
Abstract
This study aims to develop an effective and sensitive HPLC (High Performance Liquid Chromatography) method to determine the nitrate concentration in fruits and vegetables (F & V) using a C18 column (ZORBAX Eclipse XDB-C18, 80Å, 250 × 4.6 mm, 5 μm (Agilent Technologies)) maintained at 40 0 C, a mobile phase made up of methanol and buffer (pentane sulfonic acid sodium salt solution), and a Photo Diode Array Detector (PDA) at 225 nm. The developed method is validated in terms of selectivity, linearity, accuracy, precision, suitability, the limit of detection (LOD), and the limit of quantification (LOQ) according to the European Union Decision 2002/657/EC. The result revealed that a ratio of 30: 70 of the organic modifier methanol and buffer with pH 2.8 shows the highest efficiency. The calibration curve shows linearity with a correlation coefficient (r) of 0.9985. The LOD and LOQ were found to be 2.26 mg/kg and 7.46 mg/kg. The recovery was in the range of 98.96–100.21%. Moreover, the greenness assessment scores of different approaches (eco-scale score of 76, AGREE score of 0.71, and few red shades in GAPI portray) were at a very excellent level. Thus, our developed method is fully validated and can determine the nitrate content in F & V.
Similar content being viewed by others

Introduction
Nitrate is a typical compound found in the environment, which constitutes a significant part of the nitrogen cycle. It enters the human food chain through the soil, water, chemical fertilizer, and food additives [1]. Nitrate profoundly exists in fruits and vegetables and contributes to about 85% of the dietary intake of nitrate [2]. The occurrence of nitrate in the human food chain has long been acknowledged as a severe issue concerning the risk of methemoglobinemia and the formation of carcinogenic N-nitroso compounds [3,4,5,6,7].
However, in recent years, the importance of monitoring residual nitrate in the human diet, including fruits and vegetables, has changed. Some researchers are also highlighting the favorable effects of dietary intake of nitrate due to the discovery of the profound significance of nitric oxide, a derivative of nitrate, in many physiological systems. The dietary consumption of nitrate exhibits many health benefits, including reduced risk of cardiovascular disease, reduction of blood pressure, stroke, renal failure, gastric ulcer, myocardial infarction, and metabolic syndrome [8,9,10]. No epidemiological evidence is present currently to link nitrate with cancer [11, 12]. It has also been hypothesized that the antioxidants and other nutritional content found in fruits and vegetables make them less likely to have adverse effects and more likely to provide beneficial ones [13, 14].
Therefore, the effect of nitrate emerges as a bargaining issue, as is the case for so many food items and nutrients. Sometimes, nitrate can be harmful at high levels, while it can be beneficial at other levels. An ADI for dietary nitrate was established in 2002 by the Joint FAO/WHO Expert Committee on Food Additives (JECFA) at a level of 3.7 mg NO3– per kg of body weight to avoid appreciable health risks [15]. Hence the monitoring of nitrate in fruits and vegetables is apparent.
Various analytical methods have been employed to identify and quantify nitrate in fruits and vegetables, including HPLC, spectrophotometry, ion exclusion chromatography, capillary electrophoresis, liquid chromatography, and ion chromatography [16,17,18,19,20,21]. Nevertheless, many of the methods described in specialized literature are very trying, less sensitive, require many reagents, are time-consuming, and have drawbacks. In recent years, the ion chromatography (IC) approach with conductivity or UV detection has been the most widely used technique due to its speed, simplicity, and ruggedness [18, 22,23,24] and appeared as a better alternative to HPLC techniques in anion including NO3−, NO2−, Cl− etc. and different drugs & pesticides analysis [25,26,27,28].
However, some authors documented process selectivity issues, such as interfering compounds like sugar phosphate and chloride ion that have the same chromatographic behavior as the nitrate ion [29, 30]. For nitrate analysis, recent literature has reported using UPLC/MS due to their higher sensitivity [31]. However, this more sophisticated instrumentation cannot be employed for all analytical methods in routine laboratories where many analyses are usually required. Apart from this, due to its high price, lack of trained operating personnel, and complexities in the operating system make, its accessibility is very limited in different research laboratories, especially those in developing countries [32, 33]. Further, the fluorometric HPLC method requires a complicated and time-consuming sample preparation procedure to remove matrix components [34]. In addition, in this method, the long sample preparation time can introduce nitrite contamination from the environment or convert nitrate to nitrous acid due to its unstable nature [34].
UV/Vis detector is the most adaptable detector known for its wide linear range and simplicity [35, 36]. But these detectors face several flaws; for example, during the detection of nitrate, the absorbance is observed at 210 nm, which is particularly susceptible to interference from chloride that usually presents itself in fruits and vegetables [34, 37]. Though electrochemical detection is more robust than UV/Vis detection, it is subjected to chloride interference and is not practical for routine analysis due to its poor sensitivity [37]. However, a highly sensitive, selective, speedy, and accurate method for determining nitrate with minimal sample manipulation, analysis time and regents, and no interference in the detection spectra is required.
Diode-Array Detection (DAD) or Photodiode-Array Detection (PDA) is an analytical technique where the array of diodes can measure the entire wavelength spectrum in real-time [38]. Therefore, compared with the HPLC/UV/Vis detection, which generally only measures a couple of user-selectable specific wavelengths, HPLC/PDA may help distinguish analytes with different spectra. Hence, we developed and validated a rapid and sensitive HPLC method with PDA detection to determine nitrate.
Materials and methods
Experimental design
In the present study, preliminary trials were performed with the standard to optimize suitable chromatographic conditions for the determination of nitrate, which is well known as Response Surface Methodology (RSM) [39]. The trial was carried out with the organic mobile phase modifier, which plays a crucial role in separating nitrate with a symmetric peak and in determining the sensitivity of the chromatographic analytical method. In the organic modifier, the methanol and buffer ratios were chosen as 25:75, 30:70, and 35:65, respectively. The pH levels of the mobile phase were selected as 2.6, 2.8, and 3.0, respectively, and they both contributed as the independent parameters of the technique (Supplementary Table 1).
In the RSM, the dependent parameters were the peak area, theoretical plates, and tailing factor, which reduced study time and optimized the peak resolution of the system. To select the best peaks of HPLC, the peak area and the theoretical plates should be maximum, and the tailing factor should be as low as possible [39].
Chemicals and reagents
We used analytical or HPLC-grade chemicals and reagents. HPLC grade methanol, HPLC grade 1-Pentanesulfonic acid sodium salt, analytical grade potassium nitrate (Merck, Germany), and analytical grade hydrochloric acid (Sigma Aldrich) were collected from an online supplier. Deionized water was used during the experiment as the solvent.
The organic mobile phase buffer solution was prepared by dissolving 1-Pentanesulfonic acid sodium salt (1.74 g) in 950 mL deionized water in a 1000 mL beaker. Further, deionized water was added to the mark. The standard KNO3 (162.70 mg) was weighed in a 100 mL volumetric flask and then dissolved in deionized water to prepare a 1000 ppm nitrate solution. The working standard solutions were designed by further diluting the standard stock solution.
Instrumentation and chromatographic condition
Analysis was performed using HPLC (Shimadzu HPLC Prominence-i LC-2030 LT) with Photo Diode Array (PDA) detector commanded by lab solution software. The separation was carried out on a C18 column (ZORBAX Eclipse XDB-C18, 80Å, 250 × 4.6 mm, 5 μm (Agilent Technologies). The standard and sample injection volume was 10 μL with a flow rate of 1 mL/min and a wavelength of 225 nm (Chosen through the peak purity testing from a wavelength range of 200–260 nm). The column oven temperature was 40 0 C, and the run time was 10 min. The analysis was performed isocratically.
Sample preparation and extraction
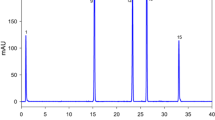
We used radish as a sample to develop the method in this study. The non-edible parts of the radish sample were removed and then subjected to cutting and homogenization. Then, the homogenized content was immediately stored at − 20 °C before the analysis. Of the homogenized content, 2 g was added to 50 mL of deionized water in a 100 mL volumetric flask to extract nitrate. The flask was then placed in a boiling water bath for 20 min at 80 °C, mixed by shaking, and kept on the table to cool down before further dilution to a final volume of 100 mL. Finally, 10 mL of the sample extract was passed through a 0.45 μ membrane filter. The first 3 mL of filtrate was discarded, and the rest was stored. The sample was analyzed within 1 h of preparation and extraction. Figure 1 shows the chromatograms of the nitrate in the radish sample. Using a standard calibration curve (Supplementary Fig. 1), the radish nitrate content was determined to be 2501 mg/kg.

Chromatogram of nitrate in radish sample
Method validation procedure
The method was validated according to the performance characteristics of European Commission Decision 2002/657/EC [40] and with the EU regulation 2017/625 [41]. The parameters such as linearity, system suitability, accuracy, precision (repeatability and reproducibility), the limit of detection (LOD), and the limit of quantification (LOQ) are evaluated [42].
Five different blank solutions (Eluent) were analyzed around the retention time of 2.018 min at wavelength 225 nm to verify the absence of other interfering compounds. The calibration curves were constructed using five different concentrations of nitrate (80, 90,100,110, 120 ppm). The linearity was calculated using the least-squares method to analyze a regression line representing the peak area as a function of the standard concentration. The linear calibration curve of the developed method can be expressed by the regression equation y = 17832.8*x + 93725.8 with the regression coefficient of 0.9999489 (see supplementary Fig. 1). The system suitability was determined by injecting 100 ppm standard solutions six times. Accuracy is defined as “the degree to which the result of a measurement conforms to the correct value or a standard” and refers to how close a measurement is to its agreed value. The accuracy study was performed at three concentration levels of 80 ppm, 100 ppm, and 120 ppm sample solution, i.e., the lower, middle, and higher concentrations. Three injections were performed for each concentration level, followed by calculating the retrieval/recovery efficiency. For preparing 100 ppm sample solution, 4 g of the radish sample encompassing 10 mg nitrate (calculated from the obtained nitrate during sample extraction) was added into 100 mL water. Similarly, for 80 ppm and 120 ppm concentration levels, 3.2 and 4.8 g of radish are added in 100 mL water, respectively.
We determined the repeatability using six injections of the sample solution having the same concentration of 0.1 mg/mL nitrate to verify the precision of the nitrate assay method. The intermediate precision-1 was determined in terms of reproducibility by injecting six sample solutions with the same concentration of 0.1 mg/mL nitrate after seven days of method precision study using the same sample and the analytical instruments used for method precision study. The intermediate precision-2 reveals the degree of reproducibility of test results obtained by analyzing the same samples under varying conditions (viz., slight change of flow rate and operating temperature) and is determined using six injections of the sample solution with the same concentration of 0.1 mg/mL nitrate by the same analyst.
We also performed the ruggedness test, which is the measurement of reproducibility performed in the same environmental and operational condition by another analyst since analyst-to-analyst results may vary. The results for this test are obtained by injecting the same solution six times (0.1 mg/mL).
In this study, the LOD and LOQ were also performed to understand the method sensitivity. LODs were calculated as the lowest observable concentration resulting in a signal-to-noise ratio of 3:1. The LOQs were estimated as the concentration giving a signal-to-noise ratio of 10:1. We calculated both the LOD and LOQ using repeated injections (n = 6) of the identical concentration (100 ppm) of standard solution.
Furthermore, we considered the greenness analytical chemistry concept and assessed the greenness of the method. This concept refers to the development of any eco-friendly technique which encompasses approaches like reducing hazardous substances, decreasing waste, increasing occupational safety, and reducing energy consumption [43]. All the mentioned approaches are usually consulted to make any analytical method inclined towards the fulfillment of sustainability aspects.
However, several tools are available to assess the greenness of any method, for example, Green Analytical Procedure Index (GAPI), National Environmental Method Index (NEMI), Analytical Eco-scale, Analytical Method Greenness Score (AMGS), and Analytical greenness (AGREE) metric [44,45,46,47]. Here, we employed the widely accepted and comprehensive analytical Eco-scale score method, GAPI and AGREE tools to evaluate the greenness of the proposed method of the present study [48,49,50]. The approach in Eco scale score involves assigning penalty points to different parameters considering their negative impact on the environment, followed by subtracting the total penalty score from 100 to obtain the Eco scale score [49, 50]. It is known/considered that when the eco scale value is closer to 100 it is considered greener, and hence criteria are set for the method in greenness analysis as ‘Ideal’ if the Eco scale score = 100, ‘Excellent’ if the score is > 50 and ‘Inadequate’ if the score is < 50. The GAPI tool facilitates qualitative assessment of greenness of any analytical method. It encompasses consideration of 15 parameters followed by representation of a five-pentagons symbol [47, 51]. The GAPI pictogram contains 3 color i.e. green, yellow and red which symbolize respectively low, medium and high impact of the respective parameters on the environment [44, 50]. In this method the greenness level usually visually appear through the color combination of the pictogram. If fewer non-green elements (i.e., reddish) and the more greenest and least green element (i.e., green and yellow tints) appear then it illustrate the excellent level of eco-friendliness of any analytical method [51].The other approach was the AGREE metric which covers all the 12 principles of green analytical chemistry [52]. It encompasses assignment of weightage to all principles to transform greenness into a unified scale of 0–1 and finally presenting the total greenness score in center after considering assessment result of each principle. The greenness of any method relies on the closeness of the score to 1 [47, 51].
Statistical analysis
We validated the method by calculating the relative standard deviation (RSD). In RSM, to optimize the best experimental conditions, we used a 3-level 2-factor face-centered central composite design (32 CCD) [53]. Variance analysis was employed in the quadratic polynomial model to determine the peak area, theoretical plate, and tailing factors. The determination coefficient (r2 statistic) was observed to evaluate the accuracy and general ability of the quadratic polynomial model. All the analysis was carried out using Minitab 17 statistical software (Minitab Inc. PA. 2010).
Results
Development and optimization of the chromatographic condition
We tried to optimize the concentration of the organic modifier and pH at various ratios. Considering the peak area, theoretical plates, and tailing factor, the highest sensitivity was observed at a ratio of 30:70 and pH of 2.7 (Fig. 2) (Data shown in Supplementary Table 2). The determination coefficient obtained (r2 statistic) indicates that the model can explain 78.71%, 99.81%, and 95.15% of the variability of the data regarding peak area, theoretical plate, and tailing factors, respectively.

Three dimensional surface response presenting the effect of ratio and pH on peak area, theoretical plates and tailing factor
Method validation results
During specificity analysis, no endogenous compounds were found to interfere with the eluent. The blank analysis does not show any peak area to reflect any other interfering compounds (Fig. 3). The data acquisition for the system suitability is measured using the retention time and peak areas of six consecutive injections. The result shows that the RSD is less than 2% in both cases, thereby indicating the system’s good performance (Supplementary Table 3). The findings of the accuracy tests are presented in Table 1 (Results shown under Supplementary Table 4).

Peak area from the blank analysis (specificity analysis)
The findings exhibit that the recovery ranged from 98.96 to 100.19%, with an RSD of 0.52%. The outputs from the precision and intermediate precision-1 tests revealed RSD of 0.38% and 0.43%, respectively (see Supplementary Tables 5 & 6). The intermediate precision-2 scored RSD < 2% with an average of 99.45% recovery (Table 2).
The outcomes obtained from the determination of the three precisions reveal that they are reproducible. The ruggedness test shows that the nitrate estimations do not notably affect the change of analysts as the values of % RSD were within the allowed limits of 2% (Table 3). We found the LOD and LOQ to be 2.26 mg/kg, and 7.46 mg/kg, respectively.
Results of greenness assessment of the method
Detailed strategies to assess the greenness of the method using analytical Eco scale are presented in Table 4. The penalty points of all potential steps of the methodology are listed and calculated. As can be seen in the table, the total calculated penalty point is 24, and so finally, the analytical eco-scale score is 76.
The associated parameters and final output in greenness assessment using GAPI tools and AGREE software are presented in Table 5. It can be observed that the AGREE score is 0.71 and in portrayed GAPI pictogram 6 fields are shaded green, 7 are in yellow and 2 are in red color.
Discussion
Response surface methodology (RSM) provides the required statistical tools for designing and analyzing experimental trials for process optimization [54]. On evaluating the effect of the independent parameters on the dependent parameters using 32 CCD, we found that the highest sensitivity was obtained at a concentration of organic modifier methanol: buffer at 30:70 and a pH of 2.8.
Through a complete validation procedure, according to the European Directives, the developed method proved to be selective, sensitive, and accurate [41]. The RSD to measure the system suitability, precision, intermediary precision, and ruggedness analysis is supposed to be less than 2%. Our study shows that RSD is within the acceptable criteria in all the cases.
This study revealed that the estimated LOD and LOQ were 2.26 mg/kg and 7.46 mg/kg, respectively, which is lower than the observed LOD and LOQ by Pardo-Marín et al. [55] (LOD = 7 mg/kg, and LOQ = 20 mg/kg) and Chung et al. [56] (LOD = 4 mg/kg, and LOQ = 20 mg/kg). These results imply that our method encompasses better detection and quantification of nitrate. Moreover, the recovery of our study is in the range of 98.9 to 100.21% which is an excellent result in comparison with the recovery result of Pardo-Marín et al. [55] (90–120%), Chung et al. [56] (83–106%), and Merino [57] (70–110%). The developed HPLC method of de Kleijn and Hoven [58] revealed an excellent LOD of 103 ng/kg; however, it had a run time of more than 20 min making it costly with high mobile phase consumption. The Capillary Ion Chromatography (CIC) analytical method was introduced by D’Amore et al. [30], which showed recovery of 93–105% and LOQ of 3.65 mg/kg. The developed method of Siu and Henshall [59] took 10 min for the analysis with a recovery of around 90% and a LOD of 500 μg/kg; thus, it can be postulated that our method is better in the aspects of sample recovery (98.9 to 100.21%) and LOD (2.26 mg/kg). Few spectrophotometric methods also exist, such as Zatar et al. [60] and Alonso et al. [61], which take more than 30 min of analysis time. Here, in our present study, we observed a retention time of 2.028 min with excellent recovery. On the contrary, the UPLC–MS technique shows a speedy separation and a very low analysis time of 1 min with the LOD and LOQ values of 0.029 and 0.088 mg/kg [31]. This might be due to the highly sensitive and selective MS detector; however, this advanced instrumentation cannot be used for routine assays where a large number of measurements are required. Furthermore, from the greenness assessment of this method, we found the analytical eco scale score is 76, which is > 50, which confirms this technique as an excellent green methodology. This findings is in concord with few previous excellent HPLC green methodology for their respective intended purpose proposed by MoHaMed and Lamie [62] having score of 86, Delhiraj and Anbazhagan [45] having score of 60, Mohamed and Fouad [63] having score of 57, Peleshok et al. [64] having score of 84, and Emam and Abdelwahab [65] having score of 90. In addition, the AGREE score of 0.71 is also indicating quite an excellent level of greenness of the method with low environmental impact. In displayed GAPI pictogram the greater number of green & yellow and few red tinted shade combinations symbolizes that this method has less effect on the environment and thereby reveals the technique as an excellent greener approach. Both the AGREE score and GAPI pictogram are conforms with the AGREE score and portrayed GAPI pictogram from the study of Chen et al. [66] (score 0.74), Chaudhari et al. [67] (score 0.68), and Jagnade et al. [68] ( score 0.69), which are assessed & presented in review article of Kannaiah et al. [44]. However, the reasons for variation in our results compared to others are mostly due to the variability in chemical usage, instrumentation, operational safety, wastage, energy utilization, and other strategies associated with the techniques [44]. So, finally, considering the sustainability aspects, we can say that this method is worth being sustainable and an excellent alternative to analyze nitrate in F & V.
Conclusion
This study presents a novel, sensitive, time-saving, and cost-effective HPLC method for analyzing nitrate content in fruits and vegetables. The method validation results, compliance with regulatory requirements, and high greenness score indicate its suitability for analysis and determination of nitrate in food products. Furthermore, the method’s precision, low solvent usage, and global availability of HPLC instruments in most research facilities emphasize its potential as an excellent alternative for routine assays for the intended purpose.
Data Availability
The dataset analyzed during the current study is available from the corresponding author on reasonable request.
References
Quijano L, Yusà V, Font G, McAllister C, Torres C, Pardo O. Risk assessment and monitoring programme of nitrates through vegetables in the region of Valencia (Spain). Food Chem Toxicol. 2017;100:42–9.
Santamaria P. Nitrate in vegetables: toxicity, content, intake and EC regulation. J Sci Food Agric. 2006;86(1):10–7.
Iammarino M, Di Taranto A. Nitrite and nitrate in fresh meats: a contribution to the estimation of admissible maximum limits to introduce in directive 95/2/EC. Int J Food Sci Technol. 2012;47(9):1852–8.
Gilchrist M, Winyard PG, Benjamin N. Dietary nitrate–good or bad? Nitric Oxide. 2010;22(2):104–9.
Cross AJ, Freedman ND, Ren J, Ward MH, Hollenbeck AR, Schatzkin A, et al. Meat consumption and risk of esophageal and gastric cancer in a large prospective study. Am J Gastroenterol. 2011;106(3):432.
Loh YH, Jakszyn P, Luben RN, Mulligan AA, Mitrou PN, Khaw K-T. N-nitroso compounds and cancer incidence: the european prospective investigation into Cancer and Nutrition (EPIC)–Norfolk Study. Am J Clin Nutr. 2011;93(5):1053–61.
Bahadoran Z, Mirmiran P, Ghasemi A, Kabir A, Azizi F, Hadaegh F. Is dietary nitrate/nitrite exposure a risk factor for development of thyroid abnormality? A systematic review and meta-analysis. Nitric Oxide. 2015;47:65–76.
Jonvik KL, Nyakayiru J, Pinckaers PJ, Senden JM, van Loon LJ, Verdijk LB. Nitrate-rich vegetables increase plasma nitrate and nitrite concentrations and lower blood pressure in healthy adults. J Nutr. 2016;146(5):986–93.
Habermeyer M, Roth A, Guth S, Diel P, Engel KH, Epe B, et al. Nitrate and nitrite in the diet: how to assess their benefit and risk for human health. Mol Nutr Food Res. 2015;59(1):106–28.
Bryan NS. Functional nitric oxide nutrition to combat cardiovascular disease. Curr Atheroscler Rep. 2018;20(5):21.
Bryan NS, Alexander DD, Coughlin JR, Milkowski AL, Boffetta P. Ingested nitrate and nitrite and stomach cancer risk: an updated review. Food Chem Toxicol. 2012;50(10):3646–65.
Eichholzer M, Gutzwiller F. Dietary nitrates, nitrites and N-nitroso compounds and cancer risk with special emphasis on the epidemiological evidence. Food safety: Contaminants and toxins. 2003:217 – 34.
Hord NG, Tang Y, Bryan NS. Food sources of nitrates and nitrites: the physiologic context for potential health benefits. Am J Clin Nutr. 2009;90(1):1–10.
Salama MF, Abbas A, Darweish MM, El-Hawwary AA, Al-Gayyar MM. Hepatoprotective effects of cod liver oil against sodium nitrite toxicity in rats. Pharm Biol. 2013;51(11):1435–43.
Authority EFS. Nitrate in vegetables-scientific opinion of the panel on contaminants in the Food chain. EFSA J. 2008;6(6):689.
Gu SH, Nicolas V, Lalis A, Sathirapongsasuti N, Yanagihara R. Complete genome sequence and molecular phylogeny of a newfound hantavirus harbored by the Doucet’s musk shrew (Crocidura douceti) in Guinea. Infection, Genetics and Evolution. 2013;20:118 – 23.
Öztekin N, Nutku MS, Erim FB. Simultaneous determination of nitrite and nitrate in meat products and vegetables by capillary electrophoresis. Food Chem. 2002;76(1):103–6.
Lopez-Moreno C, Perez IV, Urbano AM. Development and validation of an ionic chromatography method for the determination of nitrate, nitrite and chloride in meat. Food Chem. 2016;194:687–94.
Kissner R, Koppenol WH. Qualitative and quantitative determination of nitrite and nitrate with ion chromatography. Methods Enzymol. 2005;396:61–8.
Croitoru MD. Nitrite and nitrate can be accurately measured in samples of vegetal and animal origin using an HPLC-UV/VIS technique. J Chromatogr B. 2012;911:154–61.
Muhammad N, Hussian I, Ali A, Hussain T, Intisar A, Haq IU et al. A comprehensive review of liquid chromatography hyphenated to post-column photoinduced fluorescence detection system for determination of analytes. Arab J Chem. 2022:104091.
Amin M, Lim LW, Takeuchi T. Determination of common inorganic anions and cations by non-suppressed ion chromatography with column switching. J Chromatogr A. 2008;1182(2):169–75.
Muhammad N, Zhang Y, Asif M, Khan MFS, Intisar A, Mingli Y, et al. Feasibility of pyrohydrolysis and extended-steam distillation method for the extraction of two halides from zinc and lead concentrate samples followed by ion chromatography analysis. Microchem J. 2020;159:105593.
Muhammad N, Zia-ul-Haq M, Ali A, Naeem S, Intisar A, Han D, et al. Ion chromatography coupled with fluorescence/UV detector: a comprehensive review of its applications in pesticides and pharmaceutical drug analysis. Arab J Chem. 2021;14(3):102972.
Muhammad N, Subhani Q, Wang F, Guo D, Zhao Q, Wu S, et al. Application of a simple column-switching ion chromatography technique for removal of matrix interferences and sensitive fluorescence determination of acidic compounds (pharmaceutical drugs) in complex samples. J Chromatogr A. 2017;1515:69–80.
Muhammad N, Amjad A, Hussain I, Subhani Q, Dan-Dan G, Hai-Rong C, et al. Determination of fluorine and chlorine in standard steel residues and zinc sulfide concentrates by ion chromatography-matrix interference study. Chin J Anal Chem. 2023;51(2):100147.
Muhammad N, Zhang Y, Li W, Zhao YG, Ali A, Subhani Q, et al. Determination of nitenpyram and 6-chloronicotinic acid in environmental samples by ion chromatography coupled with online photochemically induced fluorescence detector. J Sep Sci. 2018;41(22):4096–104.
Muhammad N, Wang F, Subhani Q, Zhao Q, Qadir MA, Cui H, et al. Comprehensive two-dimensional ion chromatography (2D-IC) coupled to a post-column photochemical fluorescence detection system for determination of neonicotinoids (imidacloprid and clothianidin) in food samples. RSC Adv. 2018;8(17):9277–86.
Saccani G, Tanzi E, Cavalli S, Rohrer J. Determination of nitrite, nitrate, and glucose-6-phosphate in muscle tissues and cured meat by IC/MS. J AOAC Int. 2006;89(3):712–9.
D’Amore T, Di Taranto A, Vita V, Berardi G, Iammarino M. Development and validation of an analytical method for nitrite and nitrate determination in meat products by capillary ion chromatography (CIC). Food Anal Methods. 2019;12(8):1813–22.
Siddiqui MR, Wabaidur SM, ALOthman ZA, Rafiquee M. Rapid and sensitive method for analysis of nitrate in meat samples using ultra performance liquid chromatography–mass spectrometry. Spectrochim Acta Part A Mol Biomol Spectrosc. 2015;151:861–6.
Muhammad N, Subhani Q, Wang F, Lou C, Liu J, Zhu Y. Simultaneous determination of two plant growth regulators in ten food samples using ion chromatography combined with QuEChERS extraction method (IC-QuEChERS) and coupled with fluorescence detector. Food Chem. 2018;241:308–16.
Wang F, Zhang K, Wang N, Muhammad N, Wu S, Zhu Y. Ultrahigh-Performance Liquid Chromatography-Ion Chromatography System for the simultaneous determination of Vanillin, Ethyl Vanillin, and Inorganic Anions in Food samples. Food Anal Methods. 2018;11(1):243–50.
Singh P, Singh MK, Beg YR, Nishad GR. A review on spectroscopic methods for determination of nitrite and nitrate in environmental samples. Talanta. 2019;191:364–81.
Zuo Y, Wang C, Van T. Simultaneous determination of nitrite and nitrate in dew, rain, snow and lake water samples by ion-pair high-performance liquid chromatography. Talanta. 2006;70(2):281–5.
Hsu J, Arcot J, Lee NA. Nitrate and nitrite quantification from cured meat and vegetables and their estimated dietary intake in Australians. Food Chem. 2009;115(1):334–9.
Wang Q-H, Yu L-J, Liu Y, Lin L, Lu R-g, Zhu J-p, et al. Methods for the detection and determination of nitrite and nitrate: a review. Talanta. 2017;165:709–20.
SHIMADZU. UV vs Diode-Array (PDA). Detectors for (U)HPLC 2020 [Available from: https://www.ssi.shimadzu.com/sites/ssi.shimadzu.com/files/Industry/Literature/TechReport-RightInstrumentationCannabinoidsTerpenes-031620.pdfhttps://www.ssi.shimadzu.com/products/liquid-chromatography/knowledge-base/hplc-basics/uv-vs-pda-detectors.html
Ferreira SLC, Bruns RE, da Silva EGP, Dos Santos WNL, Quintella CM, David JM, et al. Statistical designs and response surface techniques for the optimization of chromatographic systems. J Chromatogr A. 2007;1158(1–2):2–14.
Commission E. Commission decision (2002/657/EC) of 12 August 2002, Brussels, Belgium. Official J Eur Communities. 2002;221:8–36.
Union EPCotE. Regulation (EU) 2017/625 of the European Parliament and of the Council of 15 March 2017. Official J Eur Union. 2017;95:1–142.
ISO. Statistical methods for use in proficiency testing by interlaboratory comparisons. International organization for standardization; 2005.
Sheldon RA. Metrics of green chemistry and sustainability: past, present, and future. ACS Sustain Chem Eng. 2018;6(1):32–48.
Kannaiah KP, Sugumaran A, Chanduluru HK, Rathinam S. Environmental impact of greenness assessment tools in liquid chromatography–A review. Microchem J. 2021;170:106685.
Delhiraj N, Anbazhagan S. Validated chromatographical methods for the simultaneous estimation of antihypertensive drugs in multicomponent formulations. Der Pharma Chemica. 2012;4(6):2416–21.
Hicks MB, Farrell W, Aurigemma C, Lehmann L, Weisel L, Nadeau K, et al. Making the move towards modernized greener separations: introduction of the analytical method greenness score (AMGS) calculator. Green Chem. 2019;21(7):1816–26.
Pena-Pereira F, Wojnowski W, Tobiszewski M. AGREE—Analytical GREEnness metric approach and software. Anal Chem. 2020;92(14):10076–82.
Gałuszka A, Migaszewski ZM, Konieczka P, Namieśnik J. Analytical Eco-Scale for assessing the greenness of analytical procedures. TRAC Trends Anal Chem. 2012;37:61–72.
Van Aken K, Strekowski L, Patiny L. EcoScale, a semi-quantitative tool to select an organic preparation based on economical and ecological parameters. Beilstein J Org Chem. 2006;2(1):3.
Płotka-Wasylka J. A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index. Talanta. 2018;181:204–9.
Aboras SI, Maher HM. Green adherent degradation kinetics study of Nirmatrelvir, an oral anti-COVID-19: characterization of degradation products using LC–MS with insilico toxicity profile. BMC Chem. 2023;17(1):23.
Gałuszka A, Migaszewski Z, Namieśnik J. The 12 principles of green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices. TRAC Trends Anal Chem. 2013;50:78–84.
Said KAM, Amin MAM. Overview on the response surface methodology (RSM) in extraction processes. J Appl Sci Process Eng. 2015;2(1).
Myers RH, Montgomery DC, Vining GG, Borror CM, Kowalski SM. Response surface methodology: a retrospective and literature survey. J Qual Technol. 2004;36(1):53–77.
Pardo-Marín O, Yusà-Pelechà V, Villalba-Martín P, Perez-Dasí J. Monitoring programme on nitrates in vegetables and vegetable-based baby foods marketed in the region of Valencia, Spain: levels and estimated daily intake. Food Addit Contam. 2010;27(4):478–86.
Chung SW, Tran JC, Tong KS, Chen MY, Xiao Y, Ho Y, et al. Nitrate and nitrite levels in commonly consumed vegetables in Hong Kong. Food Addit Contam. 2011;4(1):34–41.
Merino L. Development and validation of a method for determination of residual nitrite/nitrate in foodstuffs and water after zinc reduction. Food Anal Methods. 2009;2(3):212–20.
de Kleijn JP, Hoven K. Determination of nitrite and nitrate in meat products by high-performance liquid chromatography. Analyst. 1984;109(4):527–8.
Siu DC, Henshall A. Ion chromatographic determination of nitrate and nitrite in meat products. J Chromatogr A. 1998;804(1–2):157–60.
Zatar NA, Abu-Eid MA, Eid AF. Spectrophotometric determination of nitrite and nitrate using phosphomolybdenum blue complex. Talanta. 1999;50(4):819–26.
Alonso A, Etxaniz B, Martinez M. The determination of nitrate in cured meat products. A comparison of the HPLC UV/VIS and Cd/spectrophotometric methods. Food Addit Contam. 1992;9(2):111–7.
MoHaMed HM, Lamie NT. Analytical eco-scale for assessing the greenness of a developed RP-HPLC method used for simultaneous analysis of combined antihypertensive medications. J AOAC Int. 2016;99(5):1260–5.
Mohamed D, Fouad MM. Application of NEMI, Analytical Eco-Scale and GAPI tools for greenness assessment of three developed chromatographic methods for quantification of sulfadiazine and trimethoprim in bovine meat and chicken muscles: comparison to greenness profile of reported HPLC methods. Microchem J. 2020;157:104873.
Peleshok K, Piponski M, Ajie EA, Poliak O, Zarivna N, Denefil O, et al. Novel HPLC-UV method for simultaneous determination of valsartan and atenolol in fixed dosage form; study of green profile assessment. Pharmacia. 2021;68(1):43–51.
Emam AA, Abdelwahab NS. Stability-indicating chromatographic and chemometric methods for environmentally benign determination of canagliflozin and its major degradation product; a comparative study and greenness assessment. Biomed Chromatogr. 2019;33(10):e4612.
Chen K, Lynen F, De Beer M, Hitzel L, Ferguson P, Hanna-Brown M, et al. Selectivity optimization in green chromatography by gradient stationary phase optimized selectivity liquid chromatography. J Chromatogr A. 2010;1217(46):7222–30.
Chaudhari VS, Borkar RM, Murty US, Banerjee S. Analytical method development and validation of reverse-phase high-performance liquid chromatography (RP-HPLC) method for simultaneous quantifications of quercetin and piperine in dual-drug loaded nanostructured lipid carriers. J Pharm Biomed Anal. 2020;186:113325.
Jagnade S, Soni P, Omray LK. Development and validation of a Green Analytical Method for the determination of aspirin and Domperidone Bulk or Formulation using UV and HPLC. J Drug Delivery Ther. 2020;10(6):49–56.
Acknowledgements
Not applicable.
Funding
Authors haven’t received any funding for this study.
Author information
Authors and Affiliations
Contributions
This work was carried out in collaboration among all authors. Authors Rayhan Uddin and Mohammad Zia Uddin have performed the basic laboratory work. Rayhan Uddin and G.M. Rabiul Islam wrote the protocol and the first draft of the manuscript. G.M. Rabiul Islam and Mostak Uddin Thakur have made the concept, designed and supervised the work, and contributed to reviewing the manuscript draft.
Corresponding author
Ethics declarations
Ethical approval
This declaration is not applicable for this study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Uddin, R., Islam, G.M.R., Uddin, M.Z. et al. Development and validation of an effective and sensitive technique for nitrate determination in fruits and vegetables using HPLC/PDA. BMC Chemistry 17, 105 (2023). https://doi.org/10.1186/s13065-023-01008-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-023-01008-y