
Abstract
A facile and efficient palladium-catalyzed borylation of aryl (pseudo)halides at room temperature has been developed. Arylboronic esters were expeditiously assembled in good yields and with a broad substrate scope and good functional group compatibility. This approach has been successfully applied to the one-pot two-step borylation/Suzuki–Miyaura cross-coupling reaction, providing a concise access to biaryl compounds from readily available aryl halides. Furthermore, a parallel synthesis of biaryl analogs is accomplished at room temperature using the strategy, which enhances the practical usefulness of this method.
Similar content being viewed by others
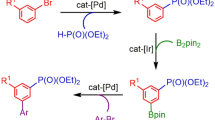
Introduction
Arylboronic acids and esters are versatile reagents in organic synthesis. They were widely used in C–C, C–O, C–N and C–S bond forming reactions [1, 2], which are essential for the construction of bioactive molecules and organic building blocks. In particular, functionalized arylboronic esters are highly valuable because they are more stable compared with arylboronic acids [3, 4]. The most common method for the synthesis of arylboronic esters is the reaction of trialkyl borates with aryllithium or Grignard reagents. The method has a problem with functional-group compatibility, and additional protection and deprotection steps are usually required [5]. A series of transition-metal-catalyzed methods for the preparation of arylboronic esters have been developed recently [6,7,8]. Particularly, palladium-catalyzed synthesis of arylboronic esters from aryl halides or pseudo-halides has opened the door for the development of efficient processes. Some improvements have been reported with respect to catalysts [9,10,11,12,13,14,15,16,17,18,19,20], ligands [12, 21,22,23,24], additives [25, 26] and reaction conditions [18, 19, 27]. However, only very few works have been reported until now on the palladium-catalyzed synthesis of arylboronic esters at room temperature from unactivated aryl chlorides [28].
Biaryl and biheteroaryl motifs are important core structures that are found in natural products, drug molecules and functionalized materials [29,30,31]. The palladium-catalyzed Suzuki–Miyaura cross-coupling reaction of arylboronic acids or esters with aryl halides has become the most common and powerful method to build such structures [28, 32,33,34]. Since one-pot two-step protocol combining borylation and Suzuki–Miyaura cross coupling steps was reported in 2004 [35], the need to prepare or purchase a boronic acid or ester could be eliminated. Growing efforts has been paid to develop the attractive method. New catalyst systems such as cyclopalladated ferrocenylimine complex [36, 37] and palladium-indolylphosphine complex [23, 38, 39] were reported successively. In 2007, the first example of borylation/cross-coupling protocol from aryl chlorides was reported [28]. With all of the advances, the one-pot two-step protocol still suffers from high catalyst loads, limited substrate scope and poor functional-group tolerance, and requires high temperature and long reaction time.
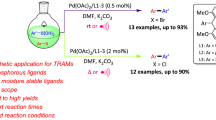
Herein, we reported a highly practical and efficient method for palladium-catalyzed borylation of aryl halides or pseudo-halides at room temperature. Furthermore, a facile single pot synthesis of biaryl and biheteroaryl compounds via sequential borylation and Suzuki–Miyaura cross coupling reaction was presented. The approach has been successfully applied in formats amenable to parallel synthesis of biaryls.
Results and discussion
Initial screening of catalytic systems for the Miyaura borylation of 4-chloroanisole (1a) were preformed using 2 mol% of palladium catalyst, 3 equiv. of B2pin2 and 3 equiv. of anhydrous KOAc or K3PO4. Various palladium catalysts and catalytic systems listed in Table 1 were tested at elevated temperature (Table 1, entries 1–10). Almost no reaction occurred when catalyst Pd(PPh3)4 [28, 40, 41] or PdCl2(dppf) [41] was used (Table 1, entries 1, 4 and 5). PdCl2(PPh3)2 [25, 42] exhibited low activity for borylation of 4-chloroanisole (Table 1, entry 3). Catalytic systems Pd(PPh3)4/PCy3 [43], Pd2dba3/PCy3 [43, 44], Pd2dba3/XPhos [28, 45], Pd2dba3/SPhos [28, 45], Pd(OAc)2/PCy3 [43, 46], Pd(OAc)2/XPhos [45, 47] gave moderate to good yields (Table 1, entries 2 and 6–10). Then we tested room temperature for the reaction of 4-chloroanisole. We discovered that these active catalytic systems for the borylation of 4-chloroanisole at elevated temperature were ineffective at room temperature. However, when Pd(OAc)2/SPhos [28] which was developed for the borylation of aryl chlorides at lower temperature were employed, the reaction proceeded very slowly, leading to 42% yield of product after 48 h (Table 1, entry 11).
Recently, activated palladium precatalysts have been developed as solutions to the problem of catalyst activation in cross coupling reactions. Many such systems, including pyridine-stabilized NHC precatalysts (PEPPSI) [48], ligated allylpalladium chloride precatalysts [49], imine-derived precatalysts [50] and palladacycle-based precatalysts [34], have been applied to C–C, C-N and C-O bond forming reactions. Since these species are pre-ligated Pd(II) source, some of which can rapidly form a requisite ligated Pd(0) species in situ even at lower temperature when exposed to base [51], we assumed that catalyzed by the species, borylation of aryl halides could proceed in an efficient manner at room temperature. After evaluated a variety of precatalysts, we selected 9 and 10 (Scheme 1), which were more stable in solution and could be readily prepared using commercially available and economical starting materials, as ideal set of precatalysts to test in the borylation reaction. SPhos and XPhos were used as supporting ligands and the μ-Cl and μ-OMs dimmers (7 or 8) as palladium sources. Following Buchwald’s protocol [51], the reaction of palladium source μ-Cl or μ-OMs dimmer with ligands rapidly afforded the desired precatalysts 9a, 9b, 10a and 10b (Scheme 1), which were directly used in our model reaction without isolation, respectively. The results clearly indicated that XPhos is the optimal ligand for this transformation, with the catalyst based on SPhos also showing some activity (Table 1, entries 12–19). Compared with the μ-Cl dimmer (7), the μ-OMs (8) is optimal as the palladium source. The use of 10b gave 93% yield of 2a in THF at room temperature for 2 h (Table 1, entry 18). The results promoted us to optimize the reaction conditions. The effects of solvents, bases and reaction time were examined, and the efficiency of 10b was further evaluated. In the presence of a sufficient amount of precatalyst (2.0 mol%) and B2pin2 (3.0 equiv), 2.0 equiv. of K3PO4 lead to 87% conversion after 1 h, while three equivalents of K3PO4 gave 98% yield (Table 1, entry 20). Finally, the optimal reaction condition was achieved as the combination of 1.0 mol% 10b, 1.2 equiv. B2pin2 and 3.0 equiv. K3PO4 in THF at room temperature for 1 h (Table 1, entry 20).

Preparation of precatalyst 9 and 10
In exploring the scope of aryl halides in the borylation reaction, we found that the reaction was broadly amenable to a range of aryl (pseudo)halides with different electronic parameters and bearing a variety of functional groups (Table 2). Electron rich and electron deficient aryl (pseudo)halides were successfully transferred to corresponding boronic esters in good to excellent yields (Table 2, 2b–2e and 2f–2m, 68–98%), as were heteroaromatic halides including indole, thiophene, pyridine and pyrazole (Table 2, 2n–2q, 71–93%). The reaction displayed excellent functional group tolerance and substrates bearing functional groups such as methyl (2b), methoxyl (2c), phenyl (2f), nitrile (2g), aldehyde (2h and 2j), trifluoromethyl (2i), carboxyl (2k), ketone (2l) and nitro (2 m) were effective units in the reaction. It is noteworthy that unprotected phenol and aniline also gave the corresponding products 2d and 2e in 70% and 84% yields, respectively. No reduced side products were observed in borylation of aldehyde (2h, 2j), ketone (2l) and nitro substrate (2m). Significantly, besides aryl bromides and iodides, less reactive aryl chlorides and triflates served as effective substrates for this process.
We subsequently examined a room-temperature tandem borylation/Suzuki–Miyaura coupling procedure to demonstrate the practical utility of the method. The result of borylation of bromobenzene and following coupling with p-chlorobenzoic acid proved to be successful under the optimized conditions shown in Table 3. In this process, the aryl halide (1) was subjected to Pd-catalyzed borylation conditions with subsequent addition of the aryl halide (3) and aqueous K3PO4. No separation of the boronic ester intermediates was required nor was catalyst added prior to conducting the cross-coupling step. As illustrated by the examples summarized in Table 3, both aryl chlorides and bromides performed well whether used as borylated substrates or electrophilic coupling partners in the reaction. Aryl halides with electron-donating groups such as hydroxyl, alkyl and methoxyl (Table 3, entries 3, 6–8), electron-withdrawing groups such as aldehyde and trifluoromethyl (Table 3, entries 4 and 5) were successfully coupled to various aryl and heteroaryl halides in one-pot to deliver a variety of diaryl compounds in 65–94% yield. The meta- and para-substituted aryl halides gave excellent to good yields (Table 3, entries 1–5). The ortho-substituted aryl halides lead to somewhat lower yields (Table 3, entries 6 and 7). However, 2-bromo-1,3-dimethylbenzene showed less reactivity, affording trace amount of the coupling product. Two methyl groups existing at the ortho-position to bromine presumably resulted in an extreme steric hindrance which precluded obtaining expected product. Heteroaryl halides employed as the borylated component or cross-coupling partner often resulted in low yield or no reaction at all in previous protocol [52]. The approach developed herein has been shown to be quite effective for heteroaromatic substrates such as pyridine and pyrazole, providing the desired products in good yield (Table 3, entries 8–10).
Arenes and heteroarenes are frequently present in medicines, agrochemicals, conjugate polymers and other functional materials. To illustrate the practicality of this approach in a medicinal chemistry setting, the chemistry was applied to parallel synthesis of biaryl scaffolds. This allows the preparation of multiple biaryl compounds in parallel from commercial aryl halides in a highly efficient manner. We chose aryl chlorides with polarity differences as electrophile in the second step of the one-opt two-step sequence. An efficient borylation/Suzuki coupling reaction can be performed, affording three distinct products in excellent yields. As shown in Scheme 2, the first chloride 4-tert-butyl-1-chlorobenzene was borylated, and the subsequent addition of aqueous K3PO4 and three aryl chlorides in equimolar amounts provided three desired products (4k–4m) in 71%, 92% and 72% yield, respectively. Heteroaryl chlorides were also successfully coupled to 4-tert-butyl-1-chlorobenzene to yield biaryl compounds (4n–4p) in good yields.

One-pot parallel synthesis of biaryl compounds
Conclusion
In conclusion, we have developed a versatile and efficient protocol for the room-temperature synthesis of arylboronic esters from aryl (pseudo)halides. This method was extended to the one-pot two-step borylation/Suzuki–Miyaura reaction that allowed the coupling of a wide range of aryl halides or heteroaryl halides with excellent functional group tolerance. The precatalyst used in the reaction can be prepared from readily available starting materials in a facile one-pot procedure and can be directly used in the reactions without isolation. The approach also displayed advantages of mild reaction conditions, good stability of catalyst and high efficiency. Further, we successfully applied the approach to parallel synthesis of biaryl compounds, which enable facile preparation of multiple biaryl analogues in a highly efficient manner from readily accessible aryl chlorides at room temperature.
References
Miura M, Nomura M (2002) Direct arylation via cleavage of activated and unactivated C–H bonds. In: Miyaura N (ed) Cross-coupling reactions, vol 219. Springer, Berlin, Heidelberg, pp 211–241
Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V (2011) Nickel-catalyzed cross-couplings involving carbon-oxygen bonds. Chem Rev 111:1346–1416
Qiu D, Jin L, Zheng ZT, Meng H, Mo FY, Wang X, Zhang Y, Wang JB (2013) Synthesis of pinacol arylboronates from aromatic amines: a metal-free transformation. J Org Chem 78:1923–1933
Merino P, Tejero T (2010) Expanding the limits of organoboron chemistry: synthesis of functionalized arylboronates. Angew Chem Int Ed 49:7164–7165
Hall DG (2005) Structure, properties, and preparation of boronic acid derivatives: overview of their reactions and applications. In: Hall DG (ed) Boronic acids: preparation, applications in organic synthesis and medicine, vol 2. Wiley-VCH, Weinheim, pp 1–99
Yang CT, Zhang ZQ, Tajuddin H, Wu CC, Liang J, Liu JH, Fu Y, Czyzewska M, Steel PG, Marder TB, Liu L (2012) Alkylboronic esters from copper-catalyzed borylation of primary and secondary alkyl halides and pseudohalides. Angew Chem Int Ed 51:528–532
Leowanawat P, Zhang N, Percec V (2012) Nickel catalyzed cross-coupling of aryl C–O based electrophiles with aryl neopentylglycolboronates. J Org Chem 77:1018–1025
Han FS (2013) Transition-metal-catalyzed Suzuki–Miyaura cross-coupling reactions: a remarkable advance from palladium to nickel catalysts. Chem Soc Rev 42:5270–5298
Ishiyama T, Murata M, Miyaura N (1995) Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: a direct procedure for arylboronic esters. J Org Chem 60:7508–7510
Ishiyama T, Itoh Y, Kitano T, Miyaura N (1997) Synthesis of arylboronates via the palladium(0)-catalyzed cross-coupling reaction of tetra(alkoxo)diborons with aryl triflates. Tetrahedron Lett 38:3447–3450
Murata M, Oyama T, Watanabe S, Masuda Y (2000) Palladium-catalyzed borylation of aryl halides or triflates with dialkoxyborane: a novel and facile synthetic route to arylboronates. J Org Chem 65:164–168
Ishiyama T, Ishida K, Miyaura N (2001) Synthesis of pinacol arylboronates via cross-coupling reaction of bis(pinacolato)diboron with chloroarenes catalyzed by palladium(0)–tricyclohexylphosphine complexes. Tetrahedron 57:9813–9816
Molander GA, Trice SLJ, Dreher SD (2010) Palladium-catalyzed, direct boronic acid synthesis from aryl chlorides: a simplified route to diverse boronate ester derivatives. J Am Chem Soc 132:17701–17703
Zhang YD, Gao J, Li WJ, Lee H, Lu BZ, Senanayake CH (2011) Synthesis of 8-arylquinolines via one-pot Pd-catalyzed borylation of quinoline-8-yl halides and subsequent Suzuki–Miyaura coupling. J Org Chem 76:6394–6400
Bello CS, Schmidt-Leithoff J (2012) Borylation of organo halides and triflates using tetrakis(dimethylamino)diboron. Tetrahedron Lett 53:6230–6235
Molander GA, Trice SLJ, Kennedy SM (2012) Stereospecific cross-coupling of secondary organotrifluoroborates: potassium 1-(benzyloxy)alkyltrifluoroborates. J Org Chem 77:8678–8688
Maluenda I, Navarro O (2015) Recent developments in the Suzuki–Miyaura reaction: 2010–2014. Molecules 20:7528–7557
Suzuki A (2011) Cross-coupling reactions of organoboranes: an easy way to construct C–C bonds (Nobel Lecture). Angew Chem Int Ed 50:6722–6737
Seechurn CCJ, Kitching MO, Colacot TJ, Snieckus V (2012) Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew Chem Int Ed 51:5062–5085
Ji H, Wu LY, Cai JH, Li GR, Gan NN, Wang ZH (2018) Room-temperature borylation and one-pot two-step borylation/Suzuki–Miyaura cross-coupling reaction of aryl chlorides. RSC Adv. 8:13643–13648
Kawamorita S, Ohmiya H, Iwai T, Sawamura M (2011) Palladium-catalyzed borylation of sterically demanding aryl halides with a silica-supported compact phosphane ligand. Angew Chem Int Ed 50:8363–8366
Li PB, Lü B, Fu CL, Ma SM (2013) Zheda-Phos for general α-monoarylation of acetone with aryl chlorides. Adv Synth Catal 355:1255–1259
Chow WK, Yuen OY, So CM, Wong WT, Kwong FY (2012) Carbon–boron bond cross-coupling reaction catalyzed by –PPh2 containing palladium–indolylphosphine complexes. J Org Chem 77:3543–3548
Huang LL, Cao Y, Zhao MP, Tang ZF, Sun ZH (2014) Asymmetric borylation of α, β-unsaturated esters catalyzed by novel ring expanded N-heterocyclic carbenes based on chiral 3,4-dihydro-quinazolinium compounds. Org Biomol Chem 12:6554–6556
Yasuike S, Dong Y, Kakusawa N, Matsumura M, Kurita J (2014) Simple base-free Miyaura-type borylation of triarylantimony diacetates with tetra(alkoxo)diborons under aerobic conditions. J Organomet Chem 765:80–85
Yamamoto Y, Nogi K, Yorimitsu H, Atsuhiro O (2017) Base-free palladium-catalyzed hydrodechlorination of aryl chlorides with pinacol borane. ChemistrySelect. 2:1723–1727
Liang Q, Xing P, Huang Z, Dong J, Sharpless KB, Li X, Jiang B (2015) Palladium-catalyzed, ligand-free Suzuki reaction in water using aryl fluorosulfates. Org Lett 17:1942–1945
Billingsley KL, Barder TE, Buchwald SL (2007) Palladium-catalyzed borylation of aryl chlorides: scope, applications, and computational studies. Angew Chem Int Ed 46:5359–5363
Liu JK (2006) Natural terphenyls: developments since 1877. Chem Rev 106:2209–2223
Mercier LG, Leclerc M (2013) Direct (hetero)arylation: a new tool for polymer chemists. Acc Chem Res 46:1597–1605
Shibahara F, Murai T (2013) Direct C-H arylation of heteroarenes catalyzed by palladium/nitrogen-based ligand complexes. Asian J Org Chem. 2:624–636
Zhang YD, Gao J, Li WJ, Lee H, Lu BZ, Senanayake CH (2011) Synthesis of 8-arylquinolines via one-pot Pd-catalyzed borylation of quinoline-8-yl halides and subsequent Suzuki–Miyaura coupling. J Org Chem 76:6394–6400
Tang WJ, Keshipeddy S, Zhang YD, Wei XD, Savoie J, Patel ND, Yee NK, Senanayake CH (2011) Efficient monophosphorus ligands for palladium-catalyzed Miyaura borylation. Org Lett 13:1366–1369
Lennox AJ, LloydJones GC (2014) Selection of boron reagents for Suzuki–Miyaura coupling. Chem Soc Rev 43:412–443
Nising CF, Schmid UK, Nieger M, Bräse S (2004) A new protocol for the one-pot synthesis of symmetrical biaryls. J Org Chem 69:6830–6833
Zou DP, Cui HM, Qin LJ, Li JY, Wu YJ, Wu YS (2011) Aminomethylation via cyclopalladated-ferrocenylimine-complexes-catalyzed Suzuki–Miyaura coupling of aryl halides with potassium N,N-dialkylaminomethyltrifluoroborates. Synlett 3:349–356
Wang LH, Cui XL, Li JY, Wu YS, Zhu ZW, Wu YJ (2012) Synthesis of biaryls through a one-pot tandem borylation/Suzuki–Miyaura cross-coupling reaction catalyzed by a palladacycle. Eur J Org Chem 2012:595–603
Yuen OY, Charoensak M, So CM, Kuhakarn C, Kwong FY (2015) A general direct arylation of polyfluoroarenes with heteroaryl and aryl chlorides catalyzed by palladium indolylphosphine complexes. Chem Asian J 10:857–861
Wong SM, Yuen OY, Choy PY, Kwong FY (2015) When cross-coupling partners meet indolylphosphines. Coordin Chem Rev. 293–294:158–186
Xie DH, Li R, Zhang DQ, Hu JN, Xiao DD, Li XY, Xiang YJ, Jin WS (2015) Palladium-catalyzed borylation of m-dibromobenzene derivative and its applications in one-pot tandem Suzuki–Miyaura arenes synthesis. Tetrahedron 71:8871–8875
Takagi J, Yamakawa T (2013) Syntheses of (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)arenes through Pd-catalyzed borylation of arylbromides with the successive use of 2,2′-bis(1,3,2-benzodioxaborole) and pinacol. Tetrahedron Lett 54:166–169
Praveenganesh N, Chavant PY (2008) Improved preparation of 4,6,6-trimethyl-1,3,2-dioxaborinane and its use in a simple [PdCl2(TPP)2]-catalyzed borylation of aryl bromides and iodides. Eur J Org Chem 27:4690–4696
Lu HT, Wang SQ, Li JY, Zou DP, Wu YS, Wu YJ (2017) Efficient synthesis of pyrazine boronic esters via palladium-catalyzed Miyaura borylation. Tetrahedron Lett 58:839–842
Sicre C, Alonso-Gómez JL, Cid MM (2006) Regioselectivity in alkenyl(aryl)-heteroaryl Suzuki cross-coupling reactions of 2,4-dibromopyridine. A synthetic and mechanistic study. Tetrahedron. 62:11063–11072
Dzhevakov PB, Topchiy MA, Zharkova DA, Morozov OS, Asachenko AF, Nechaev MS (2016) Miyaura borylation and one-pot two-step homocoupling of aryl chlorides and bromides under solvent-free conditions. Adv Synth Catal 358:977–983
Song J, Wei FL, Sun W, Li K, Tian YN, Liu C, Li YL, Xie LH (2015) Synthesis of fluoren-9-ones and ladder-type oligo-p-phenylene cores via Pd-catalyzed carbonylative multiple C–C bond formation. Org Lett 17:2106–2109
Murai M, Yanagawa M, Nakamura M, Takai K (2016) Palladium-catalyzed direct arylation of azulene based on regioselective C–H bond activation. Asian J Org Chem. 5:629–635
O’Brien CJ, Kantchev EB, Valente C, Hadei N, Chass GA, Lough A, Hopkinson AC, Organ M (2006) Easily prepared air- and moisture-stable Pd–NHC (NHC=N-heterocyclic carbene) complexes: a reliable, user-friendly, highly active palladium precatalyst for the Suzuki–Miyaura reaction. Chem Eur J 12:4743–4748
Chartoire A, Lesieur M, Slawin AMZ, Nolan SP, Cazin CS (2011) Highly active well-defined palladium precatalysts for the efficient amination of aryl chlorides. Organometallics 30:4432–4436
Bedford RB, Cazin CSJ, Coles SJ, Gelbrich T, Horton PN, Hursthouse MB, Light ME (2003) High-activity catalysts for Suzuki coupling and amination reactions with deactivated aryl chloride substrates: importance of the palladium source. Organometallics 22:987–999
Bruno NC, Tudge MT, Buchwald SL (2013) Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem Sci. 4:916–920
Kinzel T, Zhang Y, Buchwald SL (2010) A new palladium precatalyst allows for the fast Suzuki–Miyaura coupling reactions of unstable polyfluorophenyl and 2-heteroaryl boronic acids. J Am Chem Soc 132:14073–14075
Authors’ contributions
HJ designed and supervised the project and wrote the paper. JHC, NNG and ZHW performed experiments. LYW and GRL contributed for analysis of data. TY guided in data interpretation and assisted in manuscript preparation. All authors read and approved the final manuscript.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (No. 30701051), the Science and Technology Planning Project of Guangdong Province (2015A020211039), Natural Science Foundation of Guangdong Province (2018A0303130139), Scientific Research Project for Guangzhou Municipal Colleges and Universities (1201610139, 1201630263), Project for Young Innovative Talents in the Universities of Guangdong (2015KQNCX134) and Ph.D. Early Development Program of Guangzhou Medical University (2015C02).
Competing interests
The authors declare that they have no competing interests.
Associated content
Experimental procedure and characterization data of all products are reported in Additional file.
Availability of data and materials
All the main experimental and data have been presented in the form of tables and figures. General procedure, spectral data of substrates and specimen NMR spectra are given in Additional file 1.
Consent for publication
All authors consent to publication.
Ethics approval and consent to participate
Not applicable.
Funding
The research was funded by the National Natural Science Foundation of China, the Science and Technology Department of Guangdong Province, Guangzhou Education Bureau, Guangdong Provincial Department of Education and Guangzhou Medical University.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1.
Supporting Informations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ji, H., Cai, J., Gan, N. et al. Palladium-catalyzed borylation of aryl (pseudo)halides and its applications in biaryl synthesis. Chemistry Central Journal 12, 136 (2018). https://doi.org/10.1186/s13065-018-0510-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0510-6