
Abstract
Background
Methane is the primary component of natural gas and biogas. The huge abundance of methane makes it a promising alternative carbon source for industrial biotechnology. Herein, we report diamine compound, putrescine, production from methane by an industrially promising methanotroph Methylomicrobium alcaliphilum 20Z.
Results
We conducted adaptive evolution to improve putrescine tolerance of M. alcaliphilum 20Z because putrescine highly inhibits the cell growth. The evolved strain 20ZE was able to grow in the presence of 400 mM of putrescine dihydrochloride. The expression of linear pathway ornithine decarboxylase genes from Escherichia coli and Methylosinus trichosporium OB3b allowed the engineered strain to produce putrescine. A higher putrescine titer of 12.44 mg/L was obtained in the strain 20ZE-pACO with ornithine decarboxylase from M. trichosporium OB3b. For elimination of the putrescine utilization pathway, spermidine synthase (MEALZ_3408) was knocked out, resulting in no spermidine formation in the strain 20ZES1-pACO with a putrescine titer of 18.43 mg/L. Next, a genome-scale metabolic model was applied to identify gene knockout strategies. Acetate kinase (MEALZ_2853) and subsequently lactate dehydrogenase (MEALZ_0534) were selected as knockout targets, and the deletion of these genes resulted in an improvement of the putrescine titer to 26.69 mg/L. Furthermore, the putrescine titer was improved to 39.04 mg/L by overexpression of key genes in the ornithine biosynthesis pathway under control of the pTac promoter. Finally, suitable nitrogen sources for growth of M. alcaliphilum 20Z and putrescine production were optimized with the supplement of 2 mM ammonium chloride to nitrate mineral salt medium, and this led to the production of 98.08 mg/L putrescine, almost eightfold higher than that from the initial strain. Transcriptome analysis of the engineered strains showed upregulation of most genes involved in methane assimilation, citric acid cycle, and ammonia assimilation in ammonia nitrate mineral salt medium, compared to nitrate mineral salt medium.
Conclusions
The engineered M. alcaliphilum 20ZE4-pACO strain was able to produce putrescine up to 98.08 mg/L, almost eightfold higher than the initial strain. This study represents the bioconversion of methane to putrescine—a high value-added diamine compound.
Similar content being viewed by others


Background
Putrescine (1,4-diaminobutane) is a four-carbon diamine found in a wide range of organisms because it is necessary for cell growth and proliferation [1]. Putrescine has many applications in pharmaceuticals, agrochemicals, and surfactants. In chemical industry, putrescine is a chemical platform monomer used for the synthesis of high-performance bioplastic nylon-4,6 that combines the benefits of a high melting point and excellent chemical resistance [2]. There is a high demand for putrescine (about 10,000 tons per year in Europe), and this demand is predicted to increase [3]; thus, putrescine production has received significant attention. There are significant environmental and economic issues associated with the chemical synthesis routes, which requires petrochemical-based raw materials, harsh conditions, and expensive catalyst systems [4, 5]. Therefore, the development of a biotechnological process for the production of putrescine has become attractive.
To date, putrescine has been successfully produced from sugar by metabolic engineering of Escherichia coli and Corynebacterium glutamicum. Qian et al. reported a metabolically engineered E. coli K12 W3110 that produces putrescine in a glucose minimal medium [6]. The ornithine pool was enhanced by overexpression of the ornithine biosynthesis pathway and deletion of the putrescine degradation and utilization pathway. In addition, the activity of a compete pathway conversion of ornithine to arginine was reduced and ornithine decarboxylase was overexpressed. The final strain was able to accumulate 1.68 g/L of putrescine with a yield of 0.166 g/g glucose in a shake flask culture and 24.2 g/L with a productivity of 0.75 g/L h in fed-batch fermentation [6]. Metabolic engineering of C. glutamicum for putrescine production has also been reported with a yield from glucose of 0.26 g/g in a flask culture [7] and 0.166 g/g in a fed-batch fermentation [8, 9].
Methane and methanol are one-carbon (C1) substrates that have shown great potential as alternative substrates for biomanufacturing of chemicals and fuels [4, 10, 11]. Utilization of C1 substrates can reduce greenhouse gases and circumvent the social issue of using sugar for making chemicals and fuels. Methane is the cheapest carbon source based on the price per carbon. Thus, there is an increasing demand to convert methane to high value-added products using engineered strains. In recent years, methanotrophic bacteria have become favorable platform strains for industrial biotechnology. A methane- and methanol-utilizing strain, Methylomicrobium alcaliphilum 20Z, which can utilize C1 substrates as a sole carbon and energy source [12], has become an attractive model strain due to its advantages: whole genome sequenced, active in various physicochemical conditions (pH, temperature, salinity), and existing genetic tools for genetic manipulation (gene transfer, gene knockout, and replicable vector) [13].
Recently, two genome-based metabolic models of methane oxidation strains have been published [14, 15]. The genome-scale metabolic models with reconstruction of C1-carbon utilization pathway in M. buryatense 5GB1 and M. alcaliphilum 20Z provide a useful tool to obtain engineering strategies for desired products. Computational strain design has successfully improved 2,3-butandiol production using the genome-scale metabolic model of M. alcaliphilum 20Z [16]. A systematic approach for putrescine production has also been reported for C. glutamicum [17]. However, there is no report on metabolic engineering of methanotrophs using a genome-based metabolic model for methane bioconversion for putrescine production where the effect of nitrogen needs to be considered.
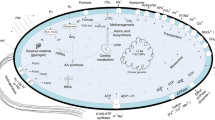
In this study, we report a development of engineered M. alcaliphilum 20Z strains for the production of putrescine from methane. The engineered M. alcaliphilum 20Z strain with inactive putrescine utilization pathway and expression of ornithine decarboxylase converted methane to putrescine. Furthermore, engineering strategies were generated by using the genome-scale metabolic model, which allowed further improvement of putrescine production from methane (Fig. 1).

Metabolic engineering of M. alcaliphilum 20Z for putrescine production. Red arrows indicate expression reactions. Green arrows indicate overexpression reactions. Yellow X indicates gene deletion. ack, acetate kinase; argA, N-acetylglutamate synthase; argB, N-acetylglutamate kinase; argC, N-acetylglutamylphosphate reductase; argD, acetylornithine aminotransferase; argJ, ornithine acetyltransferase; ldh, lactate dehydrogenase; potE, putrescine transporter; speC, constitutive ornithine decarboxylase; speE, spermidine synthase; speF, inducible ornithine decarboxylase; Agm, agmatine; Glut, glutamate; l-Arg, l-arginine; l-Orn, l-ornithine; N-Acglu, N-acetylglutamate; N-AcGluAld, N-acetylglutamate semialdehyde; N-AcGluP, N-acetylglutamylphosphate; N-AcOrn, N-acetylornithine; N-CbmP, N-carbamoylputrescine
Results
Analysis of putrescine toxicity on M. alcaliphilum 20Z
In general, putrescine is toxic to microorganisms [18]. Therefore, the impacts of extracellular putrescine inhibition on M. alcaliphilum 20Z growth was first examined. M. alcaliphilum 20Z was cultured to an exponential phase and exposed to different putrescine concentrations of 50 mM, 100 mM, 200 mM, and 400 mM (Fig. 2). M. alcaliphilum 20Z showed significantly reduced growth rates in the presence of 50 mM and higher putrescine concentrations. Approximately, 50 mM of putrescine dihydrochloride (equivalent to 4.4 g/L putrescine) caused cell lysis. Based on this observation, we concluded that low putrescine tolerance could be a main barrier for metabolic engineering of M. alcaliphilum 20Z for putrescine production.

Putrescine tolerance of M. alcaliphilum 20Z. Cells were grown at 30 °C in an NMS medium and exposed to various concentration of putrescine dihydrochloride in the exponential growth phase (OD600 of 0.4)
Enhancement of putrescine tolerance of M. alcaliphilum 20Z by adaptive laboratory evolution
Adaptive evolution to improve putrescine tolerance of M. alcaliphilum 20Z was first conducted to overcome low putrescine tolerance of M. alcaliphilum 20Z. Cells exposed to 100 mM of putrescine dihydrochloride in the tolerance test were incubated in NMS with 0.2% methanol at 30 °C, 230 rpm for 15 days. Then, 2 mL of culture broth were transferred to fresh NMS medium with 0.2% methanol to recover. A long lag phase of 3 days was observed, and active cells were transferred to fresh NMS medium. Cells were grown to an OD600 of 0.4 and exposed to 100 mM of putrescine dihydrochloride for 7 days. After 5 replication cycles (approximately more than 65 days in total), a strain with a high tolerance to putrescine was obtained (referred to as the 20ZE strain). The evolved putrescine-tolerant 20ZE strain was able to grow in the presence of 400 mM putrescine dihydrochloride. The growth rate of the evolved strain was comparable to that of wild-type strain (0.089 h−1), and the evolved strain showed even slightly better growth rate in the presence of 100 and 200 mM putrescine (0.121 h−1 and 0.111 h−1, respectively) (Fig. 3). Extension of putrescine tolerance on M. alcaliphilum 20Z allowed the evolved strain to be employed for further metabolic engineering for putrescine production from methane.

Improvement of putrescine tolerance of M. alcaliphilum 20Z by adaptive evolution. Filled circles show the wild-type strain M. alcaliphilum 20Z grown in NMS. The open circles show the evolved strain in NMS, while the filled triangles show the evolved strain exposed to 100 mM of putrescine dihydrochloride. The open triangles show the evolved strain exposed to 200 mM of putrescine dihydrochloride, and the filled squares show the evolved strain with 400 mM of putrescine dihydrochloride
Construction of a platform host strain for putrescine production from methane and methanol
Putrescine can be synthesized in vivo via ornithine by constitutive ornithine decarboxylase (speC) or inducible ornithine decarboxylase (speF) and arginine by arginine decarboxylase and agmatinase. M. alcaliphilum 20Z possesses alternative agmatine deiminase, which hydrolyzes agmatine to ammonia and N-carbamoylputrescine. The latter is subsequently hydrolyzed to putrescine, ammonia, and carbon dioxide by N-carbamoylputrescine amidohydrolase [9]. In addition, M. alcaliphilum 20Z also possesses a putrescine utilization pathway, which converts putrescine to spermidine by spermidine synthase (speE). There are five genes predicted as spermidine synthase, of which three genes have lengths of about 516 (MEALZ_1869), 825 (MEALZ_1164), and 856 (MEALZ_3010) amino acids, respectively. The two other genes are very similar to spermidine synthases of E. coli, which have protein lengths of 282 (speE1-MEALZ_3408) and 267 (speE2-MEALZ_3304) amino acids, respectively. Therefore, we tried to inactivate these two genes with similar protein lengths of E. coli spermidine synthase, and test whether putrescine could be accumulated in the mutant M. alcaliphilum 20Z strain. The spermidine synthase gene (speE1) was knocked out through unmarked allelic exchange using a sucrose counter-selection system [19]. Vector pCM433 was constructed with two flanking homology regions upstream and downstream of MEALZ_3408, resulting in vector pCSE1. Vector pCSE1 was introduced into strain 20ZE by electroporation to be integrated into the genome of 20ZE by single crossover. The single-crossover recombinants were transferred to fresh NMS medium containing 2.5% (w/v) of sucrose and were selected for the mutants that excised the vector by PCR using the primer outside the flanking regions. The speE1 mutant was obtained as strain 20ZES1, which accumulated 0.35 mg/L of putrescine after 96 h cultivation. The spermidine concentration in the culture broth was also analyzed, and there was no spermidine accumulation. This means that the knockout of the spermidine synthase (MEALZ_3408) blocked the conversion of putrescine to spermidine. Spermidine and other polyamines are involved in critical physiological processes in bacteria such as cell growth, biofilm formation, stress response, and proliferation [20], but, interestingly, spermidine was proved to be not essential for the growth of E. coli and Saccharomyces cerevisiae [21, 22]. Obviously, inactivation of putrescine degradation and utilization pathway in E. coli resulted in a higher putrescine titer [6]. Similarly, inactivation of the putrescine utilization pathway allowed putrescine to be accumulated in the engineered strain 20ZES1.
To increase putrescine production in the M. alcaliphilum 20Z, ornithine decarboxylase was expressed to directly convert ornithine to putrescine. Constitutive ornithine decarboxylase (speCEc) and a native putrescine transporter (potE) from E. coli K12 were amplified and cloned into an IncP-based broad host-range vector pAWP89 under control of a pTac promoter, resulting in a pACE vector. An ornithine decarboxylase (speCOb) from M. trichosporium OB3b, a type II methanotroph, was also cloned into the pAWP89 vector, resulting in vector pACO. Moreover, the codon adaptation index (CAI) of those genes (for predicting gene expression level) was calculated based on a M. alcaliphilum 20Z codon usage table. The CAI values of speCEc and speCOb were relatively high at 0.73 and 0.78, respectively. Vectors pACE and pACO were successfully transformed into wild-type 20Z by electroporation, resulting in strains 20Z-pACE and 20Z-pACO, respectively. Recombinant strains were cultured in a shake flask containing 50 mL of NMS medium with 50% (v/v) methane. Putrescine accumulation in the supernatant was analyzed by HPLC as described above. After 144 h of incubation, recombinant strain 20Z- pACE produced 2.27 ± 0.42 mg/L of putrescine, while the 20Z-pACO strain produced 12.44 ± 0.86 mg/L of putrescine, approximately five times higher. Enzyme ornithine decarboxylase (speCOb) from M. trichosporium OB3b showed high activity even with an alkali pH in the culture medium. Vector pACO would be used for further putrescine production experiments.
The methylated vector pACO harvested from 20Z strains was easily transformed to strain 20ZES1 with high efficiency, resulting in strain 20ZES1-pACO. Interestingly, strain 20ZES1 produced 18.43 ± 1.08 mg/L putrescine in shake flasks after 144 h (Fig. 4). This production increased by 32.5% compared to the wild-type strain harboring vector pACO.

a Growth of wild-type and engineered strain M. alcaliphilum 20Z. b Putrescine accumulation of engineered strain M. alcaliphilum 20Z with spermidine synthase knockout and strain harboring pACE and pACO vector after 144 h of cultivation. The data are represented as the mean ± SD (n = 3)
Metabolic engineering strategy for enhancing putrescine production based on in silico simulation
Genome-scale metabolic model can be employed to identify gene knockout strategies to optimize the metabolic pathway for the target product. Genome-scale metabolic models have successfully been applied to M. alcaliphilum 20Z [14, 16]. OptGene is computationally efficient, and it has been successfully applied (i) in Saccharomyces cerevisiae to improve succinate production by 30-fold [23], (ii) in Synechocystis to allow for growth-coupled biofuel production [24], and (iii) in M. alcaliphilum 20Z for improving the production of 2,3-butandiol [16]. OptGene was performed to identify gene deletion strategies to improve the biomass production-coupled yield (BPCY) as well as the yield of putrescine. In the OptGene algorithm, reaction knockouts were randomly introduced to obtain a mutant population [25]. The objective function was calculated using minimization of metabolic adjustment (MOMA), which identified the closest flux point to the wild-type point making it compatible with the gene deletion constraint [26].
Maximization of the specific growth rate and secretion of putrescine was used as the objective function with flux balance analysis (FBA) to predict the phenotype of the wild-type strain. The maximum theoretical yield and maximum productivity of putrescine production from methane were computed to be 0.579 g/g methane and 0.937 mmol/gDCW/h. Putrescine production is not favorable at the optimal biomass growth rate, as indicated by the FBA using maximization of the specific growth rate as the objective function. This resulted in no putrescine accumulation.
An in silico evolutionary programming-based method was performed using OptGene to optimize putrescine production. OptGene-derived mutations that increased BPCY for putrescine production were generated and are listed in Additional file 1: Table S1. The most common knockouts for putrescine production were acetate kinase (ACKr), serine hydroxymethyltransferase (glyA), and methylenetetrahydrofolate dehydrogenase (MTHFD). GlyA and MTHFD are two genes that evolved in the tetrahydromethanopterin (H4MPT) pathway and serine cycle. Through this pathway, formaldehyde was oxidized to formate, which played a role in formaldehyde detoxification and provided NADH for methane oxidation [27]. Knockout of these two genes was not favorable for strain growth in methane. Moreover, the predicted flux value via the H4MPT pathway was small, i.e., 0.1 compared with 11.6 via the RuMP pathway in M. alcaliphilum 20Z [14]. Knockout of ACKr gave a BPCY of 0.0027 with the highest productivity of 0.66 mmol/gDCW/h of putrescine and a yield of 0.408 g-(putrescine)/g-CH4, which is approximately 70% of the theoretical yield. The maximum specific growth rate was predicted as 0.032 h−1. Thus, it was selected as a knockout target. Subsequently, lactate dehydrogenase (LDH) was also selected as a promising target to enhance putrescine production. With the knockout mutant of ACKr, the flux redistribution toward putrescine formation from acetyl-CoA can be improved, because of the decrease in the flux from acetyl-CoA to acetate.
A triple mutant strain was successfully generated using sucrose counter-selection, resulting in strain 20ZE3. Putrescine production by the mutant strain was examined through the transformation of vector pACO to construct strain 20ZE3-pACO. Strain 20ZE3-pACO was able to accumulate 26.69 ± 1.86 mg/L of putrescine after 144 h of cultivation, which is a 37.03% improvement compared to the 20ZES1-pACO strain (Fig. 5).

Putrescine improvement in engineered strains. The data are represented as the mean ± SD (n = 3)
Furthermore, improving the ornithine pool (i.e., the direct precursor of putrescine) would be necessary to improve the putrescine titer. Ornithine was synthesized from glutamate by five sequential catalytic reactions catalyzed by N-acetylglutamate synthase (encoded by argA), acetylglutamate kinase (argB), N-acetylglutamate semialdehyde dehydrogenase (argC), and N-acetylornithine transaminase (argD) to yield N-acetylornithine. Ornithine was formed in a recycling pathway by bifunctional ornithine acetyltransferase (argJ), which catalyzed N-acetylglutamate from glutamate, and ornithine by transacetylation between N-acetylornithine and glutamate. M. alcaliphilum 20Z possesses the recycling pathway, which was recognized as an efficiency pathway for production of an ornithine-derived product such as putrescine; the recycling pathway was an efficient pathway compared to the linear pathway catalyzed by acetylornithine deacetylase (argE) found in E. coli, type II methanotrophs, and other species, which converted acetylornithine to ornithine and generated acetate as an intermediate product [28]. These genes are located in five different loci. Plasmid-based overexpression of argCJBDFRGH or argJ alone in C. crenatum SYPA 5-5 also led to enhanced arginine production, the downstream product of ornithine [29]. Overexpression of argJ increases flux from glutamate toward an ornithine biosynthesis pathway and efficient conversion of acetylornithine to ornithine.
Protein–protein association networks with speC were also investigated [30]. The interaction of argD with speC was found in a characterized methylotrophic strain, Methylobacterium extorquens AM1 [31]. Thus, argD and argJ, two native genes in M. alcaliphilum 20Z, were selected as targets for overexpression to improve the ornithine pool. N-acetylornithine transaminase (argD) and ornithine acetyltransferase (argJ) were cloned into pAWP89 under the control of a pTac promoter. Two flanking regions in the genome of M. alcaliphilum 20Z were constructed into vector pCM351 along with the pTac-argDJ fragment. Flank F2 was inserted into the SacI site, resulting in vector pCM351-F2. Flank F1 and pTac-argDJ fragments were inserted in the EcoRI and NotI sites to generate vector pCAR2. Vector pCAR2 was introduced into 20ZE3, and a double crossover allelic exchanged mutant 20ZE4A with the genotype ΔldhΔackΔspeE1::argDJ was successfully obtained. In these strains, putrescine production was improved by 21.08% and 51.57% compared to 20ZE3-pACO and 20ZES1-pACO, respectively. A maximized titer of 39.04 ± 1.35 mg/L (approximately three times higher than 20Z-pACO) was obtained (Fig. 5).
Enhanced production of putrescine with nitrogen source optimization
Although ammonia was considered to be the best inorganic nitrogen source for E. coli because it provided the fastest growth rate [32], it is not widely used for culturing methanotrophs. Currently, nitrate is the main nitrogen source for culturing methanotrophs. Nitrate is subsequently reduced to nitrite and finally to ammonia by nitrate reductase and nitrite reductase [33]. Ammonia was then assimilated by glutamate dehydrogenase (GDH) or the glutamine synthetase/glutamate synthase (GS/GOGAT) pathway. Nitrate assimilation is an energetically expensive process, which is not favorable for growth of bacteria and for glutamate-derived products. In addition, the GDH pathway was stimulated in the presence of ammonia, leading to direct assimilation of ammonia to glutamate. Therefore, we investigated the effect of different nitrogen sources on the growth of M. alcaliphilum 20Z and putrescine production from methane.
Wild-type and the engineered strains 20ZE4A-pACO were grown in nitrate mineral salt medium (NMS) with 10 mM of potassium nitrate and in an ammonium mineral salt medium (AMS) with various concentrations of ammonium chloride: 1 mM, 2 mM, 5 mM, 10 mM, and 20 mM. Unfortunately, no growth of M. alcaliphilum 20Z was observed in AMS media at any concentration of ammonium chloride. Due to the lack of substrate specificity, methane monooxygenases oxidized ammonia to hydroxylamine, which led to an incompatibility between methane oxidation and ammonia oxidation. In addition, ammonia has potential toxicity to microorganism under high pH condition [34]. Cultivation of wild-type and engineered strains in lower pH was also conducted. The wild-type and engineered strains were slowly grown in NMS at neutral pH, but they did not grow in AMS at any pH (data not shown). Therefore, ammonia toxicity is mainly caused by the incompatibility of ammonia oxidation and methane oxidation and toxicity of hydroxylamine—an ammonia oxidation product [35, 36]. Ammonia is not a suitable nitrogen source for growth in methane. However, an appropriate amount of ammonium chloride supplement during growth in NMS can enhance putrescine production. Growth of M. alcaliphilum 20Z in ammonia nitrate mineral salt medium (ANMS) with various concentrations of ammonium chloride was also examined.
The performance of wild-type and engineered strains 20ZE4A-pACO in ANMS with various concentrations of ammonium chloride (1 mM, 2 mM, 5 mM of NH4Cl) was examined. With the supplement of 1 mM ammonium chloride, the wild-type and the engineered strains 20ZE4A-pACO showed higher growth rate in comparison with growth in NMS. With the supplement of 2 mM ammonium chloride, the growth of wild-type and engineered strains showed longer lag phase in the first 48 h of growth. However, after 96 h, the engineered strains reached 1.12-fold higher optical density than wild type, and the engineered strain reached highest optical density (OD600 = 4.0) compared to that of wild type (OD600 = 2.5) in flask culture (Fig. 6a). However, no growth of either wild-type or the engineered strain was observed with 5 mM of ammonium chloride.

a Growth of M. alcaliphilum 20Z and engineered strains in NMS and ANMS media. b Putrescine production by the engineered strains 20ZE4A-pACO in NMS and ANMS with different concentrations of ammonium chloride over time
After 268 h of cultivation, the engineered strain 20ZE4A-pACO produced 39.14 ± 1.35 mg/L of putrescine in an NMS medium. On the other hand, this engineered strain was able to produce 59.46 ± 0.92 and 98.08 ± 2.86 mg/L of putrescine in ANMS with 1 mM and 2 mM of ammonium chloride, respectively (Fig. 6b). In ANMS with 2 mM of ammonium chloride, the highest titer of 98.08 mg/L putrescine with a productivity of 2.9 nmol/gDCW/h and a yield of 0.0276 g-(putrescine)/g-CH4 was obtained. This was approximately 2.5 times higher than that obtained in NMS and is the highest titer of putrescine production from methane.
Analysis of gene expression level in response to change of nitrogen sources
For further understanding how gene expression is regulated in response to the change of nitrogen sources, transcriptome shotgun sequencing (RNA-seq) of M. alcaliphilum 20Z strain 20ZE4A-pACO cultured in ANMS and NMS was analyzed. Different expression levels of genes involved in ammonia assimilation, TCA cycle (Fig. 7a), and central metabolic pathway (Fig. 7b) were estimated as logarithm base twofold change (log2FoldChange) of gene expression level in NMS versus ANMS. Log2FoldChange < 0 of the given gene means upregulation of this gene in ANMS compared to NMS.

Different gene expression of genes involves in a TCA cycle and ammonia assimilation pathway and b central metabolic pathway, estimated as log2FoldChange of NMS vs ANMS (black bar)
Among genes involved in ammonia assimilation in ANMS, only GOGAT was upregulated, while both GDH and GS were downregulated. Genes involved in ornithine biosynthesis pathway, argC, argD, and argJ, were highly upregulated in ANMS, especially, argJ was strongly upregulated with five-fold changes. Genes of the upstream of α-ketoglutarate dehydrogenase in TCA cycle were highly upregulated in ANMS, and only slight changes were observed in the downstream genes with some downregulation. Genes in the EMP pathway and methane assimilation were mostly upregulated in ANMS. Interestingly, methanol dehydrogenase large subunit mxaF and a downstream gene mxaJ were 19-fold upregulated in ANMS compared to NMS. Genes in methane assimilation, upstream reactions of TCA cycle, ammonia assimilation (GOGAT)n and ornithine biosynthesis pathway were significantly upregulated in ANMS. It can be inferred that small amount of ammonia, which reduced the incompatible inhibition of methane oxidation and ammonia oxidation, could be directly assimilated into glutamate. The saved energy consumption due to the decrease of nitrate assimilation might enhance the methane assimilation, together with 19-fold upregulation of methanol dehydrogenase large subunit in ANMS compared to NMS. As a result, a high titer of putrescine along with high growth rate and cell density was obtained in the ANMS medium.
Discussion
Methanotrophs with the ability of using methane as the sole carbon and energy source are becoming promising strains for methane bioconversion. M. alcaliphilum 20Z, a haloalkali-tolerant methanotroph, is an industrially promising biocatalyst for the conversion of methane to value-added products due to a well-characterized central metabolic pathway, rapid growth rates, high cell density cultivation, and genetic tools [37]. In addition, M. alcaliphilum 20Z possesses an efficient recycling pathway for the production of ornithine-derived product, compared to the linear ornithine biosynthesis pathway of type II methanotrophs, which forms acetate as the bypass product.
In this study, we employed M. alcaliphilum 20Z for the conversion of methane to an important industrial chemical, putrescine. However, compared to engineered E. coli, M. alcaliphilum 20Z was very sensitive to putrescine. E. coli can grow well in the presence of 250 mM putrescine dihydrochloride (22 g/L putrescine), and the growth rate significantly decreased in the presence of 500 mM putrescine dihydrochloride (44 g/L putrescine) [6]. The effect of putrescine on the growth of C. glutamicum was also reported. C. glutamicum was able to grow in the presence of up to 500 mM putrescine dihydrochloride, but the growth rate was reduced (34%) at 740 mM putrescine dihydrochloride [9]. Therefore, we have conducted the adaptive evolution to enhance putrescine tolerance of M. alcaliphilum 20Z. The evolved strain was able to grow in the presence of 400 mM putrescine dihydrochloride.
We constructed an IncP-based broad host-range plasmid for expression of ornithine decarboxylase from a well-known source, E. coli, and from a type II methanotroph, M. trichosporium OB3b, driven by a tac promoter. The expression of ornithine decarboxylase from M. trichosporium OB3b showed higher activities at an alkali pH in a cultivation medium of M. alcaliphilum 20Z.
In addition, we inactivated the putrescine utilization pathway to accumulate putrescine in M. alcaliphilum 20Z. M. alcaliphilum 20Z possesses five genes predicted to be associated with spermidine synthase. However, the knockout of one spermidine gene MEALZ_3408 completely blocked spermidine formation in the mutant strain (data not shown). Gene MEALZ_3304 could be associated with spermine synthase, which converts spermidine to spermine. We also found that a small amount of putrescine accumulation in the culture medium promoted the growth of M. alcaliphilum 20Z.
A genome-scale metabolic model of M. alcaliphilum 20Z and its application have been recently published [14]. By employing the genome-scale metabolic model, we present genetic engineering strategies to obtain the engineered M. alcaliphilum 20Z strains where putrescine production increased or growth was coupled. The ACKr gene was identified as a potential target for increase in putrescine production. With ACKr knockout, strain 20ZE3-pACO was able to produce 26.69 ± 1.86 mg/L of putrescine. For further improvement of putrescine production, we introduced key genes in the ornithine biosynthesis pathway-argDJ by integration. These genes were driven by tac promoter into the genome of M. alcaliphilum 20Z, which led to an improvement of 21.08% with a maximized titer of 39.04 ± 1.35 mg/L.
We further examined the effects of different nitrogen sources on the growth and putrescine production of the wild-type and engineered M. alcaliphilum 20ZE4A-pACO strain. Generally, M. alcaliphilum 20Z has been cultured with nitrate as the sole nitrogen source. Due to the incompatibility of ammonia oxidation and methane oxidation, methane monooxygenases (which lack substrate specificity) oxidized ammonia to hydroxylamine that inhibits methane oxidation [35, 36]. Nitrate assimilation is an energetically expensive process. However, the assimilation of ammonia via glutamate dehydrogenase, which was employed in energy-limited environment, could be favorable for a glutamate-derived product like putrescine. M. alcaliphilum 20Z was unable to grow with ammonia as the sole nitrogen source. We found that the growth of M. alcaliphilum 20Z was promoted and putrescine production was also significantly improved with up to 2 mM of ammonium chloride supplement. A maximized titer of 98.08 ± 2.86 mg/L putrescine with a productivity of 2.9 nmol/gDCW/h was obtained in a simple flask culture.
Transcriptome analysis of the engineered strains cultured on mineral salt medium with different nitrogen sources showed upregulation of most genes involved in methane assimilation, TCA cycle, and ammonia assimilation in ammonia nitrate mineral salt medium compared to nitrate mineral salt medium. Despite the incompatibility of ammonia with methane, ammonia could be used as a potential nitrogen source for growth of M. alcaliphilum 20Z and production of glutamate-related metabolites.
The maximum titer of putrescine obtained in this study was much lower than sugar-based production [6, 9]. The experimental putrescine yield was 0.0276 g/g-CH4, much lower than the yield predicted by Optgene (0.408 g/g-CH4), but the experimental specific growth rate of the engineered strain (0.048 h−1) in NMS was higher than the maximum specific growth rate predicted by Optgene (0.032 h−1). The difference in growth rates between simulated data and experimental data was due to the adaptation to putrescine by engineered strain. To enhance the productivity of putrescine from methane, the remaining obstacles of methane bioconversion including methane mass transfer limitation and methanotrophic methane oxidation efficiency [5, 38, 39] should be solved. In addition to the conventional limitations of methane bioconversion (such as low methane mass transfer, lack of genetic tools, and low carbon conversion efficiency), further studies need to be investigated for engineering methane monooxygenase to allow M. alcaliphilum 20Z to grow in ammonia, which can reduce the energy consumption by nitrate assimilation.
Conclusion
The obligate methanotrophic bacterium M. alcaliphilum 20Z has become an attractive microbial platform for methane bioconversion to a value-added product due to the development of genetic tools and new insights into methane assimilation and the central metabolic pathway. In this study, we applied a genome-scale metabolic model to identify the metabolic engineering targets, inactivated the putrescine utilization pathway in M. alcaliphilum 20Z, overexpressed the ornithine biosynthesis pathway, and optimized the nitrogen source for putrescine production. The engineered M. alcaliphilum 20ZE4A-pACO strain produced putrescine up to 98.08 ± 2.86 mg/L in simple flask culture. To the best of our knowledge, this work represents the first biotechnological application for producing putrescine from methane.
Methods
Strains and culture conditions
The bacterial strains and plasmids used in this study are listed in Table 1. Wild-type and engineered M. alcaliphilum 20Z strains were cultured in nitrate mineral salt medium (NMS), which contains 1.0 g/L MgSO4·7H2O, 0.02 g/L CaCl2·6H2O, 1.0 g/L KNO3, 15 g/L NaCl, 2 mL/L trace element solution (Na2-EDTA 0.5 g/L, FeSO4·7H2O 1 g/L, F2-EDTA 0.75 g/L, ZnSO4·7H2O 0.8 g/L, MnCl2·4H2O 0.005 g/L, H3BO3 0.03 g/L, CoCl2·6H2O 0.05 g/L, Cu-EDTA 0.4 g/L, CuCl2·2H2O 0.6 g/L, NiCl2·6H2O 0.002 g/L, and Na2MoO4·2H2O 0.05 g/L), phosphate buffer (KH2PO4 54.4 g/L, Na2HPO4·12H2O 143.4 g/L) 2 mL/L, 4.5 mL/L of a 1 M solution of NaHCO3, and 0.5 mL/L of 1 M Na2CO3; these were sterilized by filtration and added before use [40]. Liquid cultures were grown in a 500 mL baffled flask sealed with a screw cap in an atmosphere of 30%(v/v) methane in air or 0.2%(v/v) of methanol at 30 °C. Kanamycin was used at a final concentration of 50 μg/mL.
Genetic tools and plasmid construction
All the plasmids in this study were constructed using a Gibson Assembly (NEB, England). Primers were designed using Primer 3 software and synthesized by Macrogen (Seoul, South Korea) as listed in Table 2. Genomic DNA of E. coli K12 strain W3110 and M. alcaliphilum 20Z were isolated using a Wizard® genomic DNA purification kit (Promega, USA). PCR was performed using Lamp Pfu polymerase (BioFACT, Korea). Plasmids were constructed using genome compiler software. A codon usage table was calculated from nucleotide coding sequences using Emboss cusp [41]. The codon adaptation index (CAI) [42] was calculated using a CAI calculator [43]. All the plasmids and primers used in this study can be found in Tables 1 and 2, respectively. Vector pAWP89 and vector pCM433KanT were linearized by PCR, and vector pCM351 was linearized by a restriction enzyme. Ornithine decarboxylase constitutive (speCEc), putrescine transporter (potE), N-acetylornithine transaminase (argD) and ornithine acetyltransferase (argJ) from E. coli K12 strain W3110, and ornithine decarboxylase (speCOb) from M. trichosporium OB3b were amplified and assembled with the corresponding linearized backbone. The sacB counter-selection plasmids pCSE1 were constructed to delete spermidine synthase (speE1) through unmarked allelic exchange by amplifying and assembling two flanking homology regions upstream and downstream of the target gene. The pCAR2 vector was constructed by cloning the flanking region F2 in the SacI site, and the flanking region F1 and the fragment pTac-argDJ in the EcoRI and NotI sites.
Transformation of M. alcaliphilum 20Z strains
The M. alcaliphilum 20Z competent cell was prepared as follows. First, 50 mL of M. alcaliphilum 20Z strain was grown to an OD600 of 0.4–0.6 in a 500 mL flask with 0.2%(v/v) methanol. Then, the cells were harvested and washed twice with cold sterile water by centrifugation at 5000g and 4 °C for 10 min. The resulting cell pellets were resuspended in cold sterile water, and 50 µL of the cell suspension were used for electroporation. The competent cells were gently mixed with 500 ng DNA plasmids, and the mixture was then transferred to an ice-cold 1 mm cuvette (Bio-Rad). Electroporation was performed using a Gene Pulser Xcell™ Electroporation system (Bio-Rad) set at 1.3 kV, 25 μF and 200 Ω [13]. After electrical discharge, 1 mL of room temperature NMS medium was added to resuspend the mixture, and then transferred into 20 mL medium in 250 mL serum bottles with 0.2% (v/v) methanol. The cells were incubated at 30 °C for 24 h, and then the cells were harvested and spread onto selective plates in an atmosphere of 30% (v/v) methane in air for 2 weeks.
Genome-scale modeling
Genome-scale metabolic models of M. alcaliphilum 20Z [14] were used to perform a flux balance analysis using Optflux 3 [44]. Necessary reactions of putrescine biosynthesis, and exchange reactions were added to the model, resulting in the i20ZRP model. Engineering strategies were generated by performing Optgene, i.e., the “evolutionary optimization” algorithm, on the i20ZRP model [16].
Analytical methods
Cell growth was monitored by measuring the optical density at 600 nm with a Beckman spectrophotometer using 1.5 mL cuvettes with a 1 cm path length. Methane concentration was analyzed via gas chromatography (GC; Young In Chromass Corporation, Korea) [45]. Putrescine concentration was analyzed using high-performance liquid chromatography (HPLC; Jasco Corporation, Japan) equipped with a Symmetry C18 column (5 μm, Waters corporation, Massachusetts, USA) operated at 30 °C. A mobile phase (0.7 mL/min) was used for all separations with a UV spectrophotometric detector at 198 nm. Putrescine was first derivatized using a benzoyl chloride pre-column derivatization method [46]. To 500 μL of sample supernatant, 150 μL of 2 N NaOH was added to adjust the pH, followed by 5 μL of benzoyl chloride. The mixture was magnetically stirred for 20 min to complete the reaction. After mixing, 200 μL of saturated NaCl was added. The resulting mixture was extracted by 300 μL ethyl ether. Pooled ether fractions were evaporated by a stream of nitrogen, and the residue was dissolved in 200 μL 42% acetonitrile/water. Then, 40 μL of the resulting solution was subjected to HPLC separation. The standard putrescine was prepared in water and treated as described above.
Transcriptome analysis
The engineered strain 20ZE4A-pACO was grown in NMS up to OD600 of 0.9, and then cells were transferred to NMS and ANMS medium. After 12 h of incubation, cells were harvested for RNA extraction using Qiagen RNeasy kit. Transcriptome shotgun sequencing (RNA-seq) of the engineered strain in these media was performed using Illumina platform (Macrogen Co., Korea). Sequencing reads were aligned with a reference genome and assembled into full-length transcripts, and then gene expression level and different transcript-level expression of different experimental conditions were calculated [47, 48].
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Pegg AE, Casero RA Jr. Current status of the polyamine research field. Methods Mol Biol. 2011;720:3–35.
Yamanobe T, Kurihara Y, Uehara H, Komoto T. Structure and characterization of nylon 46. J Mol Struct. 2007;829(1–3):80–7.
Scott E, Peter F, Sanders J. Biomass in the manufacture of industrial products—the use of proteins and amino acids. Appl Microbiol Biotechnol. 2007;75(4):751–62.
Conrado RJ, Gonzalez R. Envisioning the bioconversion of methane to liquid fuels. Science. 2014;343:621–3.
Lee OK, Hur DH, Nguyen DTN, Lee EY. Metabolic engineering of methanotrophs and its application to production of chemicals and biofuels from methane. Biofuel Bioprod Bior. 2016;10(6):848–63.
Qian ZG, Xia XX, Lee SY. Metabolic engineering of Escherichia coli for the production of putrescine: a four carbon diamine. Biotechnol Bioeng. 2009;104:651–62.
Nguyen AQ, Schneider J, Wendisch VF. Elimination of polyamine N-acetylation and regulatory engineering improved putrescine production by Corynebacterium glutamicum. J Biotechnol. 2015;201:75–85.
Schneider J, Eberhardt D, Wendisch VF. Improving putrescine production by Corynebacterium glutamicum by fine-tuning ornithine transcarbamoylase activity using a plasmid addiction system. Appl Microbiol Biotechnol. 2012;95(1):169–78.
Schneider J, Wendisch VF. Putrescine production by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2010;88(4):859–68.
Clomburg JM, Crumbley AM, Gonzalez R. Industrial biomanufacturing: the future of chemical production. Science. 2017;355(6320):38–49.
Linton JD, Niekus HGD. The potential of one-carbon compounds as fermentation feedstocks. In: van Verseveld HW, Duine JA, editors. Microbial growth on C1 compounds. Dordrecht: Springer; 1987.
Kalyuzhnaya MG, Khmelenina V, Eshinimaev B, Sorokin D, Fuse H, Lidstrom M, Trotsenko Y. Classification of halo(alkali)philic and halo(alkali)tolerant methanotrophs provisionally assigned to the genera Methylomicrobium and Methylobacter and emended description of the genus Methylomicrobium. Int J Syst Evol Microbial. 2008;58:591–6.
Yan X, Chu F, Puri AW, Fu Y, Lidstrom ME. Electroporation-based genetic manipulation in type I methanotrophs. Appl Environ Microbiol. 2016;82:2062–9.
Akberdin IR, Thompson M, Hamilton R, Desai N, Alexander D, Henard CA, Guarnieri MT, Kalyuzhnaya MG. Methane utilization in Methylomicrobium alcaliphilum 20Z(R): a systems approach. Sci Rep. 2018;8(1):2512–24.
de la Torre A, Metivier A, Chu F, Laurens LM, Beck DA, Pienkos PT, Lidstrom ME, Kalyuzhnaya MG. Genome-scale metabolic reconstructions and theoretical investigation of methane conversion in Methylomicrobium buryatense strain 5G(B1). Microb Cell Fact. 2015;14:188–202.
Nguyen AD, Hwang IY, Lee OK, Kim D, Kalyuzhnaya MG, Mariyana R, Hadiyati S, Kim MS, Lee EY. Systematic metabolic engineering of Methylomicrobium alcaliphilum 20Z for 2,3-butanediol production from methane. Metab Eng. 2018;47:323–33.
Nguyen AQ, Schneider J, Reddy GK, Wendisch VF. Fermentative production of the diamine putrescine: system metabolic engineering of Corynebacterium glutamicum. Metabolites. 2015;5(2):211–31.
Eppelmann K, Nossin PMM, Raeven LJRM, Kremer SM, Wubbolts MG. Biochemical synthesis of 1,4-butanediamine. WO2006005603 2006.
Marx CJ. Development of a broad-host-range sacB-based vector for unmarked allelic exchange. BMC Res Notes. 2008;1:1–7.
Michael AJ. Polyamine function in archaea and bacteria. J Biol Chem. 2018;293(48):18693–701.
Hamasaki-Katagiri N, Katagiri Y, Tabor CW, Tabor H. Spermine is not essential for growth of Saccharomyces cerevisiae: identification of the SPE4 gene (spermine synthase) and characterization of a spe4 deletion mutant. Gene. 1998;210:195–201.
Xie Q-W, Tabor CW, Tabor H. Deletion mutations in the speED operon: spermidine is not essential for the growth of Escherichia coli. Gene. 1993;126:115–7.
Otero JM, Cimini D, Patil KR, Poulsen SG, Olsson L, Nielsen J. Industrial systems biology of Saccharomyces cerevisiae enables novel succinic acid cell factory. PLoS ONE. 2013;8(1):e54144.
Shabestary K, Hudson EP. Computational metabolic engineering strategies for growth-coupled biofuel production by Synechocystis. Metab Eng Commun. 2016;3:216–26.
Patil KR, Rocha I, Forster J, Nielsen J. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinform. 2005;6:308–19.
Segre D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci USA. 2002;99(23):15112–7.
Kalyuzhnaya MG, Puri AW, Lidstrom ME. Metabolic engineering in methanotrophic bacteria. Metab Eng. 2015;29:142–52.
Shin JH, Lee SY. Metabolic engineering of microorganisms for the production of l-arginine and its derivatives. Microb Cell Fact. 2014;13:166–76.
Dou W, Xu M, Cai D, Zhang X, Rao Z, Xu Z. Improvement of l-arginine production by overexpression of a bifunctional ornithine acetyltransferase in Corynebacterium crenatum. Appl Biochem Biotechnol. 2011;165(3–4):845–55.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–8.
Vuilleumier S, Chistoserdova L, Lee MC, Bringel F, Lajus A, Zhou Y, Gourion B, Barbe V, Chang J, Cruveiller S, et al. Methylobacterium genome sequences: a reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. PLoS ONE. 2009;4(5):e5584.
Reitzer L. Nitrogen assimilation and global regulation in Escherichia coli. Annu Rev Microbiol. 2003;57:155–76.
Luque-Almagro VM, Gates AJ, Moreno-Vivian C, Ferguson SJ, Richardson DJ, Roldan MD. Bacterial nitrate assimilation: gene distribution and regulation. Biochem Soc Trans. 2011;39(6):1838–43.
Kadam PC, Boone DR. Influence of pH on ammonia accumulation and toxicity in halophilic, methylotrophic methanogens. Appl Environ Microb. 1996;62(12):4486–92.
Ward N, Larsen Ø, Sakwa J, Bruseth L, Khouri H, Durkin AS, Dimitrov G, Jiang L, Scanlan D, Kang KH, et al. Genomic insights into methanotrophy: the complete genome sequence of Methylococcus capsulatus (Bath). PLoS Biol. 2004;2(10):e303.
Whittenbury R. Phillips kC, Wilkinson jF: Enrichment, isolation and some properties of methane-utilizing bacteria. J Gen Microbiol. 1970;61:205–18.
Kalyuzhnaya MG, Yang S, Rozova ON, Smalley NE, Clubb J, Lamb A, Gowda GA, Raftery D, Fu Y, Bringel F, et al. Highly efficient methane biocatalysis revealed in a methanotrophic bacterium. Nat Commun. 2013;4:2785–91.
Haynes CA, Gonzalez R. Rethinking biological activation of methane and conversion to liquid fuels. Nat Chem Biol. 2014;10(5):331–9.
Henard CA, Smith H, Dowe N, Kalyuzhnaya MG, Pienkos PT, Guarnieri MT. Bioconversion of methane to lactate by an obligate methanotrophic bacterium. Sci Rep. 2016;6:21585–93.
Ojala DS, Beck DA, Kalyuzhnaya MG. Genetic systems for moderately halo(alkali)philic bacteria of the genus Methylomicrobium. Method Enzymol. 2011;495:99–118.
Rice P, Longden I, Bleasby A. EMBOSS the European molecular biology open software suite. Trends Genet. 2000;16(6):276–7.
Sharp PM, Li W-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–95.
Puigbo P, Bravo IG, Garcia-Vallve S. CAIcal: a combined set of tools to assess codon usage adaptation. Biol Direct. 2008;3:38–45.
Rocha I, Maia P, Evangelista P, Vilaça P, Soares S, Pinto JP, Nielsen J, Patil KR, Ferreira EC, Rocha M. OptFlux: an open-source software platform for in silico metabolic engineering. BMC Syst Biol. 2010;4:45–56.
Liu X, Yang J, Ye T, Han Z. Establishment of analysis method for methane detection by gas chromatography. IOP Conf Ser Earth Environ Sci. 2018;113:012023.
Mei YH. A sensitive and fast method for the determination of polyamines in biological samples. Benzoyl chloride pre-column derivatization high-performance liquid chromatography. J Liq Chromatogr. 1994;17(11):2413–8.
Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–67.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Puri AW, Owen S, Chu F, Chavkin T, Beck DA, Kalyuzhnaya MG, Lidstrom ME. Genetic tools for the industrially promising methanotroph Methylomicrobium buryatense. Appl Environ Microbiol. 2015;81(5):1775–81.
Marx CJ, Lidstrom ME. Broad-Host-Range Cre-Lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques. 2002;33:1062–7.
Acknowledgements
Not applicable.
Funding
This research was supported by the C1 Gas Refinery Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2015M3D3A1A01064882).
Author information
Authors and Affiliations
Contributions
LTN and EYL designed the research; LTN conducted experiments; LTN wrote the manuscript and EYL revised the manuscript. EYL supervised the research. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors declare that this study does not involve human subjects, human material and human data.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1: Table S1.
Knockout strategies suggested by OptGene algorithm for the production of putrescine. Figure S1. Knockout of speE1 check by PCR using primer outside of the flanking region. Lane 1 is the mutant strain and Lane 2 is the wild-type strain. Figure S2. The insertion of pTAC-argDJ into the genome of the engineered strain 20ZE4A. Lane 1 is the 1 kbp marker Enzynomics, Lane 2 is the wild-type strain, and Lane 3 is the engineered strain.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nguyen, L.T., Lee, E.Y. Biological conversion of methane to putrescine using genome-scale model-guided metabolic engineering of a methanotrophic bacterium Methylomicrobium alcaliphilum 20Z. Biotechnol Biofuels 12, 147 (2019). https://doi.org/10.1186/s13068-019-1490-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-019-1490-z