COI sequence for this species, also from southern India, and further study is required to investigate the relationship of populations of this species across its extensive range. Culicoides innoxius is widely distributed across southern Asia [18, 97], including Bangalore in southern India [11]. This species is very similar morphologically to C. sumatrae Macfie, 1934 [18], but there is no molecular sequence data currently available for C. sumatrae so we are not able to test the validity of these species. Similarly, comparisons between C. innoxius and other Indian species of the subgenus Hoffmania of Culicoides (for example those described by Majumdar et al. [39]), would help clarify the status of these species in India.
Culicoides mesghalii and C. kepongensis are recorded for the first time in India and C. peliliouensis and C. similis for the first time in southern India, the latter having previously been recorded in West Bengal [98] and West Bengal and Jharkhand [40], respectively. This study provides the first sequence data (COI DNA barcode) for these species. Culicoides mesghalii is known to occur in the Saharo-Arabian region [38], C. peliliouensis in the Oriental region [18, 97] and C. similis from across the Afrotropical [99, 100], Saharo-Arabian [38] and Oriental [18, 101] regions, so the presence of these species in southern India is not surprising and probably reflects a paucity of collecting in southern India, rather than recent incursions. The specimens within this study identified as C. similis are consistent with the description of this species given by Nandi & Mazumdar [40]. The morphological description of C. similis by Nandi & Mazumdar [40] is, however, in contrast to that given for C. similis specimens from across the Afrotropical [99, 100], Saharo-Arabian [38] and Oriental [18, 101] regions. The specimens collected in this study have therefore tentatively been recorded as C. similis, however, the variation in diagnostic morphological characters for this species in the literature warrants further investigation to confirm the validity of this identification and whether the C. similis described by Nandi & Mazumdar [40] in fact represents a novel species or simply a morphological variant of C. similis. Culicoides peliliouensis is also considered to be morphologically similar to the Cambodian species C. pongsomiensis Chu, 1986 [18]. Collection of additional specimens and generation of DNA barcodes for the latter species would allow the exploration of the validity or potential synonymy of these taxa.
The C. huffi specimens identified in this study are consistent with the morphological description of this species by Wirth & Hubert [18]. Sequences differences [mean: 4.7 % range: 4.4–4.9 %)] were noted between C. huffi specimens collected in Karnataka and those collected in Tamil Nadu (Fig. 4). Variation has been noted in the morphological descriptions provided for C. huffi by Nandi & Mazumdar [40] and Wirth & Hubert [18], however further specimens are required to clarify the status of this species in India, and compare these with topotypic specimens from Thailand [102]. Processing additional C. huffi specimens from a wider geographic area in India and specimens from morphologically similar species including C. palpisimilis Wirth & Hubert, 1989 and C. similis [18] would further resolve the delineation of these species and aid in resolving their subgeneric placement.
This study provides further evidence of the potential utility of DNA barcodes for species identification within Culicoides, and its potential to identify areas of potential cryptic diversity, which require further investigation, e.g. C. actoni [92]. This utility is, however, proportional to the number of species represented within the reference dataset and a strong integration with morphological taxonomy [28]. Further DNA barcode data is also required across the full range of the Culicoides subgenera in order to test the ‘barcode gap’ hypothesis related to the definition of species boundaries with regard to intra- and inter-specific variation [72]. The current subgeneric classification of the Culicoides [103], however, remains largely unvalidated (for review see Harrup et al. [28]) and there is mounting evidence to suggest that at least some of the current subgenera are polyphyletic, i.e. derived from more than one common ancestor [104–107]. The additional Avaritia subgenus species sequence data presented within this study, however, supports the monophyly of this subgenus within the context of the species included in the phylogenetic analysis with the Avaritia subgenus supported by BI as a monophyletic clade (100 % posterior probability).
The UV LED-based light-suction traps tested in this study clearly outperformed the alternative green LED model in the number of Culicoides collected, with more than 16 times the number of individuals collected. Collections of C. brevitarsis, predicted to be under-represented in UV-based light-suction trap collections based on previous studies in Australia [31], were in fact collected in greater numbers in the UV LED-based trap compared to the green LED-based trap, indicating that a UV-based trap is sufficiently sensitive to collect C. brevitarsis in southern India. Also of significant importance is the absence of key potential BTV vector species, including C. imicola and C. peregrinus, on multiple nights from green LED-based light-suction trap collections when these species are present on the corresponding night in the UV LED-based light-suction trap collection. If this were to occur during surveillance activities, the reduced sensitivity of the green LED-based light-suction traps would result in pseudo-absences within the Culicoides abundance dataset potentially resulting in epidemiologically significant errors in models of Culicoides seasonality and/or distribution. Ultraviolet LED-based light-suction traps, however, did collect a greater by-catch of other insects in comparison to green LED-based traps and hence incurred a greater handling time for each sample collected. While taking into account the limited spatial-scale and temporal duration of this comparison of green and UV wavelength-based light-suction traps the increased sensitivity of detection of the UV LED-based trap makes it the preferred choice for on-going surveillance efforts in India compared to a green LED-based light-suction trap.
Ultraviolet-based light-suction traps have previously been recommended as the gold-standard for collecting Culicoides [108], based initially on the Onderstepoort Veterinary Institute (OVI) trap design. Following on from the recommendations of Mellor et al. [108], multiple UV-based light-suction traps have been utilised for the collection of Culicoides including those based on conventional fluorescent UV bulbs and LEDs [109, 110] with the numbers of Culicoides collected by the traps roughly proportional to the intensity of the UV light emitted by the trap and the power of the fan. While the LED CDC type trap tested in this study has previously been found to be outperformed by CDC type traps fitted with a conventional fluorescent UV bulb [109], this variation is likely due to variation in the light intensity between the traps. However, the significantly lower power consumption and weight of the LED-based traps in comparison to currently available fluorescent bulb-based traps increases convenience for collectors and increases the number of nights of collections which can be completed on one battery charge from one to four nights when used with a ≥10 Ah battery and photo switch. The increased power efficiency of LED-based light-suction traps in comparison to conventional fluorescent UV bulb-based light-suction traps, such as the OVI, may assist in establishing Culicoides trapping schemes in areas where there are significant logistical challenges such as limited access, limited mains electricity availability, and financial constraints with regard to the purchasing of additional batteries [35, 36].
A major consideration for establishing light-suction trapping networks is their limitations in reflecting the abundance and diversity of Culicoides feeding on ruminants. Diurnal activity has been reported in C. oxystoma [111] and C. actoni [18] indicating that any trap utilising light as an attractant has the potential to underestimate the abundance and distribution of these species in comparison to species with a principally crepuscular or nocturnal activity pattern. In addition, the preferential attraction to different light wavelength of the highly competent BTV vector C. fulvus, which is known from northern [1, 5, 6], but not southern India, remains unknown as it was not detected during this study. Further studies across different environments in this region would be useful in elucidating the distribution of this and other putative BTV vectors in this region. In addition, the continued expansion of the depth of the reference dataset of matched morphological and genetic data for Culicoides specimens from India and the surrounding regions is essential to enable accurate assessments of species abundance and diversity in relation to BTV epidemiology to be made in the future and will enable the Indian Culicoides fauna to be placed in a global context.
Conclusions
This study provides the foundation of the production of an updated inventory of valid species of Culicoides known to occur in India. The study was developed via an integrative taxonomy approach supported by publically available molecular data, in addition to new molecular data produced to strict internationally accepted metadata and quality standards [44, 112]. The study has examined the phylogenetics of Culicoides collected in southern India and placed results in context with both taxonomic status and the relatedness of global populations. The finding that morphological identification of potential vector species is relatively robust within this region has significant importance in interpreting previous studies of Culicoides carried out in southern India and in planning future studies. This taxonomic framework has the potential to be used to address a variety of areas where species-specific identification is important including relating the seasonality of adult populations to BTV outbreaks and in studies of vector capacity.
References
Prasad G, Sreenivasulu D, Singh KO, Mertens PPC, Maan S. Bluetongue in the Indian Subcontinent. In: Mellor PS, Baylis M, Mertens PPC, editors. Bluetongue. London: Elsevier - Academic Press; 2009;167–195.
Sreenivasulu D, Subba Rao MV, Reddy YN, Gard GP. Overview of bluetongue disease, viruses, vectors, surveillance and unique feature: Indian sub-continent and adjacent regions. Vet Ital. 2004;40(3):73–7.
Foster NM, Jones RH, McCrory BR. Preliminary investigation on insect transmission of bluetongue virus in sheep. Am J Vet Res. 1963;24:1195–200.
Pawan K, Prasad M, Koushlesh R, Ravinder D, Gaya P. Evidence of reassortment between eastern and western topotype strains of bluetongue virus serotype 16 (BTV-16) from India. Adv Anim Vet Sci. 2013;1(4S):14–9.
Mellor PS, Boorman J, Baylis M. Culicoides biting midges: their role as arbovirus vectors. Annu Rev Entomol. 2000;45:307–40.
Purse BV, Carpenter S, Venter GJ, Bellis G, Mullens BA. Bionomics of temperate and tropical Culicoides midges: knowledge gaps and consequences for transmission of Culicoides-borne viruses. Annu Rev Entomol. 2014;60:373–93.
Bellis G, Dyce AL. Intraspecific variation in three species of Culicoides in the Orientalis complex of the subgenus Avaritia with the Australasian zoogeograpic region. Arbovirus Res Aust. 2005;9:33–6.
Guichard S, Guis H, Tran A, Garros C, Balenghien T, Kriticos DJ. Worldwide niche and future potential distribution of Culicoides imicola, a major vector of bluetongue and African horse sickness viruses. PLoS One. 2014;9(11):e112491.
Bakhoum MT, Fall M, Fall AG, Bellis GA, Gottlieb Y, Labuschagne K, Venter GJ, Diop M, Mall I, Seck MT, et al. First record of Culicoides oxystoma Kieffer and diversity of species within the Schultzei Group of Culicoides Latreille (Diptera: Ceratopogonidae) biting midges in Senegal. PLoS One. 2013;8(12):e84316.
Holt BG, Lessard J-P, Borregaard MK, Fritz SA, Araújo MB, Dimitrov D, Fabre P-H, Graham CH, Graves GR, Jønsson KA, et al. An update of Wallace’s zoogeographic regions of the world. Science. 2013;339(6115):74–8.
Archana M, D’Souza P, Renuka Prasad C, Byregowda SM. Prevalence of different species of Culicoides in Bangalore rural and urban districts of South India. J Parasit Dis. 2014;1–14. doi:10.1007/s12639-014-0544-1.
Archana M, D’Souza PE, Renuka Prasad C, Byregowda SM. Seasonal prevalence of different species of in Bangalore rural and urban districts of South India. Vet World. 2014;7(7):517–21.
Narladkar BW, Shivpuje PR. Prevalence population dynamics and host preferences of Culicoides spp. (Diptera: Ceratopogonidae) of livestock in Marathwada region of Maharashtra State. Vet World. 2014;7(9):717–26.
Narladkar BW, Shastri UV, Shivpuje PR. Studies on Culicoides spp. (Diptera: Ceratopogonidae) prevalent in Marathwada region (Maharashtra) and their host preferences. Indian Vet J. 1993;70:116–8.
Delfinado MD, Hardy DE. A catalog of the diptera of the Oriental Region, Suborder Nematocera, vol. 1. Honolulu: The University Press of Hawaii; 1973. p. 628.
Maan S, Maan NS, Belaganahalli MN, Rao PP, Singh KP, Hemadri D, Putty K, Kumar A, Batra K, Krishnajyothi Y, et al. Full-genome sequencing as a basis for molecular epidemiology studies of bluetongue virus in India. PLoS One. 2015;10(6):e0131257.
Reddy YV, Krishnajyothi Y, Susmitha B, Devi BV, Brundavanam Y, Gollapalli SR, Karunasri N, Sonali B, Kavitha K, Patil SR, et al. Molecular typing of bluetongue viruses isolated over a decade in South India. Transbound Emerg Dis. 2015; Online Early doi: 10.1111/tbed.12320.
Wirth WW, Hubert AA. The Culicoides of Southeast Asia (Diptera: Ceratopogonidae). The American Entomological Institute: Gainesville, Florida; 1989. p. 508.
Das Gupta SK. Some Culicoides of Calcutta and the neighbouring areas. Sci Cult. 1962;28:537–9.
Das Gupta SK. Culicoides (Dipt., Ceratopogonidae) from suburbs of Calcutta. Entomol Mon Mag. 1962;98:253–4.
Ilango K. Bluetongue virus outbreak in Tamil Nadu, southern India: need to study the Indian biting midge vectors, Culicoides Latreille (Diptera: Ceratopogonidae). Curr Sci India. 2006;90(2):163–7.
Gangopadhyay D, Das Gupta SK. Glimpses of the biodiversity of Culicoides Insects (Diptera: Ceratopogonidae) in India. In: Proceedings of the National Seminar on Environmental Biology. Delhi: Daya Publishing House; 1998. p. 123–32.
Maheshwari G. Current status of bluetongue disease, its vector and pathogenesis in India. Proc Natl Acad Sci India Sect B (Biol Sci). 2012;82(4):463–75.
Hebert PD, Cywinska A, Ball SL, de Waard JR. Biological identifications through DNA barcodes. P Roy Soc B Biol Sci. 2003;270(1512):313–21.
Archana M, Placid EDS, Jalali SK, Renukaprasad C, Rakshith O. DNA barcoding of commonly prevalent Culicoides midges in South India. Indian J Anim Sci. 2015;85(1):37–9.
Bellis G, Dyce A, Gopurenko D, Yanase T, Garros C, Labuschagne K, Mitchell A. Revision of the Culicoides Avaritia Imicola complex Khamala & Kettle (Diptera: Ceratopogonidae) from the Australasian region. Zootaxa. 2014;3768(4):401–27.
Sebastiani F, Meiswinkel R, Gomulski LM, Guglielmino CR, Mellor PS, Malacrida AR, Gasperi G. Molecular differentiation of the Old World Culicoides imicola species complex (Diptera, Ceratopogonidae), inferred using random amplified polymorphic DNA markers. Mol Ecol. 2001;10(7):1773–86.
Harrup L, Bellis GA, Balenghien T, Garros C. Culicoides Latreille (Diptera: Ceratopogonidae) taxonomy: current challenges and future directions. Infect Genet Evol. 2015;30:249–66.
Calvete C, Estrada R, Miranda MA, Borràs D, Calvo JH, Lucientes J. Ecological correlates of bluetongue virus in Spain: predicted spatial occurrence and its relationship with the observed abundance of the potential Culicoides spp. vector. Vet J. 2009;182(2):235–43.
Conte A, Gilbert M, Goffredo M. Eight years of entomological surveillance in Italy show no evidence of Culicoides imicola geographical range expansion. J Appl Ecol. 2009;46(6):1332–9.
Bishop AL, Worrall R, Spohr LJ, McKenzie HJ, Barchia IM. Response of Culicoides spp. (Diptera: Ceratopogonidae) to light-emitting diodes. Aust J Entomol. 2004;43(2):184–8.
Bishop AL, Bellis GA, McKenzie HJ, Spohr LJ, Worrall RJ, Harris AM, Melville L. Light trapping of biting midges Culicoides spp. (Diptera: Ceratopogonidae) with green light-emitting diodes. Aust J Entomol. 2006;45(3):202–5.
Venter GJ, Hermanides KG. Comparison of black and white light for collecting Culicoides imicola and other livestock-associated Culicoides species in South Africa. Vet Parasitol. 2006;142(3–4):383–5.
Melville LF. Bluetongue surveillance methods in an endemic area: Australia. Vet Ital. 2004;40(3):184–7.
Cohnstaedt LW, Gillen JI, Munstermann LE. Light-emitting diode technology improves insect trapping. J Am Mosq Control Assoc. 2008;24(2):331–4.
Cohnstaedt LW, Rochon K, Duehl AJ, Anderson JF, Barrera R, Su N-Y, Gerry AC, Obenauer PJ, Campbell JF, Lysyk TJ, et al. Arthropod surveillance programs: Basic components, strategies, and analysis. Ann Entomol Soc Am. 2012;105(2):135–49.
Peel MC, Finlayson BL, McMahon TA. Updated world map of the Köppen-Geiger climate classification. Hydrol Earth Syst Sci. 2007;11:1633–44.
Boorman J. Culicoides (Diptera: Ceratopogonidae) of the Arabian peninsula with notes on their medical and veterinary importance. Fauna Saudi Arab. 1989;10:160–224.
Majumdar BC, Das Gupta SK, Gangopadhyay D. Some new species of Hoffmania Fox subgenus of Culicoides biting midges (Diptera: Ceratopogonidae) from Darjeeling. Bombay Nat Hist Soc J. 1997;16:27–34.
Nandi M, Mazumdar A. Revision of the subgenus Diphaomyia Vargas of Culicoides Latreille from India with description of a new species (Diptera: Ceratopogonidae). Zootaxa. 2014;3793:465–74.
Nandi M, Mazumdar A. The Chaetophthalmus species-group of the genus Culciodies Latreille, 1809 (Diptera: Ceratopogonidae) from India with decription of a new species. Caucasian Entomol Bull. 2014;10(1):161–4.
Bellis GA, Dyce AL, Gopurenko D, Mitchell A. Revision of the Immaculatus Group of Culicoides Latreille (Diptera: Ceratopogonidae) from the Australasian region with description of two new species. Zootaxa. 2013;3680(1):15–37.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Ratnasingham S, Hebert PDN. BOLD: The Barcode of Life Data System (www.barcodinglife.org). Mol Ecol Notes. 2007;7:355–64.
Madden T. The BLAST sequence analysis tool. In: McEntyre J, Ostell J, editors. The NCBI handbook. Bethesda: National Center for Biotechnology Information; 2003. Available at http://www.ncbi.nlm.nih.gov/books/NBK21097.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Penn O, Privman E, Landan G, Graur D, Pupko T. An alignment confidence score capturing robustness to guide-tree uncertainty. Mol Biol Evol. 2010;27(8):1759–67.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772.
Guindon S, Gascuel O. A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst Biol. 2003;52:696–704.
Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogeny. Bioinformatics. 2001;17:754–5.
Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Beard CB, Hamm DM, Collins FH. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol Biol. 1993;2(2):103–24.
Nylander JAA, Wilgenbusch JC, Warren DL, Swofford DL. AWTY: a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics. 2008;24(4):581–3.
Bellis GA, Halling L, Anderson SJ. Pictorial key to adult female Culicoides Latreille, 1809 (Diptera: Ceratopogonidae) from the Northern Territory, Western Australia and South Australia. Aust Entomol. 2014;54(1):28–59.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Fluxus-Engineering. Network version 4.6.1.2. 2014. Available at: http://www.fluxus-engineering.com/sharenet.htm.
Bandelt H-J, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Polzin T, Daneschmand SV. On Steiner trees and minimum spanning trees in hypergraphs. Oper Res Lett. 2003;31:12–20.
Brown SD, Collins RA, Boyer S, Lefort MC, Malumbres-Olarte J, Vink CJ, Cruickshank RH. Spider: an R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol Ecol Resour. 2012;12(3):562–5.
Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–90.
R Development Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2014. ISBN 3-900051-07-0, URL http://www.R-project.org. 2.9.1 edn.
Nevill H, Dyce AL. Afrotropical Culicoides: description and comparison of the pupae of seven species of the Similis supergroup (Diptera: Ceratopogonidae). Onderstepoort J Vet Res. 1994;61(1):85–106.
Bellis G. Key to females of economically important species of Culicoides subgenus Avaritia from India using characters visible under a stereomicroscope. 2014. Available from http://www.ibvnet.com/documents/Bellis_2014_Indian_Culicoides_Avaritia_Key.pdf.
Dyce AL. The recognition of nulliparous and parous Culicoides (Diptera: Ceratopogonidae) without dissection. J Aust Entomol Soc. 1969;8:11–5.
Harrup LE, Purse BP, Golding N, Mellor PS, Carpenter S. Larval development and emergence sites of farm-associated Culicoides (Diptera: Ceratopogonidae) in the United Kingdom. Med Vet Entomol. 2013;27(4):441–9.
Harrup LE. The Pirbright Institute Culicoides DNA Barcoding Protocols, Version 2. 2014. updated: 3rd July. DOI: 10.13140/RG.2.1.5026.2642.
Fournier DA, Skaug HJ, Ancheta J, Ianelli J, Magnusson A, Maunder M, Nielsen A, Sibert J. AD Model Builder: using automatic differentiation for statistical inference of highly parameterized complex nonlinear models. Optim Methods Softw. 2012;27(2):233–49.
Skaug H, Fournier D, Nielsen A, Magnusson A, Bolker B. GlmmADMB: generalized linear mixed models using AD model builder, R package version 0.7.5. 2013.
Akaike H. Information theory as an extension of the Maximum likelihood principle. In: Petrov BN, Csaksi F, editors. 2nd International Symposium on Information Theory. Budapest: Akademiai Kiado; 1973. p. 267–81.
Margalef R. Temporal succession and spatial heterogeneity in phytoplankton. In: Buzzati-Traverso AA, editor. Perspectives in marine biology. Berkeley: University of California Press; 1958. p. 323–47.
Ratnasingham S, Hebert PDN. A DNA-based registry for all animal species: the Barcode Index Number (BIN) system. PLoS One. 2013;8(7):e66213.
Meyer CP, Paulay G. DNA Barcoding: error rates based on comprehensive sampling. PLoS Biol. 2005;3(12):e422.
Meier A. DNA sequences in taxonomy - opportunities and challenges. In: Wheeler QD, editor. New taxonomy. Boca Raton: CRC Press; 2008. p. 95–127.
Ander M, Troell K, Chirico J. Barcoding of biting midges in the genus Culicoides: a tool for species determination. Med Vet Entomol. 2013;27(3):323–31.
Matsumoto Y, Tanase T, Tsuda T, Noda H. Species-specific mitochondrial gene rearrangements in biting midges and vector species identification. Med Vet Entomol. 2009;23:47–55.
Morag N, Saroya Y, Braverman Y, Klement E, Gottlieb Y. Molecular identification, phylogenetic status and geographic distribution of Culicoides oxystoma (Diptera: Ceratopogonidae) in Israel. PLoS One. 2012;7:e33610.
Calvo JH, Calvete C, Martinez-Royo A, Estrada R, Miranda MA, Borràs D, Sarto IMV, Pages N, Delgado JA, Collantes F, et al. Variations in the mitochondrial cytochrome c oxidase subunit I gene indicate northward expanding populations of Culicoides imicola in Spain. Bull Entomol Res. 2009;99(6):583–91.
Dallas JF, Cruickshank RH, Linton YM, Nolan DV, Patakakis M, Braverman Y, Capela M, Capela R, Pena I, Meiswinkel R, et al. Phylogenetic status and matrilineal structure of the biting midge, Culicoides imicola, in Portugal, Rhodes and Israel. Med Vet Entomol. 2003;17:379–87.
Nolan DV, Dallas JF, Piertney SB, Mordue (Luntz) AJ. Inclusion and range expansion in the bluetongue vector Culicoides imicola in the Mediterranean basin: a phylogeographic analysis. Med Vet Entomol. 2008;22:340–51.
Onyango MG, Michuki GN, Ogugo M, Venter GJ, Miranda MA, Elissa N, Djikeng A, Kemp S, Walker PJ, Duchemin JB. Delineation of the population genetic structure of Culicoides imicola in East and South Africa. Parasit Vectors. 2015;8(1):660.
Jacquet S, Garros C, Lombaert E, Walton C, Restrepo J, Allene X, Baldet T, Cetre-Sossah C, Chaskopoulou A, Delecolle JC, et al. Colonization of the Mediterranean Basin by the vector biting midge species Culicoides imicola: an old story. Mol Ecol. 2015;24:5707–25.
Narladkar BW, Deshpande PD, Shivpuje PR. Bionomics and life cycle on Culicoides sp. (Diptera : Ceratopogonidae). J Vet Parasitol. 2006;20(1):7–12.
Narladkar BW, Deshpande PD, Shivpuje PR, Sondge VD. Population dynamics of Culicoides sp. of domesticated animals of Marathwada region. J Vet Parasitol. 2006;20(2):171–4.
Narladkar BW, Shastri UV, Shivpuje PR. Breeding habitats of Culicodies schultzei (Enderlein, 1908) (Diptera: Ceratopogonidae). Indian J Anim Sci. 1992;62(10):944–5.
Narladkar BW, Shastri UV, Shivpuje PR. Studies on bionomics of Culicoides spp. (Diptera; Ceratopogonidae) with a note on life cycle. Indian Vet J. 1993;70:313–6.
Narladkar BW, Shastri UV, Vadlamudi VP, Shivpuje PR. Relative toxicity of some modern insecticides against larvae of Culicoides schultzei. Indian Vet J. 1993;70:766–8.
Cornet M, Brunhes J. Révision des espèces de Culicoides apparentées à C. schultzei (Enderlein, 1908) dans la région afrotropicale (Diptera, Ceratopogonidae). Bull Soc Entomol Fr. 1994;99:149–64.
Augot D, Randrianambinintsoa FJ, Gasser A, Depaquit J. Record of two species of Culicoides (Diptera : Ceratopogonidae) new for Madagascar and molecular study showing the paraphylies of the subgenus Oecacta and the Schultzei group. Bull Soc Pathol Exot. 2013;106:201–5.
Cornet M. Revision of the Culicoides species related to Culicoides schultzei (Enderlein) in the Ethiopian region-preliminary note. Isr J Entomol. 1981;13:110.
Nevill H, Venter GJ, Meiswinkel R, Nevill EM. Comparative descriptions of the pupae of five species of the Culicoides imicola complex (Diptera, Ceratopogonidae) from South Africa. Onderstepoort J Vet Res. 2007;74(2):97–114.
Desvars A, Grimaud Y, Guis H, Esnault O, Allène X, Gardès L, Balenghien T, Baldet T, Delécolle JC, Garros C. First overview of the Culicoides Latreille (Diptera: Ceratopogonidae) livestock associated species of Reunion Island, Indian Ocean. Acta Trop. 2015;142:5–19.
Gopurenko D, Bellis GA, Yanase T, Warhana AH, Thepparat A, Wang J-L, Li H-C, Mitchell A. Using integrative taxonomy to investigate species boundaries within Culicoides (Diptera: Ceratopogonidae): a case study using subgenus Avaritia from Australasia and eastern Asia. Vet Ital. 2016;51(4):345–78.
Standfast HA, Dyce AL, Muller MJ. Vectors of bluetongue virus in Australia. Prog Clin Biol Res. 1985;178:177–86.
Kim HC, Bellis GA, Kim MS, Klein TA, Gopurenko D, Cai DC, Seo HJ, Cho IS, Park JY. Species diversity and seasonal distribution of Culicoides spp. (Diptera: Ceratopogonidae) in Jeju-do, Republic of Korea. Korean J Parasitol. 2015;53(4):501–6.
Reuben R. Note on the seasonal prevalence of Culicoides Schultzei (Enderlein): Synonym Culicoides Oxystoma Kieffer (Ceratopogonidae: Diptera). J Bombay Nat Hist Soc. 1965;62(2):308–9.
Dyce AL, Bellis GA, Muller MJ. Pictorial Atlas of Australian Culicoides Wings (Diptera: Ceratopogonidae). Canberra: ABRS; 2007.
Howarth FG. Biosystematics of the Culicoides of Laos (Diptera: Ceratopogonidae). Int J Entomol. 1985;27:1–96.
Ray S, Choudhury A. Population ecology of Culicoides peliliouensis Tok. in The Hooghly Estuary, Sagar Island, India. Insect Sci Appl. 1988;9(1):17–25.
Khamala CPM, Kettle DS. The Culicoides Latreille (Diptera: Ceratopogonidae) of East Africa. Tras R Entomol Soc London. 1971;123:1–95.
Glick JI. Culicoides Biting Midges (Diptera: Ceratopogonidae) of Kenya. J Med Entomol. 1990;27:2.
Yu Y-X, Huang Y-Y. New species and records of biting midges from Macau (Diptera: Ceratopogonidae). Acta Parasit Med Entomol Sinica. 2006;13:47–50.
Causey OR. Culicoides of Siam with descriptions of new species. Am J Hyg. 1938;27:399–416.
Borkent A. The subgeneric classification of Species of Culicoides - thoughts and a warning. Salmon Arm, British Columbia, Canada: Royal British Columbia Museum, American Museum of Natural History and Instituto Nacional de Biodiversidad; 2015. Updated: 16th May 2016, available at: http://wwx.inhs.illinois.edu/files/5014/6532/8290/CulicoidesSubgenera.pdf. Accessed 2 June 2016.
Gomulski LM, Meiswinkel R, Delécolle JC, Goffredo M, Gasperi G. Phylogeny of the subgenus Culicoides and related species in Italy, inferred from internal transcribed spacer 2 ribosomal DNA sequences. Med Vet Entomol. 2006;20(2):229–38.
Linton YM, Mordue AJ, Cruickshank RH, Meiswinkel R, Mellor PS, Dallas JF. Phylogenetic analysis of the mitochondrial cytochrome oxidase subunit I gene of five species of the Culicoides imicola species complex. Med Vet Entomol. 2002;16:139–46.
Perrin A, Cêtre-Sossah C, Mathieu B, Baldet T, Delécolle JC, Albina E. Phylogenetic analysis of Culicoides species from France based on nuclear ITS1-rDNA sequences. Med Vet Entomol. 2006;20(2):219–28.
Schwenkenbecher JM, Mordue AJ, Piertney SB. Phylogenetic analysis indicates that Culicoides dewulfi should not be considered part of the Culicoides obsoletus complex. Bull Entomol Res. 2009;99(4):371–5.
Mellor PS, Tabachnick W, Baldet T, Baylis M, Bellis G, Calistri P, Delécolle JC, Goffredo M, Meiswinkel R, Mullens B, et al. Conclusions of Working Groups. Group 2. Vectors, Proceedings of the 3rd OIE Bluetongue International Symposium, 26-29th October 2003, Taormina, Italy. Vet Ital. 2004;40(4):715–7.
Hope A, Gubbins S, Sanders C, Denison E, Barber J, Stubbins F, Baylis M, Carpenter S. A comparison of commercial light-emitting diode baited suction traps for surveillance of Culicoides in northern Europe. Parasit Vectors. 2015;8:239–49.
Venter GJ, Labuschagne K, Hermanides KG, Boikanyo SN, Majatladi DM, Morey L. Comparison of the efficiency of five suction light traps under field conditions in South Africa for the collection of Culicoides species. Vet Parasitol. 2009;166(3–4):299–307.
Buckley JJC. On Culicoides as a vector of Onchocera gibsoni (Cleland and Johnson, 1910). J Helminthol. 1938;16:121–58.
Hanner R. BARCODE Data Standard v.2.4. Data standard for BARCODE records in INSDC (BRIs). 2012. updated: 28th February 2012, Accessed 10 Sept 2013, available at: http://www.barcodeoflife.org/sites/default/files/DWG_data_standards-Final.pdf.
Acknowledgements
This study was funded by the Biotechnology and Biological Sciences Research Council (BBSRC), Department for International Development (DFID) and Scottish Government grant BB/H009205/1, BB/H009167/1 and BB/H009493/1 as part of the Combating Infectious Diseases of Livestock for International Development initiative. With additional support provided by BBSRC grant BB/K021214/1; AB was also supported with funding from the London School of Hygiene and Tropical Medicine. The Indian Council of Agricultural Research supported this study through its Hisar, Chennai, Bengaluru and Hyderabad centres under the All Indian Network Programme on Bluetongue.
Authors’ contributions
LS, GB, NK and AB completed all morphological identifications assisted by LEH, KL and BM. LEH completed all molecular identifications and phylogenetic analysis. AB completed all light-suction trap comparison fieldwork. BVP completed all statistical analysis of the light-suction trap comparison data. LS, NK, YKR, YNR, SMB, KMP, SK, MP and GP facilitated the collection of specimens. LEH drafted the manuscript with contribution from SC, BVP, CG, DG, GB, JL and KL. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests. Funders had no role in study design or the collection, analysis and interpretation of data. Mention of proprietary products does not constitute an endorsement or a recommendation by the authors for their use.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
GenBank sequences used in genetic analyses of Culicoides from southern India. Table S2. Barcode Index Numbers (BINs) assigned within the Barcode of Life Database (BOLD) for specimens collected within this study. (DOCX 27 kb)
Additional file 2: Table S3.
Uncorrected percentage sequence distances, mean with range shown in parentheses. Intraspecific distances are shown in bold along the diagonal, interspecific distances are shown in the lower triangle (NA indicates comparison not possible due to singleton specimen present; number of specimens per species (n) shown in brackets with the number of specimens originating from this study; followed by the number originating from GenBank in parentheses). (XLSX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Harrup, L.E., Laban, S., Purse, B.V. et al. DNA barcoding and surveillance sampling strategies for Culicoides biting midges (Diptera: Ceratopogonidae) in southern India. Parasites Vectors 9, 461 (2016). https://doi.org/10.1186/s13071-016-1722-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-016-1722-z