
Abstract
A novel, sensitive, and green micellar UPLC method was proposed and validated for the simultaneous determination of four hypoglycemic agents used in type II diabetes mellitus treatment namely, pioglitazone, alogliptin, glimepiride, and vildagliptin. The developed UPLC method was successfully applied for quantitative analysis of these drugs in bulk, in pharmaceutical formulations, and in spiked human plasma. Chromatographic separation was carried out on a Kinetex® 1.7 μm XB-C18 100 Å (50 × 2.1 mm) column, using a degassed and filtered mixture of (0.1 M SDS- 0.3% triethyl amine- 0.1% phosphoric acid (pH 6)) and n-propanol (85:15 v/v), at a flow rate of 0.2 mL/min. The experimental conditions of the suggested method were well investigated and optimized. The newly developed micellar UPLC method is capable of determining different dosage forms at the same time with the same solvents, saving time and effort. The method was found to be efficiently applicable in spiked human plasma and could be extended to study the pharmacokinetics of the cited drugs in real human plasma samples. The greenness of the developed method was evaluated by applying the Eco-scale scoring tool, which verified the excellent greenness of the analytical method.
Similar content being viewed by others

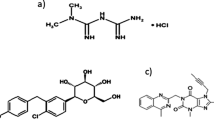
Introduction
Type 2 diabetes mellitus (T2DM) is a chronic metabolic syndrome that resulted from insulin deficiency which leads to hyperglycemia and serious complications such as micro- and macrovascular damage [1]. Management of T2DM can be achieved via treatment with oral hypoglycemic medications and by encouraging patients to change their lifestyle by following a balanced diet and regular exercise to control their blood glucose levels [2]. Oral hypoglycemic drugs can be used safely to control the blood glucose level in T2DM patients by different modes of action but due to the progressive characteristic of T2DM, a combination of oral hypoglycemic agents therapy is required [3].
Pioglitazone (PIO) Fig. 1a, 5-[[4-[2-(5-ethylpyridin-2-yl) ethoxy] phenyl]methyl]-1, 3-thiazolidine-2, 4-dione [4] is from thiazolidinediones that are indicated for the treatment of T2DM as it improves insulin sensitivity and increases glucose uptake [5]. Glimepiride (GLM) Fig. 1b, 1-[[p-[2-(3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido) ethyl] phenyl] sulfonyl]-3-(trans-4-methylcyclohexyl) urea [4], is a third-generation sulfonylurea with hypoglycemic activity, it increases the secretion of insulin by activating beta cells and is used for the management of T2DM to improve blood glucose control [6]. Alogliptin (ALO) Fig. 1c, 2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxopyrimidin-1- yl]methyl]benzonitrile [7] and vildagliptin (VLD) Fig. 1d, (2 S)-1-[2-[(3-hydroxy-1-adamantyl) amino] acetyl] pyrrolidine-2-carbonitrile [4] are Dipeptidyl Peptidase-4 (DPP-4) inhibitors that are used in the therapy of T2DM via increasing Insulin secretion and suppressing glucagon secretion [8].

Chemical structure of (a) Pioglitazone, (b) Glimepiride, (c) Alogliptin, and (d) Vildagliptin
Many clinical studies have proven that some combination therapy of hypoglycemic agents in T2DM patients can exert a superior blood glucose control than mono or single-ingredient therapy [9]. Pioglitazone (PIO) and Alogliptin (ALO) are present in the market as a combined therapy that is used for the management of T2DM under the trade name Prandaglim plus (15 mg pioglitazone/ 25 mg alogliptin). It was found that the combination of ALO and PIO gives a better effect in the treatment of T2DM than the effect of single drug treatment by improving the function of beta-cells [10]. The literature review reported a few analytical techniques for the estimation of PIO and ALO including spectrophotometric methods (area under the curve, first derivative of ratio spectra, and second order derivative) [11, 12], HPTLC [13, 14], LC-MS/MS in human plasma [15, 16] and RP-HPLC [17,18,19].
Pioglitazone (PIO) and Glimepiride (GLM) are available in the market under the trade name Zanoglide (30 mg pioglitazone / 4 mg glimepiride) combined therapy that is used for the treatment of T2DM patients who have a cardiovascular risk. Studies demonstrated that the combination treatment of pioglitazone with glimepiride can have a dual action of glycemic control and improving the body’s lipid profile and lowering cardiovascular risk [20]. Literature data reported a fewer number of analytical methods for quantitative analysis of PIO and GLM involving spectrophotometric methods (oxidative coupling and chemometric techniques) [21, 22], native spectrofluorimetry [23], LC/ tandem mass in human plasma [24] and RP-HPLC [25,26,27,28,29].
Vildagliptin (VLD) can be added to Glimepiride (GLM) for the therapy of T2DM. It is effective in improving glycemic control and well tolerated in diabetic patients when added to glimepiride as it decreases the risk of hypoglycemia and gaining weight [30, 31]. Some studies demonstrate that adding vildagliptin (VLD) to Pioglitazone (PIO) was observed to be more dynamic in glycemic control by maintaining β-cell function and lowering insulin resistance than either monotherapy component [32].
Literature data reports a few chromatographic methods for the simultaneous determination of VLD with GLM or PIO in bulk and commercially available drug products [33, 34]. Interestingly, none of the previous methods reported the simultaneous estimation of the studied drugs.
A Straightforward and well-established branch of high-performance liquid chromatography is micellar liquid chromatography (MLC). MLC has been used frequently to identify various substances in pharmaceutical medications [35, 36], biological fluids [37, 38], and food products [39].
The MLC method has multiple advantages over other conventional HPLC methods as MLC would enable concurrent separation of hydrophilic and hydrophobic solutes in the same run. MLC has rapid gradient capability, distinct separation selectivity, excellent reproducibility, reliability, robustness, improved detection, and affordability. MLC is an excellent ecological alternative for conventional HPLC since it enhances both environmental and economic influences. Also, the capacity of micellar mobile phase to dissolve sample proteins makes direct sample injection of biological samples into the column one of the principal applications of MLC [40].
In this work, a new and sensitive micellar UPLC method (MUPLC) was developed for the determination of PIO, ALO, GLM, and VLD in pure form, dosage forms, and spiked human plasma. Compared to the reported methods, the newly developed MUPLC method has advantages including better sensitivity, economic, low consumption of organic solvent, enhancement of the resolution, and eco-friendly to the environment.
Experimental
Material and reagents
Pure standards of ALO (99.85%), PIO (100.87%), GLM (99.32%), and VLD (99.91%) were kindly provided by EVA Pharma (Cairo, Egypt). Zanoglide 4/30 tablet, Batch number 2,011,283, labeled to contain 4 mg GLM and 30 mg PIO per tablet. Prandaglim plus 25/15 tablet, Batch number 2,103,041, labeled to contain 25 mg ALO and 15 mg PIO per tablet. Prandaglim 25 mg tablet, Batch number 2,001,171, each tablet contains 25 mg ALO as labeled. Gliptus 50 mg tablet, Batch number 2,010,622, labeled to contain 50 mg VLD. All tablets were manufactured by EVA Pharma (Cairo, Egypt) and purchased from the local market. Sodium dodecyl sulphate (SDS) is provided from (Merk, Germany).
Analytical grade orthophosphoric acid and triethyl amine and HPLC grade acetonitrile and n-propanol were purchased from Sigma-Aldrich (Germany). De-ionized water was freshly obtained in-house by the Millipore water purification system. Plasma samples were obtained from Al-Azhar University Hospital in Cairo, Egypt, and kept refrigerated till assayed.
Apparatus
The HPLC chromatographic system consisted of Agilent (1100 series) equipped with a quaternary pump (G 1311 A), and an automatic injector equipped with a 1 µl sample loop injector. Detection was achieved by UV-detector (model G1314 A). UPLC Core-Shell column Kinetex® 1.7 μm XB-C18 100 Å (50 × 2.1 mm) (USA). The mobile phase was degassed by a degasser (model G1322A). Jenway pH–Meter was purchased from the UK. Analytical balance (Precisa125A, Switzerland). Vortex (model IVM-300P, Taiwan), Benchtop centrifuge with (Hunan, China).
Chromatographic condition
-
Stationary phase: Kinetex® 1.7 μm XB-C18 100 Å (50 × 2.1 mm) column, the column temperature was ambient, and the injection volume was 1 µl.
-
Mobile phase: the mobile phase that was the best for chromatographic separation was consisting of solvent A: solvent B (85:15 v/v).
-
Solvent A: degassed and filtered mixture of [0.1 M SDS- 0.3% triethyl amine- 0.1% phosphoric acid (pH 6)].
-
Solvent B: n-propanol.
-
-
Flow rate: 0.2 mL/min.
-
Detection: UV detection at 225 nm.
Standard solutions
An accurately weighed amount of 10 mg of PIO, ALO, GLM, and VLD was transferred into a 100 ml volumetric flask and dissolved in 50 mL of the mobile phase then completed to volume with the same mobile phase to obtain a 100 µg/mL stock solution of each drug.
Procedures
Construction of calibration curves
Into a series of 10-mL volumetric flasks, different aliquots of PIO, ALO, GLM, and VLD were separately transferred using a micropipette from their (100 µg/mL) standard stock solutions, then the flasks were completed to volume with the mobile phase to reach a final concentration of (1–80 µg/mL) (0.1–25 µg/mL) (0.25-50 µg/mL) (0.3–50 µg/mL) for PIO, ALO, GLM, and VLD, respectively. These solutions were then transferred into vials of the autosampler and automatically injected into the column. Different calibration graphs were constructed by plotting the concentration of each drug against the corresponding peak area.
Procedure for pharmaceutical preparation
Five tablets of Zanoglid, Prendaglim plus, Prendaglim, and Gliptus, were separately weighed and ground in a mortar into a fine powder. Accurate weight of the powder of Zanoglid tablet (equivalent to 30 mg of PIO and 4 mg of GLM), Prendaglim plus tablet (equivalent to 15 mg of PIO and 25 mg of ALO), Prendaglim tablet (equivalent to 25 of ALO mg) and Gliptus tablet (equivalent to 50 mg of VLD) were separately transferred into 100-mL volumetric flask. 50 ml of the mobile phase was added followed by sonication for 30 min then the contents of the flasks were filtered. The flasks were completed to 100 mL volume with the same mobile phase to obtain stock solutions having a concentration of (300 µg/mL of PIO and 40 µg/mL of GLM), (150 µg/mL of PIO, and 250 µg/mL of ALO), (250 µg/mL of ALO) and (500 µg/mL of VLD). different dilutions with the mobile phase were done within the linear ranges. Separation was achieved as mentioned in the procedure section.
Procedure for spiked human plasma
For the calibration curve in human plasma, in a series of screw-capped tubes for centrifugation, 1 mL of human plasma was transferred into a series of centrifugation tubes and spiked with 1 mL containing different concentrations of PIO, ALO, GLM, and VLD, respectively and separately. Each centrifugation tube received 3 mL of acetonitrile (for protein denaturation), which was shaken and vortexed for 1 min. The samples were centrifuged at 4000 rpm for 30 min. The supernatants (protein-free layer) were taken and evaporated to dryness under a moderate nitrogen gas stream. The residue was dissolved in 2 ml of methanol, then transferred into a 10-mL volumetric flask and completed with methanol to the mark. 20µL of each solution was injected in triplicate using the above-mentioned chromatographic conditions, and the peak areas were calculated. The calibration curves represent the relationship between the peak areas and the corresponding concentrations in the range 5–25 µg/mL for PIO, ALO, and GLM.
Procedure for quality control samples
For the validation of the proposed MUPLC, three quality control samples: low (LQC), medium (MQC), and high (HQC) were prepared for the four studied drugs in concentrations of 5, 10, and 15 µg/mL. For the bioanalytical validation, low (LQC), medium (MQC), and high (HQC) samples were prepared in concentrations of 5, 15, and 25 µg/mL.
Results and discussions
The MUPLC method was proposed for simultaneous determination of PIO, ALO, GLM, and VLD with possible application to different dosage forms including Zanoglide (PIO/GLM), Prandaglim plus (ALO /PIO), Prandaglim (ALO), Gliptus (VLD) and in spiked human plasma.
Good separation of the cited drugs using the proposed MUPLC method was achieved at 2.05, 4.62, 6.16, and 7.35 min for PIO, ALO, GLM, and VLD, respectively with clear resolution between their peaks. Chromatograms in Figs. 2, 3, 4, 5 and 6 showed the separation of the studied drugs in their binary mixtures and spiked human plasma. As shown, there was no interference from the plasma matrix.

MUPLC chromatograms showing separation (20 µg/mL) of each PIO (Rt = 2.05 min), ALO (Rt = 4.62 min), GLM (Rt = 6.16 min), and VLD (Rt = 7.35 min) using the specified chromatographic conditions

MUPLC chromatograms of PIO (Rt = 2.05 min), and ALO (Rt = 4.62 min) in (a) Binary mixture (25:25 µg/mL) (b) Spiked plasma (25:25 µg/mL)

MUPLC chromatograms of PIO (Rt = 2.05 min), and GLM (Rt = 6.16 min) in (a) Binary mixture (25:25 µg/mL) (b) Spiked plasma (25:25 µg/mL)

MUPLC chromatograms of GLM (Rt =6.16 min) and VLD (Rt =7.35 min) in (a) Binary mixture (25:25 µg/mL) (b) Spiked plasma (25:25 µg/mL)

MUPLC chromatograms of PIO (Rt = 2.05 min), and VLD (Rt=7.35 min) in (a) Binary mixture (25:25 µg/mL) (b) Spiked plasma (25:25 µg/mL)
Advantages of the proposed method
For a company that produces these drugs alone or in a mixture, this method provides easy determination of different dosage forms at the same time and in one run with the same solvents, including saving time and effort required or column washing between runs, and the reduced amount of used solvents in the mobile phase in comparison to conventional HPLC, plus the actual advantages of using the MUPLC technique that include better sensitivity, economics, low consumption of organic solvent, enhancement of the resolution, and being eco-friendly to the environment.
Optimization of experimental conditions
In the proposed method, optimum efficient separation with fine resolution and symmetric shape peaks was achieved using a mobile phase composed of solvent A [mixture of 0.1 M SDS- 0.3% triethyl amine- 0.1% phosphoric acid (pH 6)] and solvent B (n-propanol) (85:15 v/v), on Kinetex® C18 column, with a flow rate of 0.2 mL/min and UV detection at 225 nm.
Choosing the optimum mobile phase composition
Different mobile phases were tried at different ratios to achieve optimum chromatographic separation. Best separation was achieved by using a mixture of [0.1 M SDS- 0.3% triethyl amine- 0.1% phosphoric acid (pH 6)] and n-propanol (85:15 v/v). the effect of changing the pH of the mobile phase was also tested and (pH 6) was the best for optimum separation. pH value was adjusted to 6 by adding 0.1% phosphoric acid for good resolution without tailing the peaks.
Choosing the optimum flow rate
The flow rate of the mobile phase was varied by ± 0.02 and a flow rate of 0.2 mL/ min resulted in a good resolution of peaks at a suitable retention time.
Choosing the optimum wavelength of detection
After trying several wavelengths from 210 to 240 nm, the UV detection of the studied hypoglycemic drugs was carried out at 225 nm at which the cited drugs showed good absorption.
Method validation
The proposed MUPLC was validated in accordance with the ICH guidelines [41] and the bioanalytical FDA validation guidelines [42].
Linearity
Calibration curves were constructed relating the linear relationship between the peak area and the corresponding drug concentrations (µg/ mL) in the range of (1–80 µg/mL), (0.1–25 µg/mL), (0.25-50 µg/mL), and (0.3–50 µg/mL) for PIO, ALO, GLM, and VLD, respectively for the pure drugs. Also, good linearity for the cited drugs, with correlation coefficients of 0.9999, 0.9998, 0.9997, and 0.9991 was obtained for PIO, ALO, GLM and VLD in the 5–25 µg/mL range for the spiked human plasma. The linear range of the proposed method and the regression equations parameters were shown in Tables 1&2.
Limit of detection (LOD) and limit of quantitation (LOQ)
LOD and LOQ were calculated and illustrated in Tables 1&2. Small detection and quantitation limit values indicate the sensitivity of the proposed MUPLC method. LOD and LOQ were determined from the standard deviation of the response and the slope.
Accuracy
The accuracy of the suggested method was assessed by the recovery study through the analysis of 3 different quality control concentrations of each drug (5, 10, and 15 µg/mL) for the pure form and (5, 15, and 25 µg/mL) for the spiked plasma in triplicate and calculating the mean recovery percentage (R % ±SD). The excellent recoveries indicated good accuracy of the method, Tables 1&2.
Precision
The repeatability of the suggested method was estimated by the analysis of three replicates of three quality control concentration levels of each drug (5, 10, and 15 µg/mL) for the pure form and (5, 15, and 25 µg/mL) for the spiked plasma on the same day. The values of precision or the relative standard deviation (RSD) were calculated. The intermediate precision was assessed by analysis of the same samples on three successive days. The obtained law RSD values were shown in Tables 1&2 which indicates that the suggested method is highly precise.
Stability of plasma samples
The stability of PIO, ALO, GLM, and VLD in plasma samples was investigated by triplicate analysis of the low and high-quality control samples.
Short-term stability
Short-term stability was checked by measuring the thawed LQC and HQC samples after being maintained at room temperature for 6 h.
Long-term stability
Long-term stability was assessed by analyzing LQC and HQC samples stored in the freezer at -80 °C for 14 days.
Freeze-thaw stability
Freeze-thaw stability of LQC and HQC samples was carried out after three freeze-thaw cycles where samples were frozen at -70 °C, and then samples were left to thaw spontaneously at room temperature and frozen again.
Stability was determined by calculating the % RSD as shown in Table 3.
Robustness
The robustness of the proposed method was checked by measuring of resolution after deliberating changes in the experimental method parameters including pH (± 0.5), flow rate (± 0.02 min), percent of solvent A (± 2%), and solvent B (± 2%). The induced slight changes did not result in any no significant change which indicates the method is robust and reliable, (Supplementary Table 1).
System suitability test
A system suitability test was used to evaluate the effectiveness of the developed chromatographic separation parameters. System suitability test parameters include the resolution factor, tailing factor, theoretical plates number or column efficiency, and retention time as shown in Table 4. The obtained results complied with food and drug administration (FDA) guidance.
Application to pharmaceutical tablets
The developed MUPLC method was applied for the analysis of different pharmaceutical dosage forms applying the standard addition technique. The recovery % of the studied drugs from their pharmaceutical tablets was calculated and illustrated in Table 5. No interference from excipients was observed which proved the applicability of the proposed method as shown in Figs. 2, 3, 4, 5 and 6. The results of the analysis of pharmaceutical tablets were compared to the results of the reported methods [18, 27, 43, 44] by statistical tests which showed no significant difference as shown in Table 6.
Analysis of spiked human plasma
The developed MUPLC method was tried to determine each drug concentration in spiked human plasma. The results of the estimation of PIO, GLM, and VLD in spiked plasma samples were satisfactory, as shown in Table 7. No interference from the plasma matrix was observed, as shown in Figs. 2, 3, 4, 5 and 6, and the chromatogram of the blank plasma (Supplementary Fig. 1) showed that there were no interfering substances present, so no pretreatment of the plasma is required.
Greenness assessment of the procedure using the analytical eco-scale
The Analytical Eco-scale scoring was applied to assess the greenness of the proposed MUPLC technique[45]. The analytical Eco-scale is based on the ideal green analytical procedures and has a total score of 100. Each parameter of the analytical procedures is assigned penalty points if it is deviated from the conception of green analysis [46]. Different parameters along the whole analysis procedure are to be evaluated including (hazards, type and amount of reagents used, energy consumption, and waste generation and its treatment). The sum penalty points were then subtracted from 100 to obtain the Eco-scale points of the method. Table 8 showed 85 Eco-score points which indicated an excellent green MUPLC method.
Conclusion
The newly developed MUPLC technique was successfully employed for the simultaneous estimation of PIO, ALO, GLM, and VLD in the pulk, different commercial dosage forms, and spiked human plasma. The proposed method has the advantages of being rapid, saving time and effort. Furthermore, this proposed method was sensitive enough to be used for the pharmacokinetic study of the studied drugs in human plasma. Moreover, the developed MUPLC method showed excellent greenness as it gains a high total score by using the eco-scale scoring tool in greenness assessment.
Data Availability
All data generated or analyzed during this study are included in this published article.
References
Bastaki S. Diabetes mellitus and its treatment. Dubai Diabetes and Endocrinology Journal. 2005;13:111–34.
Marín-Peñalver JJ, Martín-Timón I, Sevillano-Collantes C. del Cañizo-Gómez, Update on the treatment of type 2 diabetes mellitus. World J Diabetes. 2016;7:354–95.
Krentz AJ, Patel MB, Bailey CJ. New drugs for type 2 diabetes mellitus. Drugs. 2008;68:2131–62.
The United States pharmacopeia. National formulary. Volume 1. Rockville (MD): United States Pharmacopeial Convention; 2021.
Smith U. Pioglitazone: mechanism of action. Int J Clin Pract Suppl. 2001;121:13–8.
Meneses MJ, Silva BM, Sousa M, Sá R, Oliveira PF, Alves MG. Antidiabetic drugs: mechanisms of action and potential outcomes on cellular metabolism. Curr Pharm Design. 2015;21:3606–20.
O’Neil MJ. The Merck Index - An Encyclopedia of Chemicals, Drugs, and Biologicals. Cambridge, UK: Royal Society of Chemistry; 2013.
Christopher R, Karim A. Clinical pharmacology of alogliptin, a dipeptidyl peptidase-4 inhibitor, for the treatment of type 2 diabetes, Expert review of clinical pharmacology, 2 (2009) 589–600.
Rosenstock J, Kim SW, Baron MA, Camisasca RP, Cressier F, Outurier A, Dejager S. Efficacy and tolerability of initial combination therapy with vildagliptin and pioglitazone compared with component monotherapy in patients with type 2 diabetes. Diabetes Obes Metabolism. 2007;9:175–85.
Van Raalte DH, van Genugten RE, Eliasson B, Möller-Goede DL, Mari A, Tura A, Wilson C, Fleck P, Taskinen MR, Smith U, Diamant M. The effect of alogliptin and pioglitazone combination therapy on various aspects of β-cell function in patients with recent-onset type 2 diabetes. Eur J Endocrinol. 2014;170:565–74.
Shah DA. Estimation of pioglitazone hydrochloride and alogliptin benzoate in combination by second order derivative spectrophotometery method. Int Res J Pharm Sci. 2017;8:1–10.
Mabrouk MM, Hammad SF, Mansour FR, Amer MM. Three spectrophotometric methods for determination of alogliptin benzoate and pioglitazone HCl in combined tablet dosage form. Anal Chem Lett. 2018;8:368–78.
Sharma K, Parle A. Development and validation of HPTLC method for simultaneous estimation of alogliptin benzoate and pioglitazone hydrochloride in bulk drugs and combined dosage forms. Int J Pharm Sci Rev Res. 2015;4:35–42.
Shah D, Gajjar U, Mehta FA, Patel VB, Chhalotiya UK. Development of HPTLC method for the estimation of Pioglitazone and Alogliptin in synthetic mixture. Indian Drugs. 2017;54:44–52.
Kelani KM, Rezk MR, Badran OM, Elghobashy MR. Determination of pioglitazone, its metabolite and alogliptin in human plasma by a novel LC-MS/MS method; application to a pharmacokinetic study. J Chromatogr B. 2019;1132:121803.
D.S.S.S, Praveen S, Asha RK, Pigili. Simultaneous determination of alogliptin and pioglitazone in human plasma by a novel LC-MS/MS method. Curr Pharm Anal. 2020;16:564–77.
Abdel-Ghany MF, Abdel-Aziz O, Ayad MF, Tadros MM. New LC–UV methods for pharmaceutical analysis of novel anti-diabetic combinations. Acta Chromatographica. 2017;29:448–52.
Dhani R, Guptha HKDR, Rajamanickam D. Development of stability indicating method for the simultaneous estimation of alogliptin and pioglitazone in bulk and tablet dosage form by reversed-phase ultra-performance liquid chromatography method. J Appl Pharm Sci. 2019;9:51–6.
Kandala M, Mounika R, Sultana S, Hogue N, Ali S, Prashanthi S, Kabir S. Method Development and validation of alogliptin and pioglitazone by reversed-phase method in bulk and its marketed dosage form. World J Pharm Res. 2021;10:782–98.
Derosa G, Salvadeo SAT. Glimepiride-pioglitazone hydrochloride in the treatment of type 2 diabetes. Clin Medicine: Ther. 2009;1:835–45.
El-Zaher AA, Elkady EF, Elwy HM, Saleh MAEM. Simultaneous spectrophotometric determination of glimepiride and pioglitazone in binary mixture and combined dosage form using chemometric-assisted techniques. Spectrochim Acta Part A Mol Biomol Spectrosc. 2017;182:175–82.
Salma AT, Nawal A, Hanan AH. Kinetic and spectrophotometry methods for determination of two hypoglycemic drugs, pioglitazone hydrochloride and glimepiride in their pharmaceutical formulations. Res J Chem Environ. 2011;15:963–72.
Alarfaj NA, Al-Abdulkareem EA, Aly FA. Spectrofluorimetric determination of pioglitazone hydrochloride and glimepiride in their formulations and biological fluids. Asian J Chem. 2011;23:3441.
Ni XJ, Wang ZZ, Shang DW, Zhang M, Hu JQ, Qiu C, Wen YG. Simultaneous determination of glimepiride and pioglitazone in human plasma by liquid chromatography–tandem mass spectrometry and its application to pharmacokinetic study. J Chromatogr B. 2014;960:247–52.
Havaldar FH, Vairal DL. Simultaneous estimation of glimepiride, rosiglitazone and pioglitazone hydrochloride in the pharmaceutical dosage form. E-J Chem. 2010;7:1326–33.
Jain D, Jain S, Jain D, Amin M. Simultaneous estimation of metformin hydrochloride, pioglitazone hydrochloride, and glimepiride by RP-HPLC in tablet formulation. J Chromatogr Sci. 2008;46:501–4.
Karthik A, Subramanian G, Rao CM, Bhat K, Ranjithkumar A, Musmade P, Udupa N. Simultaneous determination of pioglitazone and glimepiride in bulk drug and pharmaceutical dosage form by RP-HPLC method. Pak J Pharm Sci. 2008;21:421–5.
Lakshmi KS, Rajesh T, Sharma S. Determination of pioglitazone and glimepiride in pharmaceutical formulations and rat plasma by RP-LC. Int J PharmTech Res. 2009;1:496–9.
Sane RT, Menon SN, Inamdar S, Mote M, Gundi G. Simultaneous determination of pioglitazone and glimepiride by high-performance liquid chromatography. Chromatographia. 2004;59:451–3.
Garber AJ, Foley JE, Banerji MA, Ebeling P, Gudbjörnsdottir S, Camisasca RP, Couturier A, Baron MA. Effects of vildagliptin on glucose control in patients with type 2 diabetes inadequately controlled with a sulphonylurea. Diabetes Obes Metabolism. 2008;10:1047–56.
Kikuchi M, Haneda M, Koya D, Tobe K, Onishi Y, Couturier A, Goodman M. Efficacy and tolerability of vildagliptin as an add-on to glimepiride in japanese patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2010;89:216–23.
Derosa G, Maffioli P, Ferrari I, Mereu R, Ragonesi PD, Querci F, Franzetti IG, Gadaleta G, Ciccarelli L, Piccinni MN, D’Angelo A, Salvadeo SAT. Effects of one year treatment of vildagliptin added to pioglitazone or glimepiride in poorly controlled type 2 diabetic patients. Horm Metab Res. 2010;42:663–9.
Inamdar HP, Mhaske AA, Sahastrabudhe SP. A revised Rp-Hplc method for simultaneous determination of Vildagliptin and Pioglitazone Hcl-Application to commercially available Drug Products. Int J Pharm Sci Res. 2013;4:847.
Amin MM, Abdel-Aziz SA, Mostafa SM, El-Adl SM. Optimization and validation of HPLC method for simultaneous determination of vildagliptin, pioglitazone hydrochloride and glimepiride in bulk and tablets. J Pharm Biol Sci. 2017;12:18–27.
Bahgat EA, Saleh H, Reda A, Fawzy MG. Development and validation of eco-friendly micellar organic solvent-free HPLC method for the simultaneous determination of some antihypertensive combinations. Microchem J. 2022;181:107740.
Bahgat EA, Hafez HM, El-Sayed HM, Kabil NAS. Development of a solvent-free micellar HPLC method for determination of five antidiabetic drugs using response surface methodology. Microchem J. 2022;179:107446.
Ibrahim F, Magdy G, Wahba ME. Utility of micellar liquid chromatography as an analytical tool for the estimation of three binary antibiotic mixtures. Application to biological fluids. J Taibah Univ Sci. 2020;14:931–42.
Belal F, Omar MA, Derayea S, Hammad MA, Zayed S, Saleh SF. Simultaneous determination of tizanidine, nimesulide, aceclofenac and paracetamol in tablets and biological fluids using micellar liquid chromatography. J Chromatogr Sci. 2018;56:233–41.
Ibrahim FA, Nasr JJM. Direct determination of ampicillin and amoxicillin residues in food samples after aqueous SDS extraction by micellar liquid chromatography with UV detection. Anal Methods. 2014;6:1523–9.
El-Shaheny RN, El-Maghrabey MH, Belal FF. Micellar liquid chromatography from green analysis perspective. Open Chem. 2015;13:877–92.
ICH. Q2 (R1): Validation of analytical procedures: text and methodology. In International Conference on Harmonization, Geneva., (2005).
Food, Administration D. FDA guidance for industry: bioanalytical method validation. Center for Drug Evaluation and Research; 2001.
Yadav PJ, Jadhav SS, Mohite SK. Development and validation of RP-HPLC method for alogliptin benzoate in bulk drug and dosage form. Int J Pharm Pharm Res. 2014;1:1–9.
Karkhanis V, Captain A. Development and validation of a Liquid Chromatographic Method for Estimation of Vildagliptin in Tablet Dosage Form. Asian J Res Chem. 2013;6:1166–8.
Dicks AP, Hent A, Koroluk KJ. The EcoScale as a framework for undergraduate green chemistry teaching and assessment. Green Chem Lett Rev. 2018;11:29–35.
Tobiszewski M. Metrics for green analytical chemistry. Anal Methods. 2016;8:2993–9.
Acknowledgements
The authors are grateful to EVA Pharma (Cairo, Egypt) for providing us with pure pioglitazone, alogliptin, glimepiride, and vildagliptin samples.
Funding
Open access funding provided by The Science, Technology & Innovation.
Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank.
(EKB).
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
M.S.E: supervision, reviewing and editing W.S.H. : supervision, review and editing H. A.M.: Review & Editing, Supervision: methodology, validation, writing original draft I. M.: Investigation, Methodology, Formal analysis, Validation, Writing - Original Draft All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All experimental protocols in the current study were approved by the Committee of Research Ethics in the Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt. All methods were carried out in accordance with relevant regulations and guidelines. Al-Azhar University Hospital waived consent as the human plasma was provided kindly by Al-Azhar University Hospital.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Elmasry, M.S., Hassan, W.S., Merey, H.A. et al. Earth-friendly micellar UPLC technique for determination of four hypoglycemic drugs in different pharmaceutical dosage forms and spiked human plasma. BMC Chemistry 17, 74 (2023). https://doi.org/10.1186/s13065-023-00983-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-023-00983-6