
Abstract
Various sets of thiazole, thiophene, and 2-pyridone ring structures containing a dimethylaniline component were synthesized. Substituted thiazoles 2–3 and thiophenes 5–7 were produced by reacting thiocarbamoyl compound 4 with α-halogenated reagents in different basic conditions. Also, a series of 2-pyridone derivatives 9a–f substituted with dimethylaniline was synthesized through Michael addition of malononitrile to α,β-unsaturated nitrile derivatives 8a–f. The synthesized products were structurally proven by spectroscopic methods such as IR, 1H NMR, 13C NMR, and MS data. Furthermore, the anti-cancer efficacy of the compounds was assessed using the MTT assay on two cell lines: hepatocellular carcinoma (HepG-2) and breast cancer (MDA-MB-231). The results showed the highest growth inhibition for derivatives 2, 6, 7, and 9c, which were further examined for their IC50 values. The IC50 for compound 2 showed equipotent activity (IC50 = 1.2 µM) against the HepG-2 cell line compared to Doxorubicin (IC50 = 1.1 µM). Compounds 2, 6, 7 and 9c showed very good ADME assessments for further drug administration. Moreover, the PASS theoretical prediction for the compounds showed high antimitotic and antineoplastic activities for compounds 2, 6, 7, and 9c, as well as potent inhibition activity for the insulysin enzyme (IDE). Molecular docking stimulations were performed on CDK1/CyclinB1/CKS2 (PDB ID: 4y72) and BPTI (PDB ID: 2ra3). When docked into (PDB ID: 4y72), all of the tested compounds showed considerable inhibition, and the 2-pyridone derivative 9d had the maximum binding affinity (− 8.1223 kcal/mol). While thiophene derivative 6 offered the maximum binding affinity (− 7.5094 kcal/mol) when docked into (PDB ID: 2ra3).
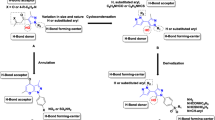
Graphical Abstract

Similar content being viewed by others


Introduction
Human death leading cause in the world is attributed to cancer [1]. Breast cancer is the most common cancer in women and the main cause of death. In Egypt, breast cancer constitutes 33% of female cancer cases, and a high frequency rate counts for hepatocellular carcinoma [2]. Moreover, two main problems in searching for a new anti-tumor drug are serious side effects and the rapid development of drug resistance [3]. Several studies have been carried out using various sulfur and/or nitrogen-containing heterocyclic compounds, including thiophene, thiazole, and pyridine, directed towards different pathologies. These thiophene, thiazole, and pyridine-containing compounds show anticancer [4,5,6,7,8], anti-inflammatory [9,10,11], antibacterial [12,13,14], antioxidant [15], anti-oxidant [16], anti-fungal [17, 18], anti-coronavirus [19, 20] properties. Thiazoles were also found to act as anti-Alzheimer [21], anti-tubercular [22], and anti-diabetic [23]. While pyridone as well exhibits antimalarial [24], anti-hepatitis B [25], cardiotonic [26], and anti-fibrosis [27] properties. Some of these thiophene, thiazole, and pyridone-containing compounds have been transferred into clinical trials and cancer therapy, acting via multiple pathways [28]. Dabrafenib and Dasatinib are examples of thiazole-containing selective drugs with tyrosine kinase inhibitory activity (Fig. 1) [29, 30]. OSI-930 is a thiophene-containing orally specific inhibitor of Kit and kinase insert domain receptor tyrosine kinases (Fig. 1) [31]. Topotecan and Tazemetostat are 2-pyridone-containing cancer drugs that act as potent topoisomerase 1 inhibitors and selective EZH2 inhibitors, respectively (Fig. 1) [32]. The cyclin-dependent kinases (CDKs) are essential proteins that play an important role in cell-cycle control [33]. CDK1 inhibition has been found to effectively break cancer cell proliferation [34]. Some studies reveal thiazole-containing compounds as potent CDK inhibitors [35]. One of the most efficient synthetic strategies for thiophenes and thiazoles is the use of α-halogenated reagents to cyclize generous thiocarbamoyl derivatives. This method creates a varied substitution at all possible sites and an easy workup [36]. Cyanoacetanilide derivative cyclization with active methylene is one of the best methods for 2-pyridone synthesis [37]. The current work is centered on the synthesis of bioactive substituted thiazoles, thiophenes, and 2-pyridone molecules substituted with dimethylaniline. The biological evaluation included anticancer activity testing and ADME assessment for future medication delivery. Furthermore, theoretical predictions for the new compounds and molecular docking studies were performed to examine their anticancer inhibition activity.

Anticancer marketed-drugs containing thiazole, thiophene and 2-pyridone
Results and discussion
Chemistry
The synthetic strategy is based on the precursor 2-cyano-N-(4-(dimethylamino)phenyl)acetamide (1). It was synthesized in good yield through the well-known cyanoacetylation reaction of N,N-dimethylbenzene-1,4-diamine with 1-(cyanoacetyl)-3,5-dimethylpyrazole in dioxane (Scheme 1). The chemical structure and conformation of cyanoacetamide compound 1 were determined through spectral analysis. The IR absorption bands detected at 3280, 2221, and 1690 cm−1 correspond to the functional groups N–H, –C≡N, and –C=O, respectively. The 1H NMR spectrum indicated a singlet for six protons (dimethylamino group) at δ 2.83 ppm, a singlet for two protons (–CH2–) at δ 3.80 ppm, two doublet signals for the para-disubstituted benzene ring at δ 6.68 and 7.34 ppm, and singlet signal for the amidic proton at δ 9.98 ppm (N–H).

Preparation of 2-cyano-N-(4-(dimethylamino)phenyl)acetamide (1)
The addition of 2-cyanoacetamide compound 1 to phenyl isothiocyanate was accomplished by stirring in DMF and potassium hydroxide to generate the non-isolated sulphide salt (intermediate A) (Scheme 2). This salt underwent in situ addition with chloroacetone to furnish the corresponding substituted thiazoline derivative 2. The 1H NMR spectrum demonstrated the presence of two singlets at δ 1.28 and 6.87 ppm for the protons of the methyl group and thiazole-C5, respectively. Meanwhile, the thiazolidine-4-one compound 3 was obtained by in situ treatment of non-isolable sulfide salt (A) with ethyl bromoacetate. 1H NMR displayed four singlet signals at δ 2.82, 3.97, and 9.34 ppm, matching six protons (dimethylamino-NMe2), two protons (thiazolidine-CH2) and N–H functions. To get the notably rich thiocarbamoyl intermediate 4 (Scheme 2), the non-isolated sulphide salt was treated with water and then neutralized with diluted HCl. The structure was validated by spectral measurements (c.f. “Experimental”).

Synthesis of substituted thiazole derivatives 2 and 3
The reactivity of thiocarbamoyl 4 towards different α-halo-ketones was investigated. Thus, nucleophilic substitution of thiocarbamoyl 4 on chloroacetone was achieved in refluxing ethanol and triethylamine. Following this, the nitrile group underwent intramolecular nucleophilic addition to produce 5-acetyl-4-aminothiophene derivative 5 (Scheme 3). Elemental studies and spectrum data confirmed the chemical structure of the newly created thiophene 5. The formation of amino functionality was confirmed in both IR and 1H NMR spectra through strong absorptions at 3443 and 3282 cm−1 in the IR spectrum and a broad singlet signal at δ 7.47 ppm in the 1H NMR spectrum. Also, the 1H NMR spectrum indicated the lack of any signal related to the protons of the methylene group.

Synthesis of 4-amino-5-substititedthiophene derivatives 5, 6 and 7
Similarly, the synthesis of 4-amino-5-benzoylthiophene derivative 6 and 4-amino-5-ethoxycarbonyl-thiophene derivative 7 was successful by treating thiocarbamoyl scaffold 4 with phenacyl bromide and ethyl bromoacetate in ethanol and triethylamine. The 1H NMR spectrum of derivative 6 indicates a characteristic singlet signal at δ 6.70 ppm and extra multiplet peaks in the region from δ 7.05 to 7.88 ppm for the protons of –NH2 group, which corresponds to phenyl rings. These new peaks appear next to the singlet signal at δ 2.84 ppm for six protons of two methyl groups (–N(CH3)2), two singlet signals at δ 9.62 and 9.82 ppm for two NH functions, and doublet signals at δ 7.04 to 7.88 ppm for the aromatic protons. In thiophene derivative 7, strong absorptions at 3352, 3293, 1740, and 1663 cm−1 verified the presence of N–H, NH2 groups, and two carbonyl groups in the IR spectrum. While the 1H NMR spectrum indicated notable triplet and quartet signals at δ 1.34 and 4.28 ppm for protons of the ethoxy group (–OCH2CH3), besides singlet at δ 2.94 ppm for six protons (–N(CH3)2), and two singlet signals at δ 8.35 and 10.83 ppm corresponding to the protons of two (N–H) groups.
The synthetic strategy of 6-amino-4-aryl-1-(4-dimethylaminophenyl)-3,5-dicyano-2-oxo-2H-pyridine derivatives 9a–f is explained in Scheme 4. Initially, the Knoevenagel condensation reaction between 2-cyanoacetanilide compound 1 and electronically different aromatic aldehydes in ethanol and piperidine offered α,β-unsaturated nitrile derivatives 8a–f. Next, the cyclized targets 3,5-dicyanopyridone derivatives 9a–f were achieved via Michael’s addition reaction of malononitrile to α,β-unsaturated nitrile compounds 8a–f in refluxing dioxane and piperidine. The cyclization occurs through the formation of the suggested intermediates B and C. The nucleophilic addition of malononitrile to the beta-carbon of α,β-unsaturated functionality yields the intermediate Michael adduct B, which then undergoes intramolecular cyclization to give intermediate C. The last step is tetrahydropyridine intermediate C oxidation to furnish the final pyridine derivatives 9a–f, whose structures were confirmed by spectroscopic examinations. We suggested that air (or the oxygen it contains) initiates the oxidation process.

Synthesis of different α,β-unsaturated nitrile derivatives 8a–f and their corresponding 2-pyridone derivatives 9a–f
Selected examples of formed structures are explored below using IR and 1H NMR spectra to prove the synthesized structures. For α,β-unsaturated nitrile 8d, new absorption bands for carbonyl (C=O) and NH functions appear next to 2-cyanoacetanilide functions at 3407, 3338, 1696, and 1670 cm−1, respectively. Also distinguished singlet signals for the protons of methyl group (Ar–CH3), methylene group (CO–CH2–O), and olefinic proton (C=CH) were observed in the 1H NMR spectrum at 2.24, 4.81, and 8.15 ppm, respectively. In the same spectrum, twelve aromatic protons resonated as six doublet signals at 6.71–8.00 ppm, and a singlet signal was observed at 2.86 ppm corresponding to the –N(CH3)2. The protons of two NH functions appeared as two singlet signals at 10.00 and 10.08 ppm. For the 2-pyridone scaffold 9d, the IR spectrum showed absorption at 3340, 3278, and 3183 cm−1 for the NH2 and N–H groups, 2213 cm−1 due to the nitrile group, and 1693 and 1659 cm−1 due to the carbonyl groups of the ester and amidic carbonyl groups, respectively. The 1H NMR spectrum recorded the singlet signals at 2.25, 2.97, and 4.78 ppm for the protons of the methyl and methylene groups (Ar–CH3, –N(CH3)2 and CO–CH2–O, respectively). Twelve aromatic protons appeared as doublet and multiplet signals at 6.83–7.50 ppm, while the N–H proton resonated at 10.07 ppm.
Anticancer activity
The cytotoxic effect of all compounds 1–9 on hepatocellular carcinoma (HepG-2) and breast cancer (MDA-MB-231) was examined using the MTT technique. The outcomes are presented in (Additional file 1: Table S4), while graphic plots of the growth inhibition curve against the compounds enable us to compare the inhibition activity for the tested compounds (Fig. 2). The results revealed that the tested drugs suppress cancer cells to varying degrees. At a 25 µM concentration after 48 h, compounds 2, 6, 7, and 9c had the most cytotoxic effect against the HepG-2 and the MDA-MB-231 cell lines. Compounds 2, 6, 7, and 9c were selected for the IC50 experiment using doxorubicin as a reference (Table 1, Additional file 1: Figs. S41–S43). The IC50 evaluation against the HepG-2 cell line showed that compound 2 containing the thiazole moiety acts as a potent anticancer with an IC50 value of 1.2 µM, which is equivalent to the doxorubicin IC50 value of 1.1 µM.

Cytotoxic activity against all compounds 1–9(a–f) against HepG-2 and MDA-MB-231 cell lines
Compounds 6 and 7 showed IC50 values of 28.7 and 6.4 µM, respectively. The highest inhibitory activity was observed for compound 9c, which contains a pyridine moiety with an IC50 value of 33.08 µM.
IC50 evaluation against the MDA-MB-231 cell line shows the lowest IC50 values for compounds 6, 7, and 9c (14.1, 21.9, and 21.73 µM), respectively. While the highest IC50 value (26.8 µM) is for compound 2. The derivative 2 showed superiority as a possible anticancer agent due to its low toxicity to normal cells (PBMC human peripheral blood mononuclear cells). (IC50 < 30 µM) compared to Doxorubicin IC50 (2 µM).
Structure activity relationship (SAR) studies
By analyzing the outcomes, we uncovered important structure–activity relationship facts (Fig. 3). Thiazole derivative 2 with a substituted methyl group at position 4 of the thiazole ring markedly recorded the strongest inhibitory effect against tested HepG2 and MDA-MB-231 cell lines (80 and 54%) with IC50 values of 1.2 and 26.8 µM, respectively. The effect of changing the methyl group at position 4 of the thiazole ring for compound 2 with the carbonyl group for compound 3 caused a decrease in inhibitory effect against tested cell lines (15 and 36%). It is clear that the cyanoacetamide derivative 1 and mercaptocrylamide derivative 4 displayed the weakest activity (14 and 6%) against the liver cell line and (32 and 28%) against the breast cell line. Also, the absence of the thiazole moiety in derivatives 1 and 4 may lead to the loss of inhibitory activity. Among the 3-amino thiophene derivatives 5–7, it was found that the presence of benzoyl and ester groups at position 2 in compounds 6 and 7 led to an increase in inhibition activity (55 and 64%) with IC50 values (28.7 and 14.1 µM) against the liver cell lines and (69 and 57%) with IC50 values (6.4 and 21.9 µM) against breast cell lines. The replacement of these groups with an acyl group at position 2 of the thiophene ring led to a decrease in the inhibitory activity. Likewise, acrylamide derivatives 8a–f showed moderate to low inhibition potency against cell lines 19–40%. Moreover, pyridine derivatives 9a–f showed moderate to high inhibition potency against cell lines 24–58%. Pyridine derivative 9c with a substituted Cl group at position 4 of the phenyl ring exhibited the strongest activity among the pyridine series (52 and 58%) against liver and breast cell lines with IC50 values of 33.08 and 21.73 µM, respectively. The effect of exchanging the chlorine atom for compounds 9c with methyl and methoxy groups for compounds 9a and 9b resulted in a decrease in inhibition growth values (37 and 34%) and (50 and 30%), respectively, towards the examined cell lines. Also, the addition of a linker (–OCH2CONHAr–) in pyridine derivatives 9d–f resulted in reduced activity compared to their direct 4-substituted phenyl ring pyridine counterparts 9a–c.

Structure–activity relationship (SAR) of thiazole, thiophene, pyrindone derivatives
Computational analysis
Prediction of in-silico ADME and oral bioavailability
During the drug discovery work, a lot of attention goes to the pharmacodynamics of the newly synthesized small molecules. While the promotion of a drug candidate focuses on the pharmacokinetics behavior of such molecules [38]. In-silico ADME screening was applied to the synthesized compounds to calculate the putative absorption, distribution, metabolism, and excretion properties [39]. Predictions for compounds 1–9 were listed in (Table 2, Additional file 1: Table S1) and processed using the SwissADME webtool provided through the Swiss Institute of Bioinformatics (SIB) (https://www.swissadme.ch) and AdmetSAR-2 online software predictors.
Generally, all the investigated compounds 1–9 possess acceptable physicochemical and pharmacokinetic properties with zero violation of Lipinski’s rule of five, except for derivatives 9d, 9e, and 9f. Regarding the parameters associated with Lipinski’s rule of five, all compounds have 4 to 10 rotatable bonds, 2 to 6 hydrogen bond acceptors, 1 to 3 hydrogen bond donors, molar refractivity (MR) 59.52 to 150.85, topological polar surface area (TPSA) between 56.13 and 137.17, and a predicted logPo/w in the range of 0.89 to 2.88 [40]. Moreover, they displayed moderate water solubility and high GIT absorption, except for derivatives 6, 7, 9d, 9e, 9f and one PAINS. These results support the oral bioavailability of the compounds [41]. Consequently, any compound showing two or more violations must be excluded from further study.
The correlation between the WLOGP and TPSA (topological polar surface area) for the newly synthesized compounds is presented in the BOILED-EGG graph [42]. Figure 4 categorizes the compounds in regions, most of them located in the region of human intestinal absorption (HIA), except 5, 6, 7, 9d, 9e, and 9f. The HIA region has no BBB permeability except for 1, 8a–8c, located in the yolk of the BOILED-EGG, which is expected to be BBB permeant. These results reflect good oral absorption for our compounds, along with poor penetration of the BBB, and hence no possible side effects on the CNS. Much more, a promising character for the compounds as they do not support a substrate for P-glycoprotein (Pgp), which is considered a drug efflux transporter [43]. In other words, all compounds can exist in the target cancer cells, simulating their cytotoxic effect. To conclude, the most active derivatives 2, 6, 7, and 9c fulfil the ADME profiles required for good distribution in the body and oral bioavailability, avoiding cancer cell resistance mediated by Pgp.

Swiss ADME boiled-egg plot for compounds 1–9(a–d)
Prediction of activity spectra using PASS online predictor
With the aid of Prediction of Activity Spectra for Substances (PASS) online software, we were able to assess the putative anticancer activity, which is presented in (Table 3, Additional file 1: Table S2) The results of the PASS prediction are based on the available biological activity of compound fragments in the database through the correlation between Pa and Pi (probability to be active or inactive) [44]. The results displayed high antimitotic and antineoplastic activities for compounds 2, 6, 7 and 9c (Table 3) in addition to other activities (Additional file 1: Table S2). Interestingly, almost all compounds displayed potent inhibition activity for the insulysin enzyme, also called the insulin-degrading enzyme (IDE).
Molecular docking
In this study, the biological targets in the studied cancer cell lines were predicted in order to define the mechanism of anticancer activities for all synthesized derivatives. The interaction of the derivatives with two different targets was inspected through molecular docking studies in order to predict their binding sites and modes on enzymes. The molecular docking study was performed using MOE “v10.2015.10 software”. The choice of targets was based on the interaction of ATP-competitive inhibitors complexed with the cyclin-dependent kinase human CDK1/CyclinB1/CKS2 (PDB ID: 4y72) [45, 46]. Also, the PASS-Online software predicts inhibition activity for the insulysin enzyme, and based on that, different related PDB codes were tested. The best interaction was found to be with Human cationic trypsin complexed with bovine pancreatic trypsin inhibitor (BPTI) (PDB ID: 2ra3). The crystallographic coordinates of the proteins were downloaded from the Protein Data Bank [47]. Markedly, all of the compounds showed significant inhibition activities against 4y72 and were higher than previously prepared compounds (Table 4, Additional file 1: Table S3) [45]. A docking study of compounds 1–9(a–d) showed that they can fit well in the ATP binding sites of CDK1 and BPTI (Additional file 1: Figs. S1–S40). The highest binding affinities for the prepared compounds with the targeted proteins 2ra3 and 4y72 are listed in (Table 4), while the binding affinities for the rest of the compounds are listed in (Additional file 1: Table S3). The compound 5 has a docking score of − 7.9160 kcal/mol. It forms strong hydrogen bonds with Asp 86. It also exhibits interactions with Gly 193, Lys 60, and Phe 41 through H-donor, H-acceptor, and π–H interactions, respectively (Fig. 5). Compound 8f has a docking score of − 7.9504 kcal/mol, which is slightly more favorable than compound 5. It forms potential strong binding interactions through hydrogen bonds with Lys 130 and Glu 12. Additionally, it shows interactions with Ile 10 through π–H, and H-acceptor interactions with benzene rings. Compound 9d exhibits the highest docking score among the three compounds, with a score of − 8.1223 kcal/mol, indicating strong binding affinity. It forms hydrogen bonds with Gln 132, Ser 84, and Lys 130 with various functional groups of the compound, including N-Amino, N-Amide, and N-dimethylamino groups. Gly 11 also interacts with compound 9d through a π–H interaction. The several hydrogen bonds and π–H interaction contribute to the strong binding affinity of compound 9d. Compound 9f has a docking score of − 8.0945 kcal/mol, which is quite favorable. It forms with Ile 10, Thr 14, and Lys 130 π–H interactions. Compound 9f primarily interacts with the protein through its benzene ring, and these π–H interactions contribute to its binding affinity. To summarize, the docking results suggest that four compounds (5, 8f, 9d, and 9f) strongly bind to the target protein due to various types of interactions. Compound 9d stands out with the highest docking score, indicating the strongest predicted binding affinity. These findings provide valuable insights into the potential of these compounds as drug candidates that could target a specific protein of interest.

2D, 3D and surface map interaction between drugs 1–9(a–f) and target proteins (4y72 and 2ra3)
Conclusion
The study aimed to synthesize thiazole, thiophene, and 2-pyridone incorporating a dimethylaniline moiety and investigate their potential for biological activity. A library of compounds 1–9(a–f) with varying structures to assess their potential as drug candidates. The cyclization reaction strategy for the thiocarbamoyl derivative using α-halogenated reagents in different reaction conditions serves to modify the chemical structures (thiazoles and thiophenes). Also, the Michael addition of malononitrile to α,β-unsaturated nitrile derivatives introduces functional groups into 2-pyridone derivatives. Anti-cancer activity evaluation on two different cancer cell lines (hepatocellular carcinoma and breast cancer) indicates potential anti-cancer properties. Compound 2 has equipotent activity for the Doxorubicin IC50 value (1.2 µM), while compounds 6, 7, and 9c had the highest cytotoxic effect. Compounds 2, 6, 7, and 9c also have favorable ADME profiles. Compounds 2, 6, 7, and 9c are identified as particularly promising anticancer agents based on PASS predictions. Molecular docking stimulations with specific biological targets (CDK1/CyclinB1/CKS2 and BPTI) demonstrated that the synthesized compounds exhibited strong binding affinities with the protein residues. Meanwhile, 2-pyridone derivatives 9d recorded an eminent docking score (-8.1223 kcal/mol) with the protein 4y72 and the thiophene 6 provided a great energy score (− 7.5094 kcal/mol) with 2ra3.
Experimental
Experimental general remarks: melting points were determined with Gallenkamp melting point apparatus and are uncorrected. The infrared (IR) spectra were recorded on Thermo Scientific Nicolet iS10 FTIR. 1H NMR and 13C NMR spectra were recorded DMSO-d6 as a solvent using JEOL’s spectrometer at 500 MHz using tetramethylsilane (TMS) as internal standard. Chemical shifts are expressed in δ, ppm. 1H NMR data are reported in order: multiplicity (br, broad; s, singlet; d, doublet; t, triplet; dd, doublet of doublet; m, multiplet), approximate coupling constant in Hertz, number of protons and type of protons. The purity of the compounds was checked by 1H NMR and thin layer chromatography (TLC) on silica gel plates using a mixture of dichloromethane and methanol or petroleum ether and ethyl acetate as eluent. UV lamp was used as a visualizing agent. Mass analyses and elemental analyses were recorded on Thermo DSQ II spectrometer at Faculty of Science, Alazhar University. The 13C NMR spectra of compounds 9b, 9c, 9d, 9e, and 9d are not recorded due to insufficient solubility in most of NMR solvents.
Preparation of 2-cyano-N-(4-(dimethylamino)phenyl)acetamide (1)
A suspension of N,N-dimethylbenzene-1,4-diamine (0.04 mol, 4.32 g) and 1-cyanoacetyl-3,5-dimethylpyrazole (0.04 mol, 6.52 g) was refluxed in dioxane for 6 h. The precipitate that obtained was collected and dried to produce cyanoacetamide compound 1.
Pale blue crystal; yield (65%); m.p. 189–191 °C. IR (ν/cm−1): 3280 (N–H), 2221 (C≡N), 1690 (C=O). 1H NMR (δ/ppm): 2.83 (s, 6H, –N(Me)2), 3.80 (s, 2H, CH2), 6.68 (d, J = 8.50 Hz, 2H, Ar–H), 7.34 (d, J = 8.50 Hz, 2H, Ar–H), 9.98 (s, 1H, NH). 13C NMR (δ/ppm): 30.94, 40.25, 40.61, 112.53 (2C), 116.87, 122.44 (2C), 128.76, 147.26, 165.38. Analysis Calcd. for C11H13N3O (203.11): C, 65.01; H, 6.45; N, 20.68%; found: C, 64.94; H, 6.40; N, 20.61%.
Preparation of 2-cyano-N-(4-(dimethylamino)phenyl)-2-(4-methyl-3-phenylthiazol-2(3H)-ylidene)acetamide (2)
Stirring of cyanoacetamide compound 1 (0.005 mol, 1.02 g) with phenyl isothiocyanate (0.005 mol, 0.61 ml) in 20 ml DMF containing KOH (0.005 mol, 0.28 g) was continued for 6 h. Then chloroacetone (0.002 mol, 0.164 ml) was added and continue stirring overnight. Pouring the mixture into ice-cold water. Finally, the solid underwent filtration, drying, and recrystallized by heating in ethanol.
Gray solid; yield (43%); m.p. 168–170 °C. IR (\(\overline{\nu }\)/cm−1): 3405 (N–H), 2176 (C≡N), 1634(C=O). 1H NMR (δ/ppm): 1.28 (s, 3H, CH3), 2.81 (s, 6H, –N(Me)2), 6.62 (d, J = 8.50 Hz, 2H, Ar–H), 6.87 (s, 1H, CH), 7.23–7.32 (m, 3H, Ar–H), 7.35–7.41 (m, 1H, Ar–H), 7.41–7.50 (m, 3H, Ar–H), 8.56 (s, 1H, NH). 13C NMR (δ/ppm): 25.76, 40.38, 40.55, 70.89, 95.53, 112.46 (2C), 116.02, 122.13 (2C), 128.58, 128.90 (2C), 129.13 (2C), 130.28 (2C), 137.52, 147.13, 163.89, 169.80. Analysis Calcd. for C21H20N4OS (376.14): C, 67.00; H, 5.35; N, 14.88%; found: C, 66.87; H, 5.30; N, 14.96%.
Synthesis 2-cyano-N-(4-(dimethylamino)phenyl)-2-(4-oxo-3-phenylthiazolidin-2-ylidene)acetamide (3)
Stirring of cyanoacetamide compound 1 (0.005 mol, 1.02 g) with phenyl isothiocyanate (0.005 mol, 0.60 ml) in 20 ml DMF containing KOH (0.005 mol, 0.28 g) was continued for 6 h. Then ethyl bromoacetate (0.002 mol, 0.84 ml) was added and continue stirring overnight. For precipitation, pouring the mixture into ice-cold water, then filtrated, and recrystallized by heating in ethanol.
Yellow crystals; yield (33%); m.p. 138–140 °C. IR (\(\overline{\nu }\)/cm−1): 3368 (N–H), 2191 (C≡N), 1714, 1622 (C=O). 1H NMR (δ/ppm): 2.82 (s, 6H, –N(Me)2), 3.97 (s, 2H, CH2), 6.63 (d, J = 9.00 Hz, 2H, Ar–H), 7.19 (t, J = 7.50 Hz, 1H, Ar–H), 7.24 (d, J = 9.00 Hz, 2H, Ar–H), 7.30 (d, J = 9.00 Hz, 2H, Ar–H), 7.33–7.39 (m, 2H, Ar–H), 9.34 (s, 1H, NH). 13C NMR (δ/ppm): 33.25, 40.47 (2C), 77.54, 112.67 (2C), 114.81, 122.06 (2C), 127.84 (2C), 128.29, 128.93, 129.75 (2C), 138.17, 148.94, 162.18, 169.33, 170.69. Analysis Calcd. for C20H18N4O2S (378.12): C, 63.47; H, 4.79; N, 14.80%; found: C, 63.68; H, 4.72; N, 14.91%.
Synthesis of 2-cyano-N-(4-(dimethylamino)phenyl)-3-mercapto-3-(phenylamino)acrylamide (4)
A suspension of cyanoacetamide compound 1 (0.005 mol, 1.02 g) and phenyl isothiocyanate (0.005 mol, 0.60 ml) was stirred in 20 ml DMF containing KOH (0.005 mol, 0.28 g) for 6 h. Firstly, the mixture was diluted with cold water, then acidified with diluted hydrochloric acid. The precipitate was filtered and washed with cold ethyl alcohol.
Pale green solid; yield (79%); m.p. 208–210 °C. IR (ν/cm−1): 3411 (N–H), 2169 (C≡N), 1641 (C=O). 1H NMR (δ/ppm): 2.98 (s, 6H, –N(Me)2), 6.85 (d, J = 8.50 Hz, 2H, Ar–H), 6.93 (t, J = 7.50 Hz, 1H, Ar–H), 7.16–7.22 (m, 4H, Ar–H), 7.45 (d, J = 8.50 Hz, 2H, Ar–H), 10.06 (s, 1H, NH), 10.75 (s, 1H, NH). 13C NMR (δ/ppm): 40.64 (2C), 83.96, 113.21 (2C), 114.42, 122.45 (2C), 124.46 (2C), 125.33, 127.36, 129.60 (2C), 136.79, 150.08, 164.80, 167.28. Mass analysis (m/z, %): 338 (M+, 32.7%). Analysis Calcd. for C18H18N4OS (338.12): C, 63.88; H, 5.36; N, 16.56%; found: C, 64.94; H, 6.41; N, 16.51%.
Preparation of 5-acetyl-4-amino-N-(4-(dimethylamino)phenyl)-2-(phenylamino)thiophene-3-carboxamide (5)
A suspension of acrylamide compound 4 (0.002 mol, 0.67 g), chloroacetone (0.002 mol, 0.16 ml), and 0.5 ml triethylamine was refluxed in ethanol for 2 h. The solid that formed was collected to produce the targeted 5-acetyl-4-aminothiophene compound 5.
Off white powder; yield (38%); m.p. 218–220 °C. IR (ν/cm−1): 3443, 3282 (NH2, N–H), 1698, 1639 (C=O). 1H NMR (δ/ppm): 2.09 (s, 3H, CH3), 2.83 (s, 6H, –N(Me)2), 6.68 (d, J = 8.50 Hz, 2H), 7.08 (t, J = 7.50 Hz, 1H), 7.30–7.45 (m, 4H), 7.43 (d, J = 9.00 Hz, 2H), 7.47 (br. s, 2H, NH2), 9.51 (s, 1H, NH), 9.77 (s, 1H, NH). 13C NMR (δ/ppm): 28.05, 40.52 (2C), 96.27, 106.40, 112.36 (2C), 119.97 (2C), 121.88 (2C), 123.74, 128.38, 129.38 (2C), 141.00, 147.33, 154.38, 156.80, 161.86, 186.70. Analysis Calcd. for C21H22N4O2S (394.15): C, 63.94; H, 5.62; N, 14.20%; found: C, 63.89; H, 5.58; N, 14.18%.
Synthesis of 4-amino-5-benzoyl-N-(4-(dimethylamino)phenyl)-2-(phenyl amino)thiophene -3-carboxamide (6)
A solution of acrylamide compound 4 (0.002 mol, 0.67 g) was refluxed with phenacyl bromide (0.002 mol, 0.39 g) and triethylamine (0.5 ml) in ethanol for 2 h. The solid that formed upon cooling was collected and dried to furnish the targeted aminothiophene compound 6.
Yellow powder; yield (89%); m.p. 198–200 °C. IR (\(\overline{\nu }\)/cm−1): 3405, 3284 (NH2 and N–H), 1650, 1616 (C=O). 1H NMR (δ/ppm): 2.84 (s, 6H, –N(Me)2), 6.70 (br. s, 2H, NH2), 7.05–7.08 (m, 1H, Ar–H), 7.28–7.35 (m, 4H, Ar–H), 7.42–7.47 (m, 5H, Ar–H), 7.56–7.58 (dd, J1 = 8.00, J2 = 2.50 Hz, 2H, Ar–H), 7.88 (s, 2H, NH2), 8.07 (s, 2H, Ar–H), 9.62 (s, 1H, NH), 9.82 (s, 1H, NH). 13C NMR (δ/ppm): 40.65 (2C), 95.83, 106.22, 112.51 (2C), 119.95 (2C), 122.10 (2C), 123.58, 128.26, 128.76 (2C), 129.40 (2C), 129.64 (2C), 132.16, 137.20, 139.94, 146.03, 153.98, 157.18, 161.78, 185.17. Analysis Calcd. for C26H24N4O2S (456.16): C, 68.40; H, 5.30; N, 12.27%; found: C, 68.25; H, 5.36; N, 12.20%.
Preparation of ethyl 4-amino-3-((4-(dimethylamino)phenyl)carbamoyl)-2-(phenylamino)thiophene-5-carboxylate (7)
Acrylamide compound 4 (0.002 mol, 0.67 g) was refluxed with ethyl bromoacetate (0.002 mol, 0.22 ml) and triethylamine (0.5 ml) in ethanol for 2 h. The formed precipitate underwent filtration, was left to dry, then recrystallized from ethyl alcohol.
Gray powder; yield (89%); m.p. 158–160 °C. IR (\(\overline{\nu }\)/cm−1): 3352, 3293 (NH2, N–H), 1740 (C=O), 1663 (C=O). 1H NMR (δ/ppm): 1.34 (t, J = 7.00 Hz, 3H, CH3), 2.94 (s, 6H, –N(Me)2), 4.28 (q, J = 7.00 Hz, 2H, CH2), 5.67 (s, 2H, NH2), 6.74 (d, J = 9.00 Hz, 2H, Ar–H), 7.10 (t, J = 7.50 Hz, 1H, Ar–H), 7.28–7.31 (m, 2H, Ar–H), 7.35–7.38 (m, 4H, Ar–H), 8.35 (s,1H, NH), 10.83 (s, 1H, NH). 13C NMR (δ/ppm): 14.58, 40.63 (2C), 59.05, 106.10, 112.46 (2C), 118.54, 119.92 (2C), 121.90 (2C), 123.67, 128.30, 129.36 (2C), 137.86, 140.98, 154.04, 157.15, 162.83, 163.51. Analysis Calcd. for C22H24N4O3S (424.16): C, 62.25; H, 5.70; N, 13.20%; found: C, 62.38; H, 5.74; N, 13.15%.
General procedure for the synthesis of α,β-unsaturated nitrile derivatives 8a–f
A suspension of cyanoacetamide scaffold 1 (0.001 mol, 0.203 g) and the appropriate aromatic aldehyde (0.001 mol) [namely; 4-methylbenzaldehyde, 4-methoxybenzaldehyde, 4-chlorobenzaldehyde, 2-(4-formylphenoxy)-N-(p-tolyl)acetamide, 2-(4-formylphenoxy)-N-(4-methoxyphenyl)acetamide, N-(4-chlorophenyl)-2-(4-formylphenoxy)acetamide] was refluxed for 2 h in absolute ethanol containing drops of piperidine. The precipitate was filtered and dried to produce the corresponding unsaturated nitriles 8a–f.
2-Cyano-N-(4-(dimethylamino)phenyl)-3-(p-tolyl)acrylamide (8a)
Red crystals; yield (81%); m.p. 198–200 °C. IR (\(\overline{\nu }\)/cm−1): 3381 (N–H), 2206 (C≡N), 1673 (C=O). 1H NMR (δ/ppm): 2.38 (s, 3H, CH3), 2.86 (s, 6H, –N(Me)2), 6.71 (d, J = 8.50 Hz, 2H, Ar–H), 7.39 (d, J = 8.50 Hz, 2H, Ar–H), 7.46 (d, J = 8.50 Hz, 2H, Ar–H), 7.88 (d, J = 8.00 Hz, 2H, Ar–H), 8.17 (s, 1H, C=CH), 10.07 (s, 1H, NH). 13C NMR (δ/ppm): 21.17, 40.48 (2C), 106.81, 112.41 (2C), 116.23, 122.13 (2C), 127.56, 128.96 (2C), 129.52 (2C), 131.63, 138.39, 147.88, 150.70, 165.08. Mass analysis (m/z, %): 305.25 (M+, 36.05%), 68.78 (69.89), 59.91 (100.0), 47.92 (51.88), 45.94 (49.28), 43.98 (51.08), 43.01 (87.65), 40.24 (71.48). Analysis Calcd. for C19H19N3O (305.15): C, 74.73; H, 6.27; N, 13.76%; found: C, 74.59; H, 6.20; N, 13.64%.
3-(p-Anisyl)-2-cyano-N-(4-(dimethylamino)phenyl)acrylamide (8b)
Orange crystals; yield (85%); m.p. 205–206 °C. IR (\(\overline{\nu }\)/cm−1): 3354 (N–H), 2206 (C≡N), 1672 (C=O). 1H NMR (δ/ppm): 2.86 (s, 6H, –N(Me)2), 3.85 (s, 3H, OCH3), 6.71 (d, J = 9.00 Hz, 2H, Ar–H), 7.15 (d, J = 9.00 Hz, 2H, Ar–H), 7.45 (d, J = 9.00 Hz, 2H, Ar–H), 7.99 (d, J = 9.00 Hz, 2H, Ar–H), 8.14 (s, 1H, C=CH), 9.99 (s, 1H, NH). 13C NMR (δ/ppm): 40.54 (2C), 56.02, 106.90, 112.63 (2C), 114.33 (2C), 116.18, 122.29 (2C), 124.92, 127.65, 130.48 (2C), 148.14, 150.58, 159.72, 164.87. Mass analysis (m/z, %): 321.20 (M+, 23.58%), 292.29 (100.0), 285.97 (97.91), 256.37 (79.44), 145.25 (75.57), 125.68 (68.39), 109.92 (73.09), 59.86 (77.72). Analysis Calcd. for C19H19N3O2 (321.15): C, 71.01; H, 5.96; N, 13.08%; found: C, C, 70.90; H, 5.89; N, 12.85%.
3-(4-Chlorophenyl)-2-cyano-N-(4-(dimethylamino)phenyl)acrylamide (8c)
Yellow powder; yield (87%); m.p. 248–250 °C. IR (\(\overline{\nu }\)/cm−1): 3321 (N–H), 2224 (C≡N), 1670 (C=O). 1H NMR (δ/ppm): 2.86 (s, 6H, –N(Me)2), 6.71 (d, J = 8.50 Hz, 2H, Ar–H), 7.46 (d, J = 9.00 Hz, 2H, Ar–H), 7.67 (d, J = 9.00 Hz, 2H, Ar–H), 7.98 (d, J = 8.50 Hz, 2H, Ar–H), 8.21 (s, 1H, C=CH), 10.14 (s, 1H, NH). 13C NMR (δ/ppm): 40.57 (2C), 106.83, 112.74 (2C), 116.09, 122.35 (2C), 127.57, 128.78 (2C), 131.11 (2C), 132.40, 133.73, 147.96, 150.64, 164.90. Mass analysis (m/z, %): 326.15 (M+, 30.94%), 324.05 (79.78), 309.90 (93.05), 268.94 (64.34), 252.80 (80.02), 142.10 (83.88), 73.07 (71.04), 71.13 (100.0). Analysis Calcd. for C18H16ClN3O (325.10): C, 66.36; H, 4.95; N, 12.90%; found: C, 66.24; H, 4.89; N, 12.81%.
2-Cyano-N-(4-(dimethylamino)phenyl)-3-(4-(2-oxo-2-(p-tolylamino)ethoxy)-phenyl)acrylamide (8d)
Orange powder; yield (95%); m.p. 258–260 °C. IR (\(\overline{\nu }\)/cm−1): 3407, 3338 (N–H), 2213 (C≡N), 1696, 1670 (C=O). 1H NMR (δ/ppm): 2.24 (s, 3H, CH3), 2.86 (s, 6H, –N(Me)2), 4.81 (s, 2H, CH2), 6.71 (d, J = 9.00 Hz, 2H, Ar–H), 7.12 (d, J = 8.50 Hz, 2H, Ar–H), 7.19 (d, J = 9.00 Hz, 2H, Ar–H), 7.45 (d, J = 9.00 Hz, 2H, Ar–H), 7.50 (d, J = 9.00 Hz, 2H, Ar–H), 8.00 (d, J = 8.50 Hz, 2H, Ar–H), 8.15 (s, 1H, C=CH), 10.00 (s, 1H, NH), 10.08 (s, 1H, NH). 13C NMR (δ/ppm): 21.15, 40.51 (2C), 66.79, 105.63, 112.51 (2C), 115.61 (2C), 116.44, 120.32 (2C), 122.17 (2C), 124.97, 127.38, 129.55 (2C), 130.25 (2C), 134.77, 136.49, 147.70, 149.88, 159.84, 161.67, 166.01. Mass analysis (m/z, %): 454.45 (M+, 6.32%), 387.22 (62.14), 212.14 (100.00), 125.09 (51.07), 118.12 (45.58), 91.12 (50.40), 77.10 (46.57), 65.11 (46.64). Analysis Calcd. for C27H26N4O3 (454.20): C, 71.35; H, 5.77; N, 12.33%; found: C, 71.30; H, 5.74; N, 12.70%.
2-Cyano-N-(4-(dimethylamino)phenyl)-3-(4-(2-((4-methoxy phenyl)amino)-2-oxoethoxy)phenyl) acrylamide (8e)
Red powder; yield (93%); m.p. 238–240 °C. IR (\(\overline{\nu }\)/cm−1): 3398 (N–H), 2199 (C≡N), 1685, 1665 (C=O). 1H NMR (δ/ppm): 2.86 (s, 6H, –N(Me)2), 3.71 (s, 3H, OCH3), 4.80 (s, 2H, CH2), 6.71 (d, J = 9.50 Hz, 2H, Ar–H), 6.89 (d, J = 9.50 Hz, 2H, Ar–H), 7.19 (d, J = 9.00 Hz, 2H, Ar–H), 7.45 (d, J = 9.50 Hz, 2H, Ar–H), 7.53 (d, J = 9.00 Hz, 2H, Ar–H), 8.00 (d, J = 9.00 Hz, 2H, Ar–H), 8.15 (s, 1H, C=CH), 10.00 (s, 1H, NH), 10.03 (s, 1H, NH). 13C NMR (δ/ppm): 40.57 (2C), 55.96, 66.87, 105.53, 112.44 (2C), 114.48 (2C), 115.54 (2C), 116.60, 122.11 (2C), 122.97 (2C), 125.05, 127.35, 130.19 (2C), 131.82, 147.76, 149.79, 159.08, 159.92, 161.84, 166.23. Mass analysis (m/z, %): 470.12 (M+, 18.62%), 374.51 (79.57), 276.61 (58.38), 256.62 (100.00), 248.14 (43.70), 213.40 (49.57), 83.70 (31.43), 75.55 (64.69). Analysis Calcd. for C27H26N4O4 (454.20): C, 68.92; H, 5.57; N, 11.91%; found: C, 68.82; H, 5.49; N, 11.87%.
3-(4-(2-((4-Chlorophenyl)amino)-2-oxoethoxy)phenyl)-2-cyano-N-(4-(dimethylamino)phenyl)acrylamide (8f)
Orange powder; yield (88%); m.p. 258–260 °C. IR (\(\overline{\nu }\)/cm−1): 3407, 3347(N–H), 2211 (C≡N), 1702, 1668 (C=O). 1H NMR (δ/ppm): 2.86 (s, 6H, –N(Me)2), 4.84 (s, 2H, CH2), 6.70 (d, J = 9.00 Hz, 2H, Ar–H), 7.19 (d, J = 8.50 Hz, 2H, Ar–H), 7.39 (d, J = 9.00 Hz, 2H, Ar–H), 7.45 (d, J = 9.50 Hz, 2H, Ar–H), 7.65 (d, J = 9.50 Hz, 2H, Ar–H), 8.00 (d, J = 9.50 Hz, 2H, Ar–H), 8.15 (s, 1H, C=CH), 10.01 (s, 1H, NH), 10.32 (s, 1H, NH). 13C NMR (δ/ppm): 40.36 (2C), 66.98, 104.28, 112.37 (2C), 115.50 (2C), 116.93, 121.22 (2C), 122.06 (2C), 125.13, 127.34, 127.76, 128.74 (2C), 132.34 (2C), 137.33, 147.64, 149.66, 160.04, 161.04, 166.15. Analysis Calcd. for C26H23ClN4O3 (474.15): C, 65.75; H, 4.88; N, 11.80%; found: C, 65.75; H, 4.88; N, 11.87%.
General procedure for the synthesis of 3,5-dicyanopyridone derivatives 9a–f
To a solution of each α,β-unsaturated nitrile derivative 8a–f (0.001 mol) in 10 ml dioxane, malononitrile (0.001 mol, 0.07 g) and two drops of piperidine were added. The mixture was refluxed for 4–6 h. The solid that produced was filtered to obtain the corresponding pyridine derivatives 9a–f.
6-Amino-3,5-dicyano-1-(4-(dimethylamino)phenyl)-2-oxo-4-(p-tolyl)-1,2-dihydropyridine (9a)
Yellow powder; yield (39%); m.p. over 300 °C. IR (\(\overline{\nu }\)/cm−1): 3272, 3184 (NH2), 2216 (C≡N), 1664 (C=O). 1H NMR (δ/ppm): 2.39 (s, 3H, CH3), 2.97 (s, 6H, –N(Me)2), 6.83 (d, J = 9.00 Hz, 2H, Ar–H), 7.11 (d, J = 9.00 Hz, 2H, Ar–H), 7.36 (d, J = 8.00 Hz, 2H, Ar–H), 7.41 (d, J = 8.00 Hz, 2H, Ar–H). 13C NMR (δ/ppm): 20.99, 39.50 (2C), 66.36, 74.91, 88.05, 113.17 (2C), 115.90, 116.66, 121.40, 127.95 (2C), 128.73 (2C), 129.19 (2C), 131.82, 140.13, 150.94, 157.72, 159.96, 161.00. Analysis Calcd. for C22H19N5O (369.16): C, 71.53; H, 5.18; N, 18.96%; found: C, 71.73; H, 5.10; N, 18.1%.
6-Amino-4-(p-anisyl)-3,5-dicyano-1-(4-(dimethylamino)phenyl)-2-oxo-1,2-dihydropyridine (9b)
Yellow powder; yield (46%); m.p. over 300 °C. IR (\(\overline{\nu }\)/cm−1): 3263, 3181 (NH2), 2214 (C≡N), 1661 (C=O). 1H NMR (δ/ppm): 2.97 (s, 6H, –N(Me)2), 3.83 (s, 3H, OCH3), 6.84 (d, J = 13 Hz, 2H, Ar–H), 7.09 (d, J = 5.5 Hz, 2H, Ar–H), 7.11 (d, J = 4.5 Hz, 2H, Ar–H), 7.49 (d, J = 12 Hz, 2H, Ar–H). Mass analysis (m/z, %): 385.83 (M+, 23.74%), 369.93 (100.00), 340.98 (65.94), 320.02 (38.82), 239.72 (41.35), 146.69 (92.94), 121.34 (44.15), 81.69 (35.74). Analysis Calcd. for C22H19N5O2 (385.15): C, 68.56; H, 4.97; N, 18.17%; found: C, 68.43; H, 4.88; N, 18.04%.
6-Amino-3,5-dicyano-4-(4-chlorophenyl)-1-(4-(dimethylamino)phenyl)-2-oxo-1,2-dihydropyridine (9c)
Yellow powder; yield (42%); m.p. over 300 °C. IR (\(\overline{\nu }\)/cm−1): 3268, 3180 (NH2), 2219 (C≡N), 1661 (C=O). 1H NMR (δ/ppm): 2.97 (s, 6H, –N(Me)2), 6.84 (d, J = 9.00 Hz, 2H, Ar–H), 7.09 (d, J = 9.00 Hz, 2H, Ar–H), 7.56 (d, J = 9.00 Hz, 2H, Ar–H), 7.65 (d, J = 9.00 Hz, 2H, Ar–H). Mass analysis (m/z, %): 391.39 (M+, 6.25%), 211.16 (36.82), 185.14 (39.83), 78.12 (42.31), 69.11 (100.00), 57.13 (55.89), 44.08 (81.77), 43.12 (54.55). Analysis Calcd. for C21H16ClN5O (389.10): C, 64.70; H, 4.14; N, 17.96%; found: C, 64.86; H, 4.21; N, 17.86%.
2-((5′-Amino-2′,6′-dicyano-4″-(dimethylamino)-3′-oxo-3′,4′-dihydro-[1,1′:4′,1″-terphenyl]-4-yl)oxy)-N-(p-tolyl)acetamide (9d)
Yellow powder; yield (48%); m.p. over 300 °C. IR (\(\overline{\nu }\)/cm−1): 3340, 3278, 3183 (NH2 and N–H), 2213 (C≡N), 1693, 1659 (C=O). 1H NMR (δ/ppm): 2.25 (s, 3H, CH3) 2.97 (s, 6H, –N(Me)2), 4.78 (s, 2H, CH2), 6.83 (d, J = 9.00 Hz, 2H, Ar–H), 7.16–7.09 (m, 6H, Ar–H), 7.54–7.50 (m, 4H, Ar–H), 10.07 (s, 1H, NH). Mass analysis (m/z, %): 518.20 (M+, 24.25%), 216.88 (60.94), 200.02 (62.47), 195.92 (65.18), 190.34 (100.00), 155.28 (80.86), 118.20 (76.35), 58.91 (53.19). Analysis Calcd. for C31H27N5O3 (517.21): C, 71.94; H, 5.26; N, 13.53%; found: C, 71.86; H, 5.20; N, 13.48%.
2-((5′-Amino-2′,6′-dicyano-4″-(dimethylamino)-3′-oxo-3′,4′-dihydro-[1,1′:4′,1″-terphenyl]-4-yl)oxy)-N-(p-anisyl)acetamide (9e)
Yellow powder; yield (47%); m.p. over 300 °C. IR (\(\overline{\nu }\)/cm−1): 3337, 3285, 3191 (NH2 and N–H), 2212(C≡N), 1661 (C=O). 1H NMR (δ/ppm): 2.97 (s, 6H, –N(Me)2), 3.71 (s, 3H, OCH3), 4.76 (s, 2H, CH2), 6.83 (d, J = 9.00 Hz, 2H, Ar–H), 6.89 (d, J = 9.00 Hz, 2H, Ar–H), 7.11 (d, J = 8.50 Hz, 2H, Ar–H), 7.16 (d, J = 9.00 Hz, 2H, Ar–H), 7.51 (d, J = 9.00 Hz, 2H, Ar–H), 7.55 (d, J = 8.50 Hz, 2H, Ar–H), 10.02 (s, 1H, NH). Mass analysis (m/z, %): 534.85 (M+, 11.25%), 208.33 (100.00), 154.20 (83.73), 125.14 (65.09), 112.28 (75.16), 106.20 (93.18), 79.56 (49.87), 45.42 (60.04). Analysis Calcd. for C31H27N5O4 (533.21): C, 69.78; H, 5.10; N, 13.13%; found: C, 69.67; H, 5.02; N, 13.01%.
2-((5′-Amino-2′,6′-dicyano-4″-(dimethylamino)-3′-oxo-3′,4′-dihydro-[1,1′:4′,1″-terphenyl]-4-yl)oxy)-N-(4-chlorophenyl)acetamide (9f)
Yellow powder; yield (54%); m.p. over 300 °C. IR (\(\overline{\nu }\)/cm−1): 3407, 3346 (NH2 and N–H), 2212 (C≡N), 1702, 1664 (C=O). 1H NMR (δ/ppm): 2.97 (s, 6H, –N(Me)2), 4.81 (s, 2H, CH2), 6.83 (d, J = 9.00 Hz, 2H, Ar–H), 7.10 (d, J = 9.00 Hz, 2H, Ar–H), 7.15 (d, J = 9.00 Hz, 2H, Ar–H), 7.38 (d, J = 9.00 Hz, 2H, Ar–H), 7.51 (d, J = 8.50 Hz, 2H, Ar–H), 7.69 (d, J = 8.50 Hz, 2H, Ar–H), 10.31 (s, 1H, NH). Mass analysis (m/z, %): 538.58 (M+, 23.63%), 277.49 (80.64), 266.16 (75.22), 177.04 (66.62), 145.70 (92.77), 79.77 (97.11), 79.26 (100.00), 69.49 (65.21). Analysis Calcd. for C30H24ClN5O3 (537.16): C, 66.98; H, 4.50; N, 13.02%; found: C, 66.90; H, 4.53; N, 13.11%.
Biological activity assays
Cell lines and reagents
HepG2 and MDA-MB-231 cell lines were purchased from Nawah Scientific Company, Egypt. Cells were grown in DMEM medium (BioWhittaker™) supplemented with bovine serum albumin (10%, Life Science Group L, UK, Cat No: S-001B-BR) and with 100 IU/ml penicillin/streptomycin (100 µg/ml) (Lonza, 17-602E). Doxorubicin was obtained from Sigma-Aldrich, solubilized in DMSO and kept at − 20 °C as a stock solution. The tested compounds were prepared in dimethyl sulfoxide (10 mM stock) (DMSO Cat. No. 20385.02, Serva, Heidelberg, Germany) and stored at − 20 °C.
Cytotoxic assay
Using the MTT assay to determine the cytotoxicity of tested compounds 1–9(a–d). MTT or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was purchased from SERVA, Germany. The cell lines were introduced into 96-well plates at a density of 4 × 104 cells/well in 100 μl of complete medium (tests were done in duplicates). These plates were incubated for 24 h, 5% CO2, at 37 °C for settle down and adhesion. The drug (Doxorubicin) solutions were earlier produced in DMSO (control) at 5 and 50 μM concentrations, these medication solutions were administered to the line cells for 48 h after adhesion. MTT (3-(4,5-dimethylthiazoyl)-2,5-diphenyl-tetrazolium bromide (MTT) (5 mg/ml (PBS) Phosphate Buffered Saline) was added, and the plate was incubated for 4 h. After that, acidified via sodium dodecyl sulfate (SDS) solution (10% SDS containing 0.01N HCl in 1× PBS) was used to solubilize formazan crystals. The absorbance was measured after 14 h of incubation at λ570–630 nm by a Biotek plate reader (Gen5™) [48, 49]. According to the initial screening, the cells underwent incubation with serial dilutions (25, 12.5, 6.25, 3.125, 1.56, and 0.78 µM) of compounds 1–9(a–d) for 48 h, then the viability was determined by using MTT reagent. The IC50% of the compounds 2, 6, 7, and 9c were calculated by using Prism 8.0 Software. The results were shown as percentage of control group (DMSO). Inhibition of cell growth was assessed using the equation:
The concentration that kills 50% of cells was identified after incubating the cells with six-point serial dilutions (50, 25, and 12.5 µM) of compounds 2, 6, 7, and 9c. Doxorubicin was used as reference. The IC50 was calculated by Prism Software.
Estimation of the pharmacokinetic parameters
The pharmacokinetics and drug-likeness properties were predicted for the synthesized compounds were predicted by online tool SwissADME predictor software (http://swissadme.ch/index.php) made by Swiss Institute of Bioinformatics. The 2D chemical structures of the tested compounds was copied into SMILEY mode from chemdraw program. The boiled-egg was generated based on the properties of each compound.
PASS online predictor for anticancer activity
The tool used for prediction is (PASS) (http://www.way2drug.com/passonline/predict.php). Prediction of activity spectra for substance. The 2D chemical structures of the tested compounds was copied into SMILEY mode from chemdraw program. Pa (probability to be active) and Pi (probability to be inactive) values were given by PASS online software for different anti-cancer activities and IDE inhibition.
Molecular docking study
All the molecular modeling studies were carried out using Molecular Operating Environment (MOE, 2015.01) software. The three-dimensional structure (3D) of the selected proteins (PDB ID 4y72), (PDB ID 2ra3) were downloaded from the PDB website. The water molecules and repeated chains were removed. Protons were added and the energy of the protein was minimized. The preparation of compounds for docking were carried out by energy minimization and potential energy calculation inside MOE program. MOE conducted the docking of the newly synthesized compounds, calculated the binding energies, and provided their binding modes.
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Lagergren P, et al. Cancer survivorship: an integral part of Europe’s research agenda. Mol Oncol. 2019;13(3):624–35. https://doi.org/10.1002/1878-0261.12428.
Ibrahim AH, Shash E, et al. General oncology care in Egypt. In: Al-Shamsi HO, et al., editors. Cancer in the Arab world. Singapore: Springer Singapore; 2022. p. 41–61.
Awasthi R, et al. Nanoparticles in cancer treatment: opportunities and obstacles. Curr Drug Targets. 2018;19(14):1696–709. https://doi.org/10.2174/1389450119666180326122831.
Metwally HM, et al. Arylidine extensions of 3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-benzenesulfonamide derivatives: synthesis, computational simulations and biological evaluation as tumor-associated carbonic anhydrase inhibitors. Bioorg Chem. 2023;135: 106492. https://doi.org/10.1016/j.bioorg.2023.106492.
Abdel-Latif E, et al. Synthesis, characterization, and anticancer activity (MCF-7) of some acetanilide-based heterocycles. J Heterocycl Chem. 2018;55(10):2334–41. https://doi.org/10.1002/jhet.3294.
Petrou A, Fesatidou M, Geronikaki A. Thiazole ring—a biologically active scaffold. Molecules. 2021;26(11):3166. https://doi.org/10.3390/molecules26113166.
Raveesha R, et al. Synthesis and characterization of novel thiazole derivatives as potential anticancer agents: molecular docking and DFT studies. Comput Toxicol. 2022;21: 100202. https://doi.org/10.1016/j.comtox.2021.100202.
Jiang W, et al. Design, synthesis, and evaluation of novel pyridone derivatives as potent BRD4 inhibitors for the potential treatment of prostate cancer. Bioorg Chem. 2022;119: 105575. https://doi.org/10.1016/j.bioorg.2021.105575.
da Cruz RMD, et al. Thiophene-based compounds with potential anti-inflammatory activity. Pharmaceuticals. 2021;14(7):692. https://doi.org/10.3390/ph14070692.
Maghraby MT, et al. Novel class of benzimidazole-thiazole hybrids: the privileged scaffolds of potent anti-inflammatory activity with dual inhibition of cyclooxygenase and 15-lipoxygenase enzymes. Bioorg Med Chem. 2020;28(7): 115403. https://doi.org/10.1016/j.bmc.2020.115403.
Fayed EA, et al. Design, synthesis, in silico studies, in vivo and in vitro assessment of pyridones and thiazolidinones as anti-inflammatory, antipyretic and ulcerogenic hits. J Mol Struct. 2022. https://doi.org/10.1016/j.molstruc.2022.132839.
Bondock S, Fadaly W, Metwally MA. Synthesis and antimicrobial activity of some new thiazole, thiophene and pyrazole derivatives containing benzothiazole moiety. Eur J Med Chem. 2010;45(9):3692–701. https://doi.org/10.1016/j.ejmech.2010.05.018.
Borcea AM, et al. An overview of the synthesis and antimicrobial, antiprotozoal, and antitumor activity of thiazole and bisthiazole derivatives. Molecules. 2021;26(3):624. https://doi.org/10.3390/molecules26030624.
Hwang J, et al. Optimization of peptide-based inhibitors targeting the HtrA serine protease in Chlamydia: design, synthesis and biological evaluation of pyridone-based and N-Capping group-modified analogues. Eur J Med Chem. 2021;224: 113692. https://doi.org/10.1016/j.ejmech.2021.113692.
Metwally HM, et al. Synthesis, DFT investigations, antioxidant, antibacterial activity and SAR-study of novel thiophene-2-carboxamide derivatives. BMC Chem. 2023;17(1):6. https://doi.org/10.1186/s13065-023-00917-2.
Sudha D, et al. 2-Methylimidazolium pyridine-2,5-dicarboxylato zinc(II) dihydrate: synthesis, characterization, DNA interaction, anti-microbial, anti-oxidant and anti-breast cancer studies. J Coord Chem. 2021;74:2701–19. https://doi.org/10.1080/00958972.2021.1981302.
de Araújo Neto LN, et al. Thiophene-thiosemicarbazone derivative (L10) exerts antifungal activity mediated by oxidative stress and apoptosis in C. albicans. Chemico-Biol Interact. 2020;320: 109028. https://doi.org/10.1016/j.cbi.2020.109028.
Saravana Kumar P, et al. Rapid isolation of ricinine, a pyridone alkaloid from Ricinus communis (L.) with antifungal properties. J Biol Act Prod Nat. 2022;12(1):33–41. https://doi.org/10.1080/22311866.2021.2021985.
Seck I, Nguemo F. Triazole, imidazole, and thiazole-based compounds as potential agents against coronavirus. Results Chem. 2021;3: 100132. https://doi.org/10.1016/j.rechem.2021.100132.
Anwar M, et al. Chapter 13—Pyridone and SARS-CoV-2. In: Niaz K, editor., et al., Application of natural products in SARS-CoV-2. London: Academic Press; 2023. p. 293–311.
Zhu H, et al. Sulfur-containing therapeutics in the treatment of Alzheimer’s disease. Med Chem Res. 2021;30(2):305–52. https://doi.org/10.1007/s00044-020-02687-1.
Scarim CB, Pavan FR. Thiazole, triazole, thio- and semicarbazone derivatives—promising moieties for drug development for the treatment of tuberculosis. Eur J Med Chem Rep. 2021;1: 100002. https://doi.org/10.1016/j.ejmcr.2021.100002.
Chen K, et al. Thiazole-based and thiazolidine-based protein tyrosine phosphatase 1B inhibitors as potential anti-diabetes agents. Med Chem Res. 2021;30(3):519–34. https://doi.org/10.1007/s00044-020-02668-4.
Audu O, et al. In silico design, chemical synthesis and biological screening of novel 4-(1H)-pyridone-based antimalarial agents. Chem Biol Drug Des. 2022;99(5):674–87. https://doi.org/10.1111/cbdd.13987.
Alizadeh SR, Ebrahimzadeh MA. Antiviral activities of pyridine fused and pyridine containing heterocycles, a review (from 2000 to 2020). Mini Rev Med Chem. 2021;21(17):2584–611. https://doi.org/10.2174/1389557521666210126143558.
Xu X, et al. Cobalt(III)-catalyzed regioselective C6 olefination of 2-pyridones using alkynes: olefination/directing group migration and olefination. Org Lett. 2021;23(12):4624–9. https://doi.org/10.1021/acs.orglett.1c01368.
Chen Y, et al. Design, synthesis and anti-fibrosis evaluation of imidazo[1,2-a]pyridine derivatives as potent ATX inhibitors. Bioorg Med Chem. 2021;46: 116362. https://doi.org/10.1016/j.bmc.2021.116362.
Leoni A, et al. Novel thiazole derivatives: a patent review (2008–2012; part 1). Expert Opin Ther Pat. 2014;24(2):201–16. https://doi.org/10.1517/13543776.2014.858121.
Puszkiel A, et al. Clinical pharmacokinetics and pharmacodynamics of dabrafenib. Clin Pharmacokinet. 2019;58(4):451–67. https://doi.org/10.1007/s40262-018-0703-0.
Keating GM. Dasatinib: a review in chronic myeloid leukaemia and Ph+ acute lymphoblastic leukaemia. Drugs. 2017;77(1):85–96. https://doi.org/10.1007/s40265-016-0677-x.
Pathania S, Chawla PA. Thiophene-based derivatives as anticancer agents: an overview on decade’s work. Bioorg Chem. 2020;101: 104026. https://doi.org/10.1016/j.bioorg.2020.104026.
Zhang Y, Pike A. Pyridones in drug discovery: recent advances. Bioorg Med Chem Lett. 2021;38: 127849. https://doi.org/10.1016/j.bmcl.2021.127849.
Pavletich NP. Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol. 1999;287(5):821–8. https://doi.org/10.1006/jmbi.1999.2640.
Zhang M, et al. CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res. 2021;11(5):1913–35.
Ayati A, et al. Thiazole in the targeted anticancer drug discovery. Future Med Chem. 2019;11(15):1929–52. https://doi.org/10.4155/fmc-2018-0416.
Metwally MA, Abdel-Latif E, Amer FA. Synthesis and reactions of some thiocarbamoyl derivatives. Sulfur Lett. 2003;26(3):119–26. https://doi.org/10.1080/0278611031000095322.
Wang K, et al. Cyanoacetamide multicomponent reaction (I): parallel synthesis of cyanoacetamides. J Comb Chem. 2009;11(5):920–7. https://doi.org/10.1021/cc9000778.
Tuntland T, et al. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front Pharmacol. 2014;5:174. https://doi.org/10.3389/fphar.2014.00174.
Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br J Pharmacol. 2007;152(1):21–37. https://doi.org/10.1038/sj.bjp.0707306.
Daina A, Michielin O, Zoete V. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model. 2014;54(12):3284–301. https://doi.org/10.1021/ci500467k.
Elmorsy MR, et al. Synthesis, biological evaluation and molecular docking of new triphenylamine-linked pyridine, thiazole and pyrazole analogues as anticancer agents. BMC Chem. 2022;16(1):88. https://doi.org/10.1186/s13065-022-00879-x.
Daina A, Zoete V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11(11):1117–21. https://doi.org/10.1002/cmdc.201600182.
Martin YC. A bioavailability score. J Med Chem. 2005;48(9):3164–70. https://doi.org/10.1021/jm0492002.
Lagunin A, et al. PASS: prediction of activity spectra for biologically active substances. Bioinformatics. 2000;16(8):747–8. https://doi.org/10.1093/bioinformatics/16.8.747.
Farghaly TA, et al. Discovery of thiazole-based-chalcones and 4-hetarylthiazoles as potent anticancer agents: synthesis, docking study and anticancer activity. Bioorg Chem. 2020;98: 103761. https://doi.org/10.1016/j.bioorg.2020.103761.
Brown NR, et al. CDK1 structures reveal conserved and unique features of the essential cell cycle CDK. Nat Commun. 2015;6(1):6769. https://doi.org/10.1038/ncomms7769.
Brenk R, et al. Probing molecular docking in a charged model binding site. J Mol Biol. 2006;357(5):1449–70. https://doi.org/10.1016/j.jmb.2006.01.034.
El-Senduny FF, et al. Urea-functionalized organoselenium compounds as promising anti-HepG2 and apoptosis-inducing agents. Future Med Chem. 2021;13(19):1655–77. https://doi.org/10.4155/fmc-2021-0114.
Skehan P, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–12. https://doi.org/10.1093/jnci/82.13.1107.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at Northern Border University, Arar, KSA for funding this research work through the project number “NBU-FFR-2023-0105”.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
HMM: conceptualization, computational study, writing original draft, data analysis, proofreading, manuscript handling. NMY: synthesis, methodology, characterization of compounds. EA-L and AER: supervision, initial corrections, biological evaluation, data curation. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Table S1. Pharmaceutical prediction of in silico ADMET properties compounds 1-9f. Table S2. Computational prediction of the biological activity spectrum for compounds 1-9f.Fig. S1. The binding interaction of 1 with (PDB ID: 4y72). Fig. S2 The binding interaction of 2 with (PDB ID: 4y72).Fig. S3 The binding interaction of 3 with (PDB ID: 4y72).Fig. S4 The binding interaction of 4 with (PDB ID: 4y72).Fig. S5 The binding interaction of 5 with (PDB ID: 4y72). Fig. S6 The binding interaction of 6 with (PDB ID: 4y72).Fig. S7 The binding interaction of 7with (PDB ID: 4y72). Fig. S8 The binding interaction of 8awith (PDB ID: 4y72).Fig. S9 The binding interaction of 8b with (PDB ID: 4y72). Fig. S10 The binding interaction of 8c with (PDB ID: 4y72). Fig. S11 The binding interaction of 8d with (PDB ID: 4y72). Fig. S12 The binding interaction of 8e with (PDB ID: 4y72). Fig. S13 The binding interaction of 8f with (PDB ID: 4y72). Fig. S14 The binding interaction of 9awith (PDB ID: 4y72). Fig. S15 The binding interaction of 9b with (PDB ID: 4y72). Fig. S16 The binding interaction of 9c with (PDB ID: 4y72). Fig. S17 The binding interaction of 9d with (PDB ID: 4y72). Fig. S18 The binding interaction of 9e with (PDB ID: 4y72). Fig. S19 The binding interaction of 9f with (PDB ID: 4y72). Fig. S20 The binding interaction Doxorubicin with (PDB ID: 4y72). Fig. S21 The binding interaction of 1 with (PDB ID: 2ra3). Fig. S22 The binding interaction of 2 with (PDB ID: 2ra3). Fig. S23 The binding interaction of 3 with (PDB ID: 2ra3). Fig. S24 The binding interaction of 4 with (PDB ID: 2ra3). Fig. S25 The binding interaction of 5 with (PDB ID: 2ra3). Fig. S26 The binding interaction of 6 with (PDB ID: 2ra3). Fig. S27 The binding interaction of 7with (PDB ID: 2ra3). Fig. S28 The binding interaction of 8awith (PDB ID: 2ra3). Fig. S29 The binding interaction of 8b with (PDB ID: 2ra3). Fig. S30 The binding interaction of 8c with (PDB ID: 2ra3). Fig. S31 The binding interaction of 8d with (PDB ID: 2ra3). Fig. S32 The binding interaction of 8e with (PDB ID: 2ra3 ). Fig. S33 The binding interaction of 8f with (PDB ID: 2ra3). Fig. S34 The binding interaction of 9awith (PDB ID: 2ra3). Fig. S35 The binding interaction of 9b with (PDB ID: 2ra3). Fig. S36 The binding interaction of 9c with (PDB ID: 2ra3). Fig. S37 The binding interaction of 9d with (PDB ID: 2ra3). Fig. S38 The binding interaction of 9e with (PDB ID: 2ra3). Fig. S39 The binding interaction of 9f with (PDB ID: 2ra3). Fig. S40 The binding interaction Doxorubicin with (PDB ID: 2ra3). Table S3. Interaction between drugs 1-9(a-f) and target proteins (4y72 and 2ra3) and their docking scores. Table S4. Cell viability and growth inhibition percent after treatment of cells with 25 µM of the tested compounds. Fig S41. IC50% for the compound 2 against HepG2, MDA-MB-231 cell lines. Fig. S42 Microscopic images of HepG2 cells following 48 h of exposure to compounds 2, 6, 7, and 9c with different concentrations (50, 25, and 12.5 µM). Figure S43. Microscopic images of MDA-MB-231 cells following 48 h of exposure to exposure to compounds 2, 6, 7, and 9c with different concentrations (50, 25, and 12.5 μM). Fig. S44 1H-NMR spectrum of compound 1. Fig. S45 13C-NMR spectrum of compound 1. Fig. S46 1H-NMR spectrum of compound 2. Fig. S47 13C-NMR spectrum of compound 2. Fig. S48 1H-NMR spectrum of compound 3. Fig. S49 13C-NMR spectrum of compound 3. Fig. S50 1H-NMR spectrum of compound 4. Fig. S51 13C-NMR spectrum of compound 4. Fig. S52 1H-NMR spectrum of compound 5. Fig. S53 13C-NMR spectrum of compound 5. Fig. S54 1H-NMR spectrum of compound 6. Fig. S55 13C-NMR spectrum of compound 6. Fig. S56 1H-NMR spectrum of compound 7. Fig. S57 13C-NMR spectrum of compound 7. Fig. S58 1H-NMR spectrum of compound 8a. Fig. S59 13C-NMR spectrum of compound 8a. Fig. S60 1H-NMR spectrum of compound 8b. Fig. S61 13C-NMR spectrum of compound 8b. Fig. S62 1H-NMR spectrum of compound 8c. Fig. S63 13C-NMR spectrum of compound 8c. Fig. S64 1H-NMR spectrum of compound 8d. Fig. S65 13C-NMR spectrum of compound 8d. Fig. S66 1H-NMR spectrum of compound 8e. Fig. S67 13C-NMR spectrum of compound 8e. Fig. S68 1H-NMR spectrum of compound 8f. Fig. S69 13C-NMR spectrum of compound 8f. Fig. S70 1H-NMR spectrum of compound 9a. Fig. S71 1H-NMR spectrum of compound 9b. Fig. S72 1H-NMR spectrum of compound 9c. Fig. S73 1H-NMR spectrum of compound 9d. Fig. S74 1H-NMR spectrum of compound 9e. Fig. S75 1H-NMR spectrum of compound 9f.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Metwally, H.M., Younis, N.M., Abdel-Latif, E. et al. New thiazole, thiophene and 2-pyridone compounds incorporating dimethylaniline moiety: synthesis, cytotoxicity, ADME and molecular docking studies. BMC Chemistry 18, 52 (2024). https://doi.org/10.1186/s13065-024-01136-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-024-01136-z