
Abstract
Fraxinus hubeiensis is a plant endemic to China and widely used as folk medicine to treat various diseases. However, its chemical constituents have never been reported sufficiently. Thus, the primary objective of this study was to investigate the phytochemical constituents and biological activities of F. hubeiensis leaves. Hence, combined column chromatographic and spectroscopic techniques were used to identify and characterize the secondary metabolites such as a pair of 3-keto-glycoside epimers (1) and (2), along with five known compounds (3 ~ 7). The results of α-glucosidase inhibitory activity exhibited that 1 and 2 had moderate activity with IC50 values of 359.50 and 468.43 µM, respectively, compared to a positive control acarbose with the IC50 value of 164.08 µM. However, Compounds 1–6 were shown to be inactive against the tested microbes.
Similar content being viewed by others

Introduction
Fraxinus hubeiensis belongs to the family Oleaceae which comprises about 27 genera and more than 400 species in the world [1, 2]. About 30 species of the Fraxinus genus native to China and some of which were used in traditional Chinese Medicines as Cortex Fraxini [3], commonly known as ‘Qin-Pi’ in Chinese. Because of its very narrow distribution and relatively long growth cycle, F. hubeiensis, as a rare species, has received little attention [4]. In folk applications, it has also been widely used in bonsai due to its exquisite shape. However, the phytochemical study for F. hubeiensis has barely been reported [5,6,7], which showed that five coumarins, one flavonoid, one steroid, one iridoid glycosides and two fatty acids have been found from the leaves and barks of F. hubeiensis [5] to the best of our knowledge. Besides, the previous research was mainly focused on the essential oil and its antimicrobial effects. In continuing our search for diversity of phytochemistry and bioactivities from F. hubeiensis [5], we have investigated the constituents of the leaves of this plant, which led to the isolation of seven compounds, including a pair of 3-keto-glycoside epimers ethyl α-D-ribo-hex-3-ulopyranoside (1) and ethyl β-D-ribo-hex-3-ulopyranoside (2), as well as five known compounds (3–7). In this paper, we described the isolation and structural elucidation of these compounds by spectroscopic analysis and comparison with those of literature values. The α-glucosidase inhibitory activity of compounds 1 and 2 were evaluated. Meanwhile, compounds 1–6 were screened for antimicrobial activity. Thus, all of these could lay the foundation for discussing the chemotaxonomic research on Fraxinus and elucidating its active ingredients, and indicate great potential for the development of novel plant-derived hypoglycemic drugs.
Results and discussion
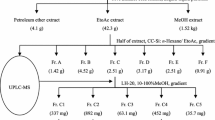
The ethyl acetate (EtOAc) extract (189.0 g, 3.78% yield) form a 5.0 kg sample of F. hubeiensis was loaded to column chromatography (CC) on silica gel to yield eight fractions, which were purified further by repeated CC and semi-preparative HPLC to afford a mixture of 1 (1.5 mg) and 2 (2.1 mg), 3 (5.3 mg), 4 (4.0 mg), 5 (7.0 mg), 6 (5.2 mg) and 7 (21.4 mg) (Fig. 1).

Chemical structures of 1–7 isolated from F. hubeiensis
Initially, the mixture (1 and 2) was obtained and exhibited a single spot in TLC and one peak detected over a rountine HPLC equipment. Its 1 H and 13 C NMR spectra showed two sets of resonances respectively, and can be assigned clearly to each compound, which suggested the mixture was a racemic. The above deduction was in accordance with its optical activity which was close to zero. To confirm, subsequent chiral HPLC of the mixture resulted in the purification of two enantiomers in a ratio of 1:2, with opposite optical rotation values.
The molecular formula of α-D-ribo-hex-3-ulopyranoside (1) was determined as C8H14O6 based on its positive HR-ESI-MS data of [M + Na]+ peak at m/z 229.0683 (calcd for 229.0688) and supported by the 13 C-NMR data (Table 1), indicating two degrees of unsaturation. The IR spectrum of 1 exhibited the presence of hydroxy at 3512 cm-1, carbonyl at 1652 cm-1. The 1 H-NMR data of 1 exhibited the presence of one methyl group resonating at δH 1.20 (t, J = 7.1 Hz); two methylene groups at δH 3.80 (dd, J = 12.0, 4.7 Hz, H-6a), 3.86 (d, J = 12.0, 2.2 Hz, H-6b), and 3.54 (dq, J = 9.8, 7.1 Hz, H-9a), 3.78 (dq, J = 9.8, 7.1 Hz, H-9b); four methine group at δH 5.16 (d, J = 4.3 Hz, H-1), 4.39 (dd, J = 4.3, 1.6 Hz, H-2), 4.22 (dd, J = 9.7, 1.6 Hz, H-4) and 3.69 (ddd, J = 9.7, 4.7, 2.2 Hz, H-5), combined with its DEPT and HSQC spectrum. All eight carbons were well resolved in the 13 C NMR spectrum, which indicated the presence of a carbonyl carbon in the downfield region. The connectivity of CH (4)- CH (5)-CH2 (6) could be well deduced from the cross-peaks of H-4/H-5/H-6 in the 1 H-1 H COSY spectrum (Fig. 2). Meanwhile, the linkage of CH (1)-CH (2) was established based on the cross-peaks of H-1/H-2. The detailed analysis of 1D and 2D NMR spectra implied 1 had a 3-keto-glycoside skeleton, along with their comparison with those of the reported glycoside derivatives. The remaining ethoxy group based on the peak shapes from the 1 H-NMR and 1 H-1 H COSY spectrum, was linked with C-1 to form the glycoside, considering the molecular formula and the unsaturation degrees. For the stereochemistry, the anomeric proton exhibited the relatively small coupling constant (J = 4.3 Hz), supporting the α-glycosidic configuration. Thus, the structure of 1 was elucidated as α-D-ribo-hex-3-ulopyranoside as shown in Fig. 1.

Key 1 H-1 H COSY and HMBC correlations of compounds 1 and 2
β-D-ribo-hex-3-ulopyranoside (2), was also obtained as colorless gum. The molecular formula was determined to be C8H14O6 based by HR-ESI-MS peak at [M + Na]+ peak at m/z 229.0697 (calcd for 229.0688). Interesting that, the 1 H and 13 C-NMR signals were different from those of its enantiomer 1, even when they were mixed together. Meanwhile, the HMBC cross-peaks confirmed the structure of 2 (Fig. 2), which was previously found as a biotransformation product in the Coleus forskohlii root culture induced from Agrobacterium rhizogenes [8]. The large coupling constant (J = 7.9 Hz) of the anomeric proton indicated it had β-glycosidic configuration. Therefore, the structure of 2, as a new natural product, was determined as β-D-ribo-hex-3-ulopyranoside.
The known compounds (Fig. 1) were characterized as methylinsitol (3) [9], umbelliferone (4) [10], esculetin (5) [11], scopoletin (6) [12], salicylic acid (7) [13], by comparison of their NMR data with those of the reported literature.
The α-glucosidase inhibition assay was carried out for a pair of 3-keto-glycoside epimers 1 and 2. Compounds 1 and 2 exhibited moderate activities with their IC50 values of 359.50 and 468.43µM respectively, compared to acarbose with the IC50 value of 164.08µM (Table 2). Meanwhile, the antimicrobial activities of compounds 1–6 were tested, while none of them exhibited activities.
Experimental
General experimental procedures
Optical rotations were measured on an Autopol-III automatic polarimeter (Rudolph Research Analytical, USA). IR spectra were recorded on an Agilent Technologies Cary 630 FTIR spectrophotometer with KBr pellets. NMR spectra were acquired on Bruker AV-600 spectrometer using methanol-d4 (CD3OD) or chloroform-d1 (CDCl3) as references, locked to the deuterium signal of the solvent. Chemical shifts were given in parts per million (ppm), and coupling constants in hertz (Hz). HRESIMS were measured on a Waters UHPLC-H-CLASS/XEVO G2-XS Q-TOF mass spectrometer (Waters, USA). Semi-preparative HPLC was performed using an Agilent 1260 Infinity II liquid chromatograph system equipped with UV-VIS detector. And a reversed-phase C18 column (Agilent Eclipse XDB, 250 × 9.4 mm., 5 μm) and a chiral-phase column (NQ (2)-RH, 250 × 4.6 mm, 5 μm) were used for purification and enantioselective analysis. Thin-layer chromatography (TLC) was conducted using silica gel GF254 (Qingdao Haiyang Chemical Co. Ltd.) plates. Open column chromatography was performed using silica gel (Qingdao Haiyang Chemical Co. Ltd., 200–300 mesh) and Sephadex LH-20 (Pharmacia Biotech Ltd.) and reverse-phase C18 silica gel ODS (Merck & Co., Inc. USA). All the solvents were of analytical grade and were purchased from Beijing Chemical Company Ltd. TLC spots were observed under UV light or by spraying with 5% sulfuric acid vanillin solution.
α-glucosidase and acarbose from Saccharomyces cerevisiae, p-nitrophenyl-α-glucopyranoside (pNPG) were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA), and they were all stored at − 20 °C before using. Staphylococcus aureus [S. aureus, CMCC (B) 26,003], was purchased from Shanghai Luwei Technology Co., Ltd.). Escherichia coli (E. coli, ATCC 25,922), Pseudomonas aeruginosa (P. aeruginosa, ATCC 27,853) and Candida albicans [C. albicans, CMCC (F) 98,001] and Bacillus subtilis (B. subtilis), were all purchased from Shanghai Bioresource Collection Center.
Plant materials
The dried leaves of F. hubeiensis were collected in the Bonsai garden of Jingzhou in China, in June 2017. Plant materials were identified by one of the authors (Prof. Qing-Lai Wu). A voucher specimen (No. DJBL201706) was deposited at TCM and Ethnomedicine Innovation & Development International Laboratory in Hunan University of Chinese Medicine.
Extraction and isolation
The dried leaves of F. hubeiensis (5.0 kg) were pulverized and immersed in 40 L 95% aqueous EtOH for three times at room temperature, each time 10 days. The solvent was evaporated under vacuum to obtain a crude extract (1.01 g), and the extract was suspended in 2 L H2O and partitioned successively with petroleum ether (PE, 60 ~ 90 °C) and ethyl acetate (EtOAc), to yield PE (311.0 g), EtOAc (189.0 g), and water (420.1 g) fractions. The EtOAc fraction was isolated by column chromatography (CC) using silica gel (200–300 mesh) and eluted with PE/CH2Cl2 (9:1), PE/EtOAc (9:1), CH2Cl2/EtOAc (9:1), CH2Cl2/MeOH (9:1), CH2Cl2/MeOH (1:1) gradientally, to yield eight fractions (Fr.A-H).
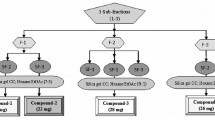
Fr. A (2.1 g) was loaded to silica gel CC and eluted with CH2Cl2/EtOAc (1:0 ~ 0:1) to obtain twelve subfractions (Fr. A1-A12), and subfraction Fr. A9 was absorbed to CC with Sephadex LH-20, eluting with CH2Cl2/MeOH (1:1) to obtain fourteen subfractions and yield compound 4 (4.0 mg) and 6 (5.2 mg). Fr. C (5.1 g) was absorbed to silica gel CC (CH2Cl2/EtOAc,1:0 ~ 0:1) to obtain eight major subfractions (Fr.C1-C8). And then Fr.C5 was loaded to a Sephadex LH-20 CC (CH2Cl2/MeOH, 1:1) and recrystallized to yield compound 5 (7.0 mg).
Fr. G (40.3 g) was isolated by CC on silica gel, eluting with CH2Cl2/EtOAc (1:0 ~ 0:1) to get ten subfractions (Fr. G1-G10), and Fr.G9 (1.2 g) was loaded on a Sephadex LH-20 CC, eluting with MeOH to obtain eight subfractions (Fr.G9.1-G9.8). Fr. G9.4 was purified on Sephadex LH-20 CC, eluting with MeOH to yield compound 3 (5.3 mg). Fr. G9.2 was purified by semi-preparative HPLC (MeOH/H2O, 35:65, 3 mL/min) to yield a mixture of 1 and 2 (4.0 mg, tR: 3.5 min), exhibiting a single peak, whose value of optical rotation was close to zero. Therefore, the mixture of 1 and 2 was separated on a Chiral NQ (2)-RH Column (MeOH/H2O, 75:25, 0.7 mL/min) to obtain 1(1.5 mg), tR1:4.7 min and 2 (2.1 mg), tR2:5.2 min.
Compound (1): white, amorphous gum; \([{{\upalpha }]}_{D}^{25}\) + 5.8 (c 0.06, MeOH); UV (MeOH) λmax (log ε): 198 (1.43) ,282 (0.38) nm; IR (KBr) νmax3512, 2958, 1652, 1506, 1032, 1018 cm−1; 1 H and 13 C NMR data (CD3OD, 600/151 MHz), see Table 1; HRESIMS m/z 229.0683 [M + Na]+ (calcd for C8H14O6Na+, 229.0688).
Compound (2): white, amorphous gum; \([{{\upalpha }]}_{D}^{25}\) – 16.0 (c 0.08, MeOH); UV (MeOH) λmax (log ε): 198 (1.19), 286 (0.13) nm; IR (KBr) νmax3512, 2958, 1652, 1506, 1032, 1018 cm− 1; 1 H and 13 C NMR data (CD3OD, 600/151 MHz), see Table 1; HRESIMS m/z 229.0697 [M + Na]+ (calcd for C8H14O6Na+, 229.0688).
α-glucosidase inhibitory assay
The α-glucosidase inhibition assay was carried out on the basis of the method reported with minor modifications [14, 15]. Briefly, 2µL of test sample (0.1, 0.25, 0.5, 1.0 mmol/L) dissolved in 98µL of PBS buffer (0.1 mol/L, pH 6.8) and 25µL α-glucosidase (0.2 U/mL) solution were mixed and pre-incubated at 37 °C for 20 min in a 96-well microplate. Then, 25µL p-nitrophenyl-α-D-glucopyranoside (pNPG, 4 mmol/L) solution was added to initiate the reaction. The 96-well microplate was incubated at 37 °C for an additional 15 min before being stopped with 50µL Na2CO3 (0.2 mol/L). The absorbance of each well was measured at a wavelength of 405 nm, and the data were recorded and measured in parallel for three times. In this experiment, acarbose was used as the positive control, PBS buffer was used as the blank group, and DMSO solution was used as the negative control group. Other reagents were consistent with the sample experimental group. The absorbance at 405 nm was measured by microplate reader, and the inhibition rate was calculated.
The α-glucosidase inhibitory activity was expressed as percent inhibition and was calculated as follows:
Antimicrobial activity
Compounds 1–6 were screened for antimicrobial activities against the tested microbes (S. aureus, B. subtilis, E. coli, P. aeruginosa and C. albicans) by the previously reported method [16, 17]. Regrettably, compounds 1–6 exhibited no activities. Perhaps for these isolates, the rang of strains should be expanded to test for the activity. However, because of no sufficient of these compounds isolated from this plant, it was not able to screen them more broadly for antimicrobial activities.
Conclusion
In summary, seven compounds were isolated from the leaves of F. hubeiensis including two ketoglycosides, one methylinsitol, three coumarins and salicylic acid. All compounds except 5 were isolated from F. hubeiensis for the first time. This study is the first reported of successful isolation of a pair of 3-ketoglycoside isomers by chiral-phase HPLC, which are undescribed compounds from nature. The results of α-glucosidase inhibitory indicated that 1 and 2 possessed moderate activities. It was worth mentioning that the activity of 1 with α-configuration was better than that of 2 with β-configuration, so the subsequent work on structural modification based on the ketose-type skeleton and their structure-activity relationship deserves further study. According to traditional uses of the plant, compounds 1–6 were evaluated for antimicrobial effect. Unfortunately, these isolates had no activity. Therefore, more in-depth phytochemistry and pharmacological research is worth continuing in the future.
Data Availability
All data generated or analysed during this study are included in this current article and its supplementary information files.
References
Qu SZ, Xiang QB, Su PL. A new specie of the genus Fraxinus Linn. From Hubei province [J]. J Nanjing Forestry Univ (Natural Sci Edition). 1979;21:146–8.
Zeng XB, Wang JC, Wu WX. Fraxinus hupehensis—A new specie of the genus Fraxinus Linn. [J]. For Sci Technol. 1979;12:6–7.
Chinese Pharmacopoeia Commission. Pharmacopoeia of the people’s Republic of China [M]. Beijing: China Medical Science Press; 2020.
Cheng JR, Zhou MQ. The distribution status and conservation strategy of Fraxinus Hupehensis germplasm resources [J]. J Yangtze Univ (Natural Sci Edition). 2016;13(09):7–9.
Zhao CN, Yao ZL, Yang D, Ke J, Wu QL, Li JK, Zhou XD. Chemical constituents from Fraxinus hupehensis and their antifungal and herbicidal activities [J]. Biomolecules, 2020, 10(1).
Zhang LJ, Xu ZH, Xu HH, Li JK, Wu QL. GC–MS analysis and preliminary study on structures of extracts from Fraxinus hupehensis leaves [J]. Non-wood for Res. 2017;35(03):186–92.
Zhang LJ, Xu ZH, Xu HH, Li JK, Wu QL. GC–MS analysis and fungicidal activities determination of volatile components from the leaves of Fraxinus hupehensis [J]. J Henan Agricultural Sci. 2017;46(06):80–3.
Li W, Koike K, Asada Y, Yoshikawa T, Nikaido A. Biotransformation of low-molecular-weight alcohols by Coleus Forskohlii hairy root cultures [J]. Carbohydr Res, 2003, 338(8).
Zafer U, Nazli B, Elçin T, Omer K, Ihsan Y, Süheyla K. Flavonoid glycosides and methylinositol from Ebenus haussknechtii [J]. Nat Prod Res, 2006, 20(11).
Atsumi S, Hiroshi U, Masanori I, Hitoshi Y. Glutaminase inhibitory activity of umbelliferone isolated from kabosu (Citrus Sphaerocarpa Hort. Ex Tanaka) [J]. Nat Prod Res, 2020, 36(2).
Xu W, Wang JH, Ju BZ, Lan XJ, Ying XX, Didier S. Seven compounds from Portulaca oleracea L. and their anticholinesterase activities [J]. Nat Prod Res, 2021, 36(10).
Mamoon-Ur-Rashid, Saqib A, Muhammad A, John I, Carol C, Syed Qaiser S, Valerie AF, Alexander Irvine G. Muhammad Rafiullah K. Phytochemical and antitrypanosomal investigation of the fractions and compounds isolated from Artemisia elegantissima [J]. Pharm Biol, 2014, 52(8).
Chang CC, Ku AF, Tseng YY, Yang WB, Fang JM, Wong CH. 6,8-Di-C-glycosyl flavonoids from Dendrobium huoshanense [J]. J Nat Prod, 2010, 73(2).
Omar R, Li L, Yuan T, Seeram NP. α-Glucosidase inhibitory hydrolyzable tannins from Eugenia jambolana seeds [J]. J Nat Prod. 2012;75(8):1505–9.
Feng J, Yang XW, Wang RF. Bio-assay guided isolation and identification of α-glucosidase inhibitors from the leaves of Aquilaria sinensis [J]. Phytochemistry. 2011;72(2–3):242–7.
Lin XC, Cao S, Sun J, Lu D, Zhong B, Chun J. The chemical compositions, and antibacterial and antioxidant activities of four types of citrus essential oils [J]. Molecules, 2021, 26(11).
Fu C, Lan XH, Yuan JQ, Li CH, Li LM, Yu ZL, Tan TT, Yuan MQ, Du FG. Research on the optimization, key chemical constituents and antibacterial activity of the essential oil extraction process of Thuja Koraiensis Nakai [J]. J Microbiol Methods. 2022;194:106435.
Acknowledgements
We would like to express our appreciation to Prof. Pei-Wu Cui and Dr. Qing-Ling Xie for constructive suggestions in the biological assays and the NMR experiment of these isolates.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81903514), the Opening Project for the first-class disciplines of pharmacy (2021YX11) and the Innovation Training program (2022-86) for college students in Hunan University of Chinese Medicine.
Author information
Authors and Affiliations
Contributions
XDZ and HXT designed the overall experiments. XDZ, XYL and HXT contributed to the isolation and structure determination of these compounds with support of QLW collected the plant material. HXT, QJL, XZG, LXM, and WBS performed the biological study with support of YPY supervised the biological study. XDZ, XYL and HXT wrote the manuscript. XDZ and WW supervised the study. XDZ, YQJ and WW revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. The authors have obtained the permission to collect Fraxinus hubeiensis.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests to others.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, XY., Tang, HX., Sheng, WB. et al. Glycosides from the leaves of Fraxinus Hubeiensis. BMC Chemistry 17, 182 (2023). https://doi.org/10.1186/s13065-023-01070-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-023-01070-6