
Abstract
Background
Lignin peroxidases catalyze a variety of reactions, resulting in cleavage of both β-O-4′ ether bonds and C–C bonds in lignin, both of which are essential for depolymerizing lignin into fragments amendable to biological or chemical upgrading to valuable products. Studies of the specificity of lignin peroxidases to catalyze these various reactions and the role reaction conditions such as pH play have been limited by the lack of assays that allow quantification of specific bond-breaking events. The subsequent theoretical understanding of the underlying mechanisms by which pH modulates the activity of lignin peroxidases remains nascent. Here, we report on combined experimental and theoretical studies of the effect of pH on the enzyme-catalyzed cleavage of β-O-4′ ether bonds and of C–C bonds by a lignin peroxidase isozyme H8 from Phanerochaete chrysosporium and an acid stabilized variant of the same enzyme.
Results
Using a nanostructure initiator mass spectrometry assay that provides quantification of bond breaking in a phenolic model lignin dimer we found that catalysis of degradation of the dimer to products by an acid-stabilized variant of lignin peroxidase isozyme H8 increased from 38.4% at pH 5 to 92.5% at pH 2.6. At pH 2.6, the observed product distribution resulted from 65.5% β-O-4′ ether bond cleavage, 27.0% Cα-C1 carbon bond cleavage, and 3.6% Cα-oxidation as by-product. Using ab initio molecular dynamic simulations and climbing-image Nudge Elastic Band based transition state searches, we suggest the effect of lower pH is via protonation of aliphatic hydroxyl groups under which extremely acidic conditions resulted in lower energetic barriers for bond-cleavages, particularly β-O-4′ bonds.
Conclusion
These coupled experimental results and theoretical explanations suggest pH is a key driving force for selective and efficient lignin peroxidase isozyme H8 catalyzed depolymerization of the phenolic lignin dimer and further suggest that engineering of lignin peroxidase isozyme H8 and other enzymes involved in lignin depolymerization should include targeting stability at low pH.
Similar content being viewed by others

Background
Lignocellulosic biomass, which is composed mainly of cellulose, hemicellulose, and lignin, is the most abundant source of renewable carbon on Earth [1], and the aromatic biopolymer lignin is the most abundant source of renewable aromatics [2]. Currently, most lignin is either stockpiled or burned to generate power, but techno-economic models of lignocellulosic biorefineries show that diverting lignin from on-site combustion saves costs [3] and converting lignin to value-added biofuels and bioproducts greatly improves the economics of biorefineries and reduces greenhouse gas (GHG) emissions [4]. As a first step in the process of converting lignin to valuable products, depolymerization of lignin to biologically available intermediates is critical. Unfortunately, efficient enzymatic depolymerization of lignin to biologically available intermediates at high yields has been impeded by a limited fundamental understanding of how lignin-degrading enzymes function and, more importantly, how we might control the activity of these enzymes to produce the most biologically useful lignin fragments.
In nature, fungi and bacteria produce an array of enzymes, including laccases, lignin peroxidases (LiP), versatile peroxidases (VP), and manganese peroxidases (MnP), that are secreted into the environment and catalyze depolymerization of lignin [5, 6]. In general, numerous physio-chemical factors such as pH, ionic strength, enzyme concentration, and redox potentials are potential driving forces for depolymerization of lignin but in some cases may also drive the re-polymerization of the lignin fragments produced during depolymerization. Some microorganisms involved in biomass degradation actively modify the pH of their environment via secretion of acids and bases, and fungi produce organic acids, resulting in significant acidification of conditions in their microenvironment, which is thought to play many key roles in nature [7,8,9]. Notably, two Basidiomycota known to degrade lignin, Phanerochaete chrysosporium, and Trametes menziesii, have been shown to acidify their environment to pH 2 or lower, implicating the pH of the fungal environment as a key driving force for the biological breakdown of biomass and specifically for depolymerization of lignin catalyzed by LiP, MnP, VP and laccases [10].
The fungi P. chrysosporium and T. menziesii employ extracellular peroxidases including LiP, VP, and MnP to catalyze bond cleavage in lignin [11,12,13]. One particular well-studied enzyme is the lignin peroxidase isozyme H8 (LiPH8) from P. chrysosporium, which harbors a surface exposed catalytic Trp171 amino acid known to play a vital role in the oxidation of high-redox-potential mediators such as veratryl alcohol (VA) and nonphenolic lignin derivatives [14]. The VA cation radical has been proposed as a radical mediator to oxidize polymeric substrates with which LiP presumably cannot interact directly [15,16,17]. The mediator radical withdraws an electron from lignin resulting in a cationic radical intermediate and ultimately producing a wide variety of ligninolytic breakdown products through non-specific bond-cleavage occurrences [18, 19].
LiPs have been shown to have higher activity toward oxidation of compounds such as 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid (ABTS) [20], VA [21,22,23,24,25] and the azo dye reactive black 5 [20] at lower pH.
Stopped-flow techniques have been used to investigate the kinetics of the reaction of LiP compounds I and II (LiPI and LiPII) with VA, and the formation of LiPI and LiPII in the presence of VA was strongly dependent on pH [25,26,27]. In addition, LiPH8 from P. chrysosporium has been shown to catalyze oxidative cleavage of both β-O-4′ ether and C–C bonds in aryl ether dimers [28] and to catalyze breaking of β-O-4′ ether, C–C, and C-H bonds in trimeric lignin model compounds [29]. Combined, these studies suggest LiPH8 should catalyze β-O-4′ ether and C–C at higher rates as pH is reduced, but studies aimed at understanding bond cleavage as a function of pH and describing potential pH-dependent mechanisms have not been performed.
To partially address this lack of knowledge for LiP, the work presented here is focused on developing a fundamental understanding of LiPH8– catalyzed depolymerization of a phenolic lignin-like dimer compound as a function of pH. We present detailed studies of the effect of pH on the enzyme-catalyzed cleavage of β-O-4′ ether bonds and C–C bonds by LiPH8 from P. chrysosporium using a recently developed assay based on model lignin-like dimers and nanostructure initiator mass spectrometry (NIMS) [30]. Using the NIMS assay, we quantified dimer conversion and the distribution of products formed by breaking C–C and β-O-4′ ether bonds upon incubation of a phenolic lignin-like model compound with LiPH8. We show the distribution of products is pH-dependent, suggesting pH adjustments can be used to control LiPH8 catalyzed breaking of β-O-4′ ether bonds and C–C bonds and production of lignin-derived compounds. We also report on our use of quantum calculations and ab initio molecular dynamic (AIMD) simulations to generate a fundamental understanding of how pH assist bond cleavage and show the roles of low pH are to i) provide a thermodynamically favorable condition for the formation of a cation radical intermediate required for energy-favorable degradation of lignin, ii) pH-assisted formation of protonated cationic radical intermediate at both phenolic and aliphatic hydroxyl groups, resulting in higher cleavage frequencies for various bond types in lignin, especially β-O-4′ ether bonds.
Results and discussion
pH-controlled catalysis of bond cleavage by the lignin peroxidase from P. chrysosporium
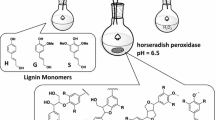
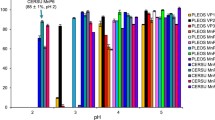
Lignin is an unusual biopolymer because of its structural heterogeneity, which has limited detailed studies of lignin-degrading enzymes in terms of their ability to catalyze the breaking of specific bond types. The most predominant linkage between phenylpropane units in both softwood and hardwood lignin is the β-O-4′ ether bond, comprising 45 – 60% [31, 32] of the bonds in lignin. It was found that LiPH8 directly interacts with the synthesized lignin macromolecules, which was supported by kinetic analysis of its binding affinity [33]. However, quantitative detection of phenolic products has not been reported for in vitro depolymerization of lignin by LiPH8. It is hypothesized that repolymerization of degraded lignin fragments spontaneously occurs and presents a barrier to in vitro depolymerization as well as to the detection of fragments produced and subsequent analysis of bond cleavage specificity. Thus, devising schemes to control depolymerization of lignin to produce defined intermediates requires a detailed understanding of reaction mechanisms and conditions, which are not well understood for this enzyme and, in general, for enzymes shown to be involved in lignin depolymerization. Herein, we study catalysis of bond cleavage events in a phenolic lignin dimer by quantitative analysis of product formation during LiPH8-catalyzed degradation of a GGE model compound (GGE-NIMS compound) using nanostructure-initiator mass spectrometry (GGE-NIMS dimer structure shown in Fig. 1 [30]. The enzyme assay was experimentally measured at four pH levels between 2.6 and 5.0, which are levels previously determined to result in the high activity of VA oxidation for wild-type LiPH8 and a low pH stabilized variant of it [23]. Products from the oxidation of the GGE-NIMS dimer through a one-electron reaction step were quantified using the NIMS assay and resulted from catalysis of four main reactions: Cα-oxidation, Cα-Cβ bond cleavage, β-O-4 ether bond cleavage, and Cα-C1 carbon bond (ring A) cleavage (Fig. 1). The data presented in Fig. 2a show that wildtype LiPH8 exhibited its highest catalytic capability at pH 3.0, with greater than 90% of the GGE-NIMS dimer being converted to products. At pH < 3.0 (pH = 2.6), wildtype LiPH8 lignin peroxidase was essentially inactivated, resulting in more than 75% of the GGE-NIMS substrate being unmodified (Fig. 2a). At all pH levels, β-O-4′ ether bond cleavage released phenolic product (m/z 954.3, Fig. 1), which was the dominate catalytic event, followed by Cα-C1 carbon bond cleavage and Cα oxidation.

Representative products from bond cleavage events of the GGE-NIMS dimer used to assay LiPH8 from Phanerochaete chrysosporium

Product distribution from bond cleavage of fluorous-tagged GGE by wildtype lignin peroxidase (a) and acid-stabilized mutant of the same peroxidase (b). The reaction contained 1 mM of NIMS-tagged phenolic lignin dimer, 5 µM of LiPH8 enzyme, 20 mM of veratryl alcohol as mediator was performed in sodium acetate buffer pH 2. 6–5.0. The reaction was started by adding H2O2 fed at a rate of 250 uM every 30 min up to 3 h. Error bars are the standard deviation for three replicates, and the first column at each pH condition in Fig. 1 is the mean and standard deviation of the products from both β-O-4′ ether and Cα-C1 bond cleavage combined. The means and standard deviations for β-O-4′ bond cleavage were 65.5 ± 0.8 (pH 2.6), 60.0 ± 1.3 (pH 3.0), 38.4 ± 1.0 (pH 4.0) and 26.5 ± 0.4 (pH 5.0). The means and standard deviations for Cα-C1 bond cleavage were 27.0 ± 4.3 (pH 2.6), 26.24 ± 4.5 (pH 3.0), 13.0 ± 5.0 (pH 4.0 and 11.9 ± 3.0 (pH 5.0)
To further investigate the effects of acidic conditions on bond cleavage frequency and specificity, a previously reported acid-stabilized variant of LiPH8 was assayed over pH ranging from 2.6 to 5.0 [23]. The triple mutant A55R-N156E‑H239E LiPH8 with 2 additional salt-bridges on the solvent-exposed regions showed excellent stability and oxidation activity under extremely acidic conditions down to pH 2.6 [23]. The stabilized mutant showed higher activity levels at all three pH levels tested and had the highest activity at pH 2.6 (Fig. 2b). Comparison of the residual GGE-NIMS substrate (Fig. 2b, purple bars) shows a clear upward trend of increased conversion of lignin dimer, converting 96.1% and 45.3% of the dimer at pH 2.6 and pH 5.0, respectively. The higher conversion of substrate resulted in more products from various bond cleavage events under the catalysis of the acid-stable mutant. For instance, β-O-4 ether bond cleavage was elevated from 26.5% to 65.5%, and products from Cα-C1 carbon bond cleavage were also increased from 11.9% to 27.0% when pH decreased from 5.0 to 2.6.
While very acidic pH conditions drove the reaction to be more complete, the distributions of products produced from β-O-4′ ether bond and Cα-C1 carbon bond cleavage at any of the pH levels studied were very similar. The ratio of product from β-O-4′ ether bond cleavage to product from Cα-C1 carbon bond was distributed over a narrow range between 2.7 and 2.2 when acidifying pH from 5.0 to 2.6, while Cα oxidation events decreased from 6.8% to 3.6% between pH 5.0 and 2.6, respectively. These results suggested pH affects overall bond-cleavage frequency but does not determine product fragmentation pathways. Further study with various lignin dimer types may be needed to better understand the structural effect on product fragmentation pathways.
Exactly how in nature fungi alter the pH of their environment is not exactly know, but white-rot basidiomycetes have been shown to secret organic acids into their microenvironment, which could assist extracellular ligninase catalyzed lignin degradation [10]. This is in agreement with other studies showing that LiP and VP catalyze VA and non-phenolic lignin dimer oxidation with a pH optimum of 3.0 [20, 24, 25, 34,35,36]. It has been suggested through experimental study and quantum mechanics/molecular mechanics calculations that the redox potential of heme peroxidases increases at low pH, providing LiP and VP the capability to oxidize the recalcitrant lignin polymer at low pH [25, 37, 38]. Another explanation put forth is that the acidic residues surrounding the surface Trp171 that interacts with either lignin or mediators may be involved in the composition of an acidic environment, which acts to stabilize radical cation intermediates of small phenolic compounds [14]. Herein, we propose a pH-dependent mechanism in which the lignin dimer is protonated to different degrees at different pH values and study these proposed mechanisms using quantum calculation and AIMD simulation study.
Low pH conditions drive reaction equilibrium toward the favorable formation of the active cationic radical intermediate
We proposed that bond cleavage in the GGE dimer occurs via either cationic or phenoxy radical intermediates and investigated the relative free energies for each pathway. The cationic radical intermediate formed from LiPH8/H2O2–catalyzed 1-electron oxidation of GGE dimer is capable of undergoing a variety of reactions such as side-chain oxidation, C–C bond, and β-O-4′ ether bond cleavage. The intermediates were predicted from a heterolytic bond cleavage reaction mechanism when the first step 1-electron oxidation takes place at lower redox potential–Ring A (Fig. 3). The deprotonation of the short-lived cationic radical resulted in the formation of the phenoxy radical which sequentially cleaved into fragments (Fig. 3).

The proposed scheme of LiPH8/H2O2 catalyzed depolymerization and mechanisms and bond cleavage pathways from phenolic lignin dimer via either a cationic radical (left figure) or a phenoxy radical intermediate (right figure)
The relative calculated Gibbs free energy for the occurrence of bond cleavage via either the cationic radical intermediate or the phenoxy radical intermediate indicated that much high energy is required for bond cleavage to occur from the phenoxy radical intermediate compared to the free energy required when starting from the cationic radical intermediate (Fig. 4). For instance, the calculated Gibbs free energy required for β-O-4′ ether bond cleavage from the cationic radical is 32.8 kcal/mol versus 81.2 kcal/mol from the phenoxy radical intermediate. These results indicate that stabilizing the radical cation and decreasing the deprotonation rate will drive bond-cleavage occurrence via the cationic radical intermediate, suggesting an approach for controlling lignin depolymerization rates. The position of the equilibrium will depend on the countercation and solvent, and herein we suggest an approach of H+ concentration-controlled protonation/deprotonation equilibrium between cationic radicals and phenoxy radicals. Simply, according to LeChâtelier’s principle, the addition of H+ ions (as in a low pH solution) drives the equilibrium to the left and the protonated cationic radical predominates. This finding is consistent with the natural modulation of the significant acidic condition in the microenvironment around fungi cells which was previously reported by Liaud et al. [10].

Relative Gibbs free energy required for breaking various bond types in lignin via the cationic radical intermediate (left) and the phenoxy radical intermediate (right). Gibbs free energy was normalized to reactants at each reaction step
Protonation of hydroxyl group under acidic conditions is a key step in bond-cleavage pathways
Regarding lignin’s functional groups, aliphatic hydroxyl and phenolic hydroxyl groups have been shown to significantly affect the reactivity of lignin by nucleophilic aromatic substitution reactions including amination, nitration, and esterification [39,40,41]. Many studies have reported on the effects of these functional groups on depolymerization mechanisms. In reductive depolymerization, Wang and co-workers showed that the aliphatic alcohol moieties (CαH − OH) in lignin itself can act as the hydrogen donor [42]. In oxidative depolymerization, under acidic conditions, stabilization of aliphatic hydroxyl groups by formic acid and formaldehyde resulted in increased product yield of monomers [43, 44]. Herein, we hypothesized that under more acidic conditions formation of protonated hydroxyl groups drives the reaction to further completion by lowering bond dissociation energies. Under acidic conditions, hydronium cations react with aliphatic OH groups (Cα-OH, Cγ-OH), and phenolic-OH groups to form protonated Cα-OH GGE, protonated Cγ-OH GGE and protonated phenolic-OH GGE intermediates, respectively. Gibbs free energies were calculated from single-point energies at the B3LYP/6-311G**/SMDwater level of theory for the formation of the protonated hydroxyl GGE cationic radicals formed through either a pre-protonation-oxidation route (Additional file 1: Figure S1) or a pre-oxidation protonation route (Additional file 1: Figure S2). The calculated Gibbs free energies suggest that oxidation of GGE and subsequent bond cleavage via the pre-protonation route required much lower energies compared with the pre-oxidation protonation route. Specifically, these data strongly suggest that the formation of protonated Cα-OH/Cγ-OH GGE intermediates is more favorable than the formation of a protonated phenolic-OH GGE intermediate (Additional file 1: Figure S1). The protonated Cα-OH GGE and protonated Cγ-OH GGE can spontaneously form under acidic conditions, while the formation of protonated phenolic-OH GGE intermediates requires either more energy or strong acids. Three kinds of protonated aliphatic-OH GGE intermediates can be formed through the pre-protonation oxidation route, and under the catalysis of the acid-stabilized LiPH8 mutant protonated hydroxyl GGE cationic radicals are formed and further cleaved into product fragments (Fig. 5).

Proposed routes to bond-cleavage through a pre-protonation mechanism and an oxidation route and their respective protonated intermediates
For each protonated hydroxyl GGE cationic radical intermediate, we carried out 5 ps of AIMD simulations and counted bond-breaking events to determine the frequency of each type of bond-breaking event as a function of protonation of GGE hydroxyl groups at different positions on GGE (Fig. 5). In the absence of protonation, only β-O-4′ ether bond cleavage was observed for the non-protonated GGE cationic radical with an 11.42% bond cleavage efficiency. As hypothesized above, protonated GGE cationic radical intermediates are formed when the solution is acidified to lower pH and a higher frequency of β-O-4′ ether bond cleavages was observed for protonated GGE intermediates at Cα-OH or Cγ-OH position. The highest β-O-4′ ether bond cleavage frequency (33.66%) was observed for the GGE cationic radical intermediate in which our calculations show protonation would have occurred at both the Cα-OH and Cγ-OH groups. This trend in β-O-4′ ether bond cleavage frequency is consistent with the experimental data reported in Fig. 2, which shows an increase in the β-O-4′ ether bond cleavage for the acid-stable LiPH8 as the pH drops from 5 to 2.6. The AIMD simulations also showed additional Cα-C1 bond-breaking (3.86%) in the protonated hydroxyl GGE intermediates, and these were not observed for AIMD simulation of inert non-protonated OH GGE cationic radical intermediates. In comparison with the experiment, the content of the product from Cα-C1 carbon bond increased from 11.9% to 27.0% when pH was shifted from 5.0 to 2.6 when using the acid-stable lignin peroxidase.
For Cα-Cβ bond cleavage, it is more energetically favorable than Cα-C1 carbon bond cleavage; however, Cα-Cβ bond cleavage products were not detected experimentally (Figs. 2 and 4). The AIMD simulations also showed higher bond-cleavage frequency for Cα-Cβ bonds than for Cα-C1 bonds (Fig. 6). This can be explained by noting the product formed from Cα-Cβ cleavage in Fig. 1 is not stable and would likely decompose rapidly to the structure obtained from β-O-4′ ether bond cleavage (Fig. 1). At the current state of the assay, we do not have a tool to quench reactions and trap these unstable products from Cα-Cβ bond cleavage; however, quantum calculations and AIMD simulation data provided an understanding of the existence of different pathways and interconversion of product intermediates.

Bond-cleavage frequency computed by AIMD simulation of non-protonated and protonated OHs GGE cationic radical for different linkages
We also found that deprotonation at Cα and Cγ occurs in the AIMD simulations of protonated Cα-OH/ Cγ-OH GGE intermediates, suggesting it may play a role in catalysis of C–C bond and β-O-4′ ether bond breaking. To further investigate this, we computed the energetic barriers for the conversion of the GGE cationic radical intermediate to products via β-O-4 ether bond cleavage events observed in the AIMD trajectory analysis. Cα-OH2 or Cγ-OH2 bond cleavage was initiated and deprotonation of Cγ-OH or Cβ-H was found in the reaction path of β-O-4′ ether bond cleavage (Additional file 1: Figure S3, S4). These structures were used as intermediates in NEB TS searches, which were computed using TeraChem at the unrestricted B3LYP/6-311G**/PCMwater level of theory (Fig. 7). Although the procedure of NEB calculation does not guarantee convergence to an exact transition state, it nonetheless provides the saddle points connecting two minima through pre-defined intermediates. It was found that the maximum relative energy for the transition state of non-protonated GGE cationic radicals was 2.41 eV. Protonation at either aliphatic Cα-OH or Cγ-OH resulted in a lower energy barrier for β-O-4′ ether bond cleavage occurrences. The lowest energy barrier of 0.36 eV was for the protonated intermediate at both Cα-OH and Cγ-OH position. A proposed mechanism for β-O-4′ ether bond cleavage from protonated Cα-OH/ Cγ-OH GGE cationic radical was suggested in Additional file 1: Figure S5, S6. In this proposed mechanism, an aliphatic hydroxyl Cα-OH/ Cγ-OH is protonated by H+ from the acid reagent, forming an alkyloxonium ion + OH2. This ion then, which is primed to acts as a good leaving group, leaves to form a carbocation at either the Cα or the Cγ position. The benzyl radical then acts as an electron-withdrawing group, which induces electron reorganization and C-O bond-cleavage. This simulation data matched with AIMD simulation where protonation at either Cα-OH or Cγ-OH resulted in higher cleavage frequencies for various bond types. Again, the combination of AIMD simulation and NEB-based TS searches matched our experimental data where protonated hydroxyl GGE intermediates were formed by acidifying reaction solution and sequentially resulted in a lower energy barrier for bond cleavages especially β-O-4′ ether, and higher conversion yield of dimer degradation.

Reaction path computed with NEB for β-O-4′ ether bond cleavage from non-protonated and protonated hydroxyl GGE cationic radical intermediates
Conclusion
We investigated the effects of pH on the activity of the lignin peroxidase isozyme H8 from P. chrysosporium using a novel nanostructure-initiator mass spectrometry assay for studying catalysis of bond breaking in a fluorous-tagged β-O-4′ phenolic lignin dimer model compound synthesized for use with this assay. Results from these assays showed increases in catalysis of β-O-4′ ether and Cα-C1 carbon bond-breaking events and little change in oxidation at the Cα position as the reaction conditions became more acidic. To better understand how lower pH enhances bond-breaking reactions in the β-O-4′ lignin model compound we ran a series of computations to examine bond cleavage via either a cationic radical intermediate or a phenoxy radical intermediate. Our calculated relative Gibbs free energy for the occurrence of bond cleavage indicated that much high energy is required for bond cleavage to occur from the phenoxy radical intermediate compared to the Gibbs free energy required when starting from the cationic radical intermediate. Data from AIMD simulations and climbing-image NEB-based transition state searches suggested that under more acidic conditions formation of protonated hydroxyl groups drives the reaction to further completion by lowering energetic barriers for bond-cleavage, especially β-O-4′ bonds. This work generates a more robust fundamental understanding of how pH assists bond cleavage and provides insights into the optimal design of reaction conditions to improve the efficiency of lignin peroxidase-catalyzed phenolic lignin degradation.
Materials and methods
Materials
Hydrogen peroxide, hemin, oxidized glutathione, ampicillin, isopropyl-β-d-thiogalactopyranoside, guanidine hydrochloride, dibasic potassium phosphate, citric acid, trizma hydrochloride, and guaiacol used in this study were purchased from the Sigma Chemical Co., U.S and were used without any further purification.
Synthesis of phenolic β-O-4 ether model compound (3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecyl (19-(4-((1,3- dihydroxy-1-(4-hydroxy-3-methoxyphenyl)propan-2-yl)oxy)-3- methoxyphenyl)-3-imino-2-methyl-9,17-dioxo-2,4,10,16-tetraazanonadecan8-yl)carbamate) followed previously reported methods [30].
Recombinant enzyme preparation
The gene coding for the wild-type lignin peroxidase LiPH8 (UniprotKB ID: P06181) and for the acid stabilized variant with three mutated residues (A55R/N156E‑H239E) was retrieved from a previously published report [23]. The expression in E. coli, refolding and purification procedures for both the wild-type enzyme and the pH stabilized enzyme were performed as previously reported [45].
Enzyme reaction with flourous-tagged phenolic β-O-4 dimeric model compound
Enzyme reaction with NIMS-tagged phenolic lignin dimer (1 mM) was performed in sodium acetate buffer pH 2. 6–5.0 in the presence of 20 mM of veratryl alcohol as mediator. 5 µM enzyme was used in this reaction and the reaction was started by adding H2O2 fed at a rate of 250 uM every 30 min up to 3 h. The reported data are the mean and standard derivations for n = 3 replicates. The first column at each pH condition in Fig. 1 is the mean and standard of the products from both β-O-4′ ether and Cα-C1 bond cleavage combined.
Nanostructure‑initiator mass spectrometry
A protocol for analyzing fluorous-tagged guaiacylglycerol-β-guaiacyl ether (GGE) using NIMS and reaction products was performed as previously described [30]. Briefly, 0.2 µL of the quenched reaction sample was spotted onto the NIMS surface and removed after 30 s. The spotting and identification of sample spots in the spectrometer were manually drawn on the NIMS chip using a diamond-tip scribe. The modified matrix-assisted laser desorption (MALDI) plate containing chip was loaded into Bruker Ultraflextreme MALDI TOF/TOF mass analyzer. Signal intensities were identified for the ions of the products and ~ 5000 laser shots were collected for each sample.
Simulation method
The Gibbs free energy of oxidation of the GGE dimer into the cationic radical intermediate and bond dissociation for all bond types in GGE was defined as the standard-state Gibbs free energy change for the reaction at a specified temperature, here at 298 °K. Simulations were performed in the Gaussian 09 software package [46] with unrestricted density functional theory (DFT), Becke three-parameter exchange and Lee–Yang–Parr correlation (B3LYP) [47, 48] using the 6-311G** basis set [49, 50] and implicit solvation model based on density (SMD) [51]. The effect of solvent was modeled using the Born-Haber Cycle as shown in the following scheme 1:

Born-Haber thermodynamic cycle for calculating the Gibbs free energy of Guaiacylglycerol-β-guaiacyl ether (GGE) dimer oxidation
Where \({G}_{ox (g)}\) is Gibbs free energy for oxidation in the gas phase at 298 °K, \({G}_{sol (I)}\) is the energy with solvent effect for the resting-state dimer; \({G}_{sol (II)}\) is the energy with solvent effect for the cationic radical dimer; \({G}_{sol (III)}\) is the energy with solvent effect for electron, which was obtained from the absolute protonation free energy of the solvated electron in water as − 35.5 kcal/mol using a reliable computational protocol of first-principles solvation-included electronic structure calculations [52].
AIMD simulations were carried out using the TeraChem quantum chemistry package (Petachem LLC, CA) [53,54,55,56]. The simulations were performed with unrestricted density functional theory (DFT) using the long-range corrected ωPBEh exchange–correlation functional [57] and the 6-31 g basis set. Simulations were run for 5 ps using a 1 fs time step. An unrestricted, radical solution, level-shifting, and a 1.0 eV and 0.0 eV shift were applied to α states and β states, respectively. TeraChem uses v6.0 of the NBO package for a full natural bond orbitals (NBO) analysis. Bond breaking events were defined as a separation distance between two atoms from fragments being longer than the corresponding bond length of initial model structure, and bond cleavage frequency was counted over 5000 AIMD time steps for the following linkages in the lignin dimer: Cα-OH bond, Cα-Cβ bond, β-O-4′ ether bond and Cα-C1 carbon bond.
The climbing-image Nudge Elastic Band (NEB) method was used to calculate the relative energy of the transition state through a reaction path defined with a set of images (initial, intermediate, and final images) taken from the analysis of AIMD simulation trajectories. The number of NEB images in the transition state search calculation was set to 100 and the min and max spring constants between NEB images were set to 0.005 and 0.05, respectively. The solvation model based on the conductor-like solvation model (COSMO) [58] was performed with a water dielectric of ε = 78.39.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Protein sequence: UniprotKB ID P06181. E. coli strains containing the genes of interest are available from the.
Joint BioEnergy Institutes strain archive. Strain JBx_137395: E. coli DH5α contains pET-21b( +) plasmid cloned with lignin peroxidase isozyme H8 wildtype gene. Strain JBx_137394: E. coli DH5α contains pET-21b( +) plasmid cloned with lignin peroxidase isozyme H8 triple mutant with the following mutations: A55R, N156E and H239E.
Abbreviations
- LiP:
-
Lignin peroxidase
- VP:
-
Versatile peroxidase
- MnP:
-
Manganese peroxidase
- LiPH8:
-
Lignin peroxidase isozyme H8
- VA:
-
Veratryl alcohol
- NIMS:
-
Nanostructure initiator mass spectrometry
- AIMD:
-
Ab initio Molecular dynamic
- GGE:
-
Guaiacylglycerol-β-guaiacyl ether
- MALDI:
-
Matrix-assisted laser desorption
- DFT:
-
Density functional theory
- SMD:
-
Solvation model based on density
- PCM:
-
Polarizable continuum model
- COSMO:
-
Conductor-like solvation model
- NEB:
-
Nudge Elastic Band
References
Zakzeski J, Bruijnincx PCA, Jongerius AL, Weckhuysen BM. The catalytic valorization of lignin for the production of renewable chemicals. Chem Rev. 2010;110(6):3552–99.
Xu Z, Lei P, Zhai R, Wen Z, Jin M. Recent advances in lignin valorization with bacterial cultures: microorganisms, metabolic pathways, and bio-products. Biotechnol Biofuels. 2019;12:32.
Baral NR, Sundstrom ER, Das L, Gladden J, Eudes A, Mortimer JC, Singer SW, Mukhopadhyay A, Scown CD. Approaches for more efficient biological conversion of lignocellulosic feedstocks to biofuels and bioproducts. ACS Sustainable Chem Eng. 2019;7(10):9062–79.
Scown CD, Gokhale AA, Willems PA, Horvath A, McKone TE. Role of lignin in reducing life-cycle carbon emissions, water use, and cost for United States cellulosic biofuels. Environ Sci Technol. 2014;48(15):8446–55.
Janusz G, Pawlik A, Sulej J, Swiderska-Burek U, Jarosz-Wilkolazka A, Paszczynski A. Lignin degradation: microorganisms, enzymes involved, genomes analysis and evolution. FEMS Microbiol Rev. 2017;41(6):941–62.
Lee S, Kang M, Bae J-H, Sohn J-H, Sung BH: Bacterial valorization of lignin: strains, enzymes, conversion pathways, biosensors, and perspectives. Front Bioeng Biotechnol 2019, 7(209).
Galkin S, Vares T, Kalsi M, Hatakka A. Production of organic acids by different white-rot fungi as detected using capillary zone electrophoresis. Biotechnol Tech. 1998;12(4):267–71.
Moreira MT, Feijoo G, Mester T, Mayorga P, Sierra-Alvarez R, Field JA. Role of organic acids in the manganese-independent biobleaching system of Bjerkandera sp. strain BOS55. Appl Environ Microbiol. 1998;64(7):2409–17.
Munir E, Yoon JJ, Tokimatsu T, Hattori T, Shimada M. A physiological role for oxalic acid biosynthesis in the wood-rotting basidiomycete Fomitopsis palustris. Proc Natl Acad Sci. 2001;98(20):11126–30.
Liaud N, Giniés C, Navarro D, Fabre N, Crapart S, Gimbert IH, Levasseur A, Raouche S, Sigoillot J-C. Exploring fungal biodiversity: organic acid production by 66 strains of filamentous fungi. Fungal Biol Biotechnol. 2014;1(1):1.
Fernandez-Fueyo E, Ruiz-Duenas FJ, Miki Y, Martinez MJ, Hammel KE, Martinez AT. Lignin-degrading peroxidases from genome of selective ligninolytic fungus Ceriporiopsis subvermispora. J Biol Chem. 2012;287(20):16903–16.
Fernández-Fueyo E, Ruiz-Dueñas FJ, Martínez MJ, Romero A, Hammel KE, Medrano FJ, Martínez AT. Ligninolytic peroxidase genes in the oyster mushroom genome: heterologous expression, molecular structure, catalytic and stability properties, and lignin-degrading ability. Biotechnol Biofuels. 2014;7(1):2.
Lundell TK, Makela MR, Hilden K. Lignin-modifying enzymes in filamentous basidiomycetes–ecological, functional and phylogenetic review. J Basic Microbiol. 2010;50(1):5–20.
Smith AT, Doyle WA, Dorlet P, Ivancich A. Spectroscopic evidence for an engineered, catalytically active Trp radical that creates the unique reactivity of lignin peroxidase. Proc Natl Acad Sci USA. 2009;106(38):16084–9.
Recabarren R, Fuenzalida-Valdivia I, Alzate-Morales J. Studying the binding mechanisms of veratryl alcohol to P. chrysosporium lignin peroxidase: insights from theoretical approaches. Theoretical Chem Accounts. 2016;135(3):71.
Goodwin DC, Aust SD, Grover TA. Evidence for veratryl alcohol as a redox mediator in lignin peroxidase-catalyzed oxidation. Biochemistry. 1995;34(15):5060–5.
Christian V, Shrivastava R, Shukla D, Modi H, Vyas BRM. Mediator role of veratryl alcohol in the lignin peroxidase-catalyzed oxidative decolorization of Remazol Brilliant Blue R. Enzyme Microb Technol. 2005;36(2):327–32.
Snook ME, Hamilton GA. Oxidation and fragmentation of some phenyl-substituted alcohols and ethers by peroxydisulfate and Fenton’s reagent. J Am Chem Soc. 1974;96(3):860–9.
Kersten PJ, Tien M, Kalyanaraman B, Kirk TK. The ligninase of Phanerochaete chrysosporium generates cation radicals from methoxybenzenes. J Biol Chem. 1985;260(5):2609–12.
Fernández-Fueyo E, Ruiz-Dueñas FJ, Martínez AT. Engineering a fungal peroxidase that degrades lignin at very acidic pH. Biotechnol Biofuels. 2014;7(1):114.
Tuisel H, Sinclair R, Bumpus JA, Ashbaugh W, Brock BJ, Aust SD. Lignin peroxidase H2 from Phanerochaete chrysosporium: purification, characterization and stability to temperature and pH. Arch Biochem Biophys. 1990;279(1):158–66.
Marquez L, Wariishi H, Dunford HB, Gold MH. Spectroscopic and kinetic properties of the oxidized intermediates of lignin peroxidase from Phanerochaete chrysosporium. J Biol Chem. 1988;263(22):10549–52.
Pham LTM, Seo H, Kim K-J, Kim YH. In silico-designed lignin peroxidase from Phanerochaete chrysosporium shows enhanced acid stability for depolymerization of lignin. Biotechnol Biofuels. 2018;11(1):325.
Sáez-Jiménez V, Fernández-Fueyo E, Medrano FJ, Romero A, Martínez AT, Ruiz-Dueñas FJ. Improving the pH-stability of versatile peroxidase by comparative structural analysis with a naturally-stable manganese peroxidase. PLoS ONE. 2015;10(10):e0140984–e0140984.
Sollewijn Gelpke MD, Lee J, Gold MH. Lignin peroxidase oxidation of veratryl alcohol: effects of the mutants H82A, Q222A, W171A, and F267L. Biochemistry. 2002;41(10):3498–506.
Wariishi H, Huang J, Dunford HB, Gold MH. Reactions of lignin peroxidase compounds I and II with veratryl alcohol. Transient-state kinetic characterization. J Biol Chem. 1991;266(31):20694–9.
Khindaria A, Yamazaki I, Aust SD. Veratryl alcohol oxidation by lignin peroxidase. Biochemistry. 1995;34(51):16860–9.
Baciocchi E, Fabbri C, Lanzalunga O. Lignin peroxidase-catalyzed oxidation of nonphenolic trimeric lignin model compounds: fragmentation reactions in the intermediate radical cations. J Org Chem. 2003;68(23):9061–9.
Miki K, Renganathan V, Gold MH. Mechanism of beta-aryl ether dimeric lignin model compound oxidation by lignin peroxidase by Phanerochaete chrysosporium. Biochemistry. 1986;25(17):4790–6.
Deng K, Zeng J, Cheng G, Gao J, Sale KL, Simmons BA, Singh AK, Adams PD, Northen TR. Rapid characterization of the activities of lignin-modifying enzymes based on nanostructure-initiator mass spectrometry (NIMS). Biotechnol Biofuels. 2018;11:266.
Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Ralph SA, Christensen JH, et al. Lignins: Natural polymers from oxidative coupling of 4-hydroxyphenyl-propanoids. Phytochem Rev. 2004;3(1):29–60.
Adler E. Lignin chemistry—past, present and future. Wood Sci Technol. 1977;11(3):169–218.
Johjima T, Itoh N, Kabuto M, Tokimura F, Nakagawa T, Wariishi H, Tanaka H. Direct interaction of lignin and lignin peroxidase from Phanerochaete chrysosporium. Proc Natl Acad Sci. 1999;96(5):1989–94.
Pham LT, Kim SJ, Kim YH. Improvement of catalytic performance of lignin peroxidase for the enhanced degradation of lignocellulose biomass based on the imbedded electron-relay in long-range electron transfer route. Biotechnol Biofuels. 2016;9:247–57.
Martínez ÁT, Ruiz-Dueñas FJ, Martínez MJ, del Río JC, Gutiérrez A. Enzymatic delignification of plant cell wall: from nature to mill. Curr Opin Biotechnol. 2009;20(3):348–57.
Gonzalez-Perez D, Mateljak I, Garcia-Ruiz E, Ruiz-Dueñas FJ, Martinez AT, Alcalde M. Alkaline versatile peroxidase by directed evolution. Catal Sci Technol. 2016;6(17):6625–36.
Ayuso-Fernandez I, De Lacey AL, Canada FJ, Ruiz-Duenas FJ, Martinez AT. Increase of redox potential during the evolution of enzymes degrading recalcitrant lignin. Chemistry. 2019;25(11):2708–12.
Castro L, Crawford L, Mutengwa A, Gotze JP, Buhl M. Insights into structure and redox potential of lignin peroxidase from QM/MM calculations. Org Biomol Chem. 2016;14(8):2385–9.
Huang J, Zhang L. Effects of NCO/OH molar ratio on structure and properties of graft-interpenetrating polymer networks from polyurethane and nitrolignin. Polymer. 2002;43(8):2287–94.
Garbern JC, Hoffman AS, Stayton PS. Injectable pH- and temperature-responsive Poly(N-isopropylacrylamide-co-propylacrylic acid) copolymers for delivery of angiogenic growth factors. Biomacromol. 2010;11(7):1833–9.
Sivasankarapillai G, McDonald AG. Synthesis and properties of lignin-highly branched poly (ester-amine) polymeric systems. Biomass Bioenerg. 2011;35(2):919–31.
Luo N, Wang M, Li H, Zhang J, Hou T, Chen H, Zhang X, Lu J, Wang F. Visible-light-driven self-hydrogen transfer hydrogenolysis of lignin models and extracts into phenolic products. ACS Catal. 2017;7(7):4571–80.
Rahimi A, Ulbrich A, Coon JJ, Stahl SS. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature. 2014;515(7526):249–52.
Shuai L, Amiri MT, Questell-Santiago YM, Heroguel F, Li Y, Kim H, Meilan R, Chapple C, Ralph J, Luterbacher JS. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science. 2016;354(6310):329–33.
Doyle WA, Smith AT. Expression of lignin peroxidase H8 in Escherichia coli: folding and activation of the recombinant enzyme with Ca2+ and haem. Biochem J. 1996;315(Pt 1):15–9.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H et al: Gaussian 09, Revision A.02. In. Wallingford CT; 2016.
Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Physics. 1993;98(7):5648–52.
Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter. 1988;37(2):785–9.
Davidson ER, Feller D. Basis set selection for molecular calculations. Chem Rev. 1986;86(4):681–96.
Wiberg KB: Ab Initio Molecular Orbital Theory by W. J. Hehre, L. Radom, P. v. R. Schleyer, and J. A. Pople, John Wiley, New York, 548pp. Price: $79.95 (1986). Journal of Computational Chemistry 1986, 7(3):379–379.
Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B. 2009;113(18):6378–96.
Zhan C-G, Dixon DA. The nature and absolute hydration free energy of the solvated electron in water. J Phys Chem B. 2003;107(18):4403–17.
Ufimtsev IS, Martinez TJ. Quantum chemistry on graphical processing units. 3. analytical energy gradients, geometry optimization, and first principles molecular dynamics. J Chem Theory Comput. 2009;5(10):2619–28.
Ufimtsev IS, Martinez TJ. Quantum chemistry on graphical processing units. 2. direct self-consistent-field implementation. J Chem Theory Comput. 2009;5(4):1004–15.
Ufimtsev IS, Martínez TJ. Quantum chemistry on graphical processing units. 1. strategies for two-electron integral evaluation. J Chem Theory Comput. 2008;4(2):222–31.
Luehr N, Jin AG, Martinez TJ. Ab initio interactive molecular dynamics on graphical processing units (GPUs). J Chem Theory Comput. 2015;11(10):4536–44.
Ernzerhof M, Scuseria GE. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J Chem Phys. 1999;110(11):5029–36.
Klamt A. Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena. J Phys Chem. 1995;99(7):2224–35.
Acknowledgements
This work conducted by the Joint BioEnergy Institute was supported by the Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC, a wholly-owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525.
Funding
Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
LTMP and KLS conceived the study. LTMP designed and performed experiments and simulations. KD analyzed mass spectrometry. LTMP and KLS wrote the manuscript. TRN, SWS, BAS, and PDA discussed, revised, and checked the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing financial interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Relative Gibbs free energy for the formation of protonated intermediates at phenolic OH (blue bar), Cα-OH (green bar) and Cγ-OH (orange bar) positions through a pre-protonation – oxidation route. Gibbs Free energy was normalized to reactants on each reaction step. Figure S2. Relative Gibbs free energy for the formation of protonated intermediates at phenolic OH (blue bar), Cα-OH (green bar) and Cγ-OH (orange bar) positions through a pre-oxidation – protonation route. Gibbs Free energy was normalized to reactants on each reaction step. Figure S3. Snapshots of intermediates from AIMD simulation of the protonated Cα-OH, GGE cationic radical for β-O-4′ ether bond cleavage. Figure S4. Snapshots of intermediates from AIMD simulation of protonated Cγ-OH, GGE cationic radical for β-O-4′ ether bond cleavage. Figure S5. Proposed mechanism for β-O-4′ ether bond cleavage from protonated Cα-OH, GGE cationic radical. Figure S6. Proposed mechanism for β-O-4′ ether bond cleavage from protonated Cγ-OH, GGE cationic radical.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pham, L., Deng, K., Northen, T.R. et al. Experimental and theoretical insights into the effects of pH on catalysis of bond-cleavage by the lignin peroxidase isozyme H8 from Phanerochaete chrysosporium. Biotechnol Biofuels 14, 108 (2021). https://doi.org/10.1186/s13068-021-01953-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-021-01953-7