
Abstract
An efficient one pot method for the synthesis of α-alkoxymethylphosphonium iodides is developed by using PPh3/I2 combination at room temperature. Reaction conditions are found general to synthesize wide range of structurally variant alkoxymethylphosphonium iodides in high yield (70–91%). These new functionalized phosphonium salts are further used in stereoselective synthesis of vinyl ethers as well as in carbon homologation of aldehydes.

Similar content being viewed by others

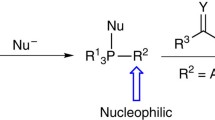
Introduction
Functionalized phosphonium salts are gaining much attention for their diverse applications in organic synthesis [1,2,3,4,5]. α-Alkoxymethyl phosphonium salts are largely used for carbon homologation to carbonyl compounds [6,7,8,9,10] and also as significant synthetic intermediates [11,12,13,14,15,16,17]. Recently, unique reactivity of this class has been explored in nucleophilic substitution [18,19,20] and in novel phenyl transfer reactions [21, 22]. Methoxymethyltriphenylphosphonium chloride is commercially available salt from this class, but problem associated with its preparation involve toxic intermediate, higher temperature and long reaction time [9, 11, 23]. In perspective of alternative derivatives; α-methoxymethyl triphenylphosphonium iodide was reported by reaction of bis-methoxymethane (1a) with TMSI, followed by phosphination of methoxymethyl iodide in benzene (Scheme 1a) [24]. This only available method for iodide analogue also involves sensitive and toxic; reagent, solvent as well as intermediate along with difficult purification of product. In past few years, PPh3/I2 combination has successfully facilitated many functional groups inter-conversions [25,26,27,28,29,30,31,32]. Therefore, we decided to explore reactivity of PPh3/I2 with bis-alkoxymethanes (1) and herein efficient synthesis of a broad range of structurally diverse α-alkoxymethyl triphenylphosphonium iodides (2) is being reported (Scheme 1b). To best of our knowledge, this is the first report on general one pot synthesis of O,P-acetals, directly from dioxacetals on employing PPh3/I2 combination (Scheme 1b).

Synthesis of α-alkoxymethyl triphenylphosphonium iodides 2
Results and discussion
Current study was initiated from the model reaction of bis-butoxy methane (1a) with PPh3/I2 combination under different conditions (Table 1). Our preliminary attempt was encouraging, where 27% desired conversion (2a) was observed on refluxing equal molar amounts of acetal (1a) and PPh3/I2 in toluene for an hour (Table 1, entry 1). To improve the yield, reaction time was increased up to 3 h but only 33% required conversion was observed (Table 1, entry 2). Low yield might be associated with the sublimation of iodine at high temperature therefore, it was considered to decrease the reaction temperature. To our delight, yield was increased to 55% when the same experiment was performed at room temperature (Table 1, entry 3). Increasing the amount of PPh3 to 2 equivalent and reaction time up to 5 h further improved the yield (80%) (Table 1, entry 4). However, further attempts with increase in reaction time and replacing toluene with acetonitrile or solvent free conditions, were not effectual (Table 1, entry 5–8).

To explore the substrate scope of this reaction, optimized conditions were employed to structurally different bis-alkoxy methanes (1a–j, see Additional file 1) [33]. The method was found equally efficient to obtain broad range of alkoxymethylphosphonium iodides (2a–j, Table 2) based on primary, secondary, tertiary and benzylic alkoxy groups. Acetals having simple methoxy, ethoxy, benzoxy and phenylethoxy groups provided desired O,P-acetals 2b–e in 75–87%. Similarly, when acetal of (S)-2-butanol was reacted with PPh3/I2, corresponding salt 2f was obtained in 90% yield with retention in configuration, which was ultimately confirmed by X-ray diffraction analysis (Fig. 1).

ORTEP diagram of (S)-2-sec-butoxymethyltriphenylphosphonium iodide 2f
Optimized reaction conditions were further extended to cyclic chiral alkoxy groups including fenchyl, menthyl and borneyl, where respective chiral phosphonium salts 2g–i were obtained in good yields (Table 2).
Here, (+)-menthoxymethyltriphenylphosphonium iodide 2h is worth mentioning as its chloride analogue was prepared by tedious methodology with long reaction time [12]. Interestingly, the reaction was also successful with acetal of t-butanol where corresponding salt 2j was produced in 77% yield (Table 2).
In terms of mechanism, we envision that initially I2 and PPh3 generate phosphonium intermediate (i), which reacts with bis-alkoxymethane 1 to provide oxonium intermediate (ii) (Scheme 2). Another equivalent of PPh3 attack on oxonium intermediate (ii) to transform it into the target O,P-acetal 2 (Scheme 2).

Plausible mechanism for the preparation of alkoxymethylphosphonium iodides 2
After having a range of alkoxymethylphosphonium iodides in hand, we further explored their applications in organic synthesis. Vinyl ethers also known as enol ethers are considered important synthetic targets for the organic chemists. They itself are part of many natural products and also involve as intermediate in their total synthesis [34,35,36]. They act as key intermediates in many important organic reactions like Diels–Alder reaction [37], Coupling reaction [38,39,40,41,42,43], Olefin metathesis [44], Claisen rearrangement [45, 46] and Nazarov cyclization [47, 48]. They are also used in materials sciences due to their polymerization ability through cationic mechanism [49]. Despite extensive applications of enol ethers, still there is lack of general and direct method for their synthesis. Metal-catalyzed couplings are the most common available method [50,51,52,53,54], along with some other indirect methodologies [55,56,57,58,59,60,61,62]. Direct synthesis of enol ethers by a Wittig reaction with alkoxymethylphosphonium salt is though an evident concept but no systematic study is found in literature. Most often commercially available methoxymethylphosphonium chloride is used [63, 64], whereas effect of other alkoxy groups as well as counter anions is still need to explore. For this purpose, at first ethoxymethyltriphenylphosphanium iodide 2c was reacted with benzaldehyde and its derivatives in the presence of n-BuLi, which afforded corresponding vinyl ethers 3a–d (Table 3) in good yield (67–71%) and selectivity (69–73% trans).

Providentially, trans isomer 3e′ was obtained almost exclusively (99% selectivity) with (+)-menthoxymethyltriphenylphosphonium iodide 2h. Earlier, Fuwa and Sasaki obtained same isomer 3e′ in 9% yield along with 36% cis isomer 3e through Pd coupling [40].
Further, cost effective n-butoxymethylphosphonium iodide 2a was employed for carbon homologation, where both aliphatic and aromatic aldehydes were successfully converted to higher analogous 4 in good yield (Table 4). Results show that these directly prepared and environmentally benign salts are good alternative to their chloride analogues.

To evaluate catalytic potential of chiral phosphonium salts in asymmetric reduction of acetophenone, initially 10 mol% of 2g with NaBH4 provided (R)-1-phenylethanol with 92% yield and 4% ee (Scheme 3).

α-Alkoxymethylphosphonium iodide 2g in asymmetric reduction
Detailed study and further investigation on the application of these structurally unique α-alkoxymethylphosphonium salts in stereoselective synthesis of enol ethers carrying chiral auxillaries as well as in other related fields are currently underway in our laboratory.
Conclusion
In conclusion, a facile general method for the synthesis of α-alkoxymethyl triphenylphosphonium iodides is developed under very mild conditions. This protocol demonstrates PPh3/I2 mediated green route to functionalized phosphonium salts. Major advantage of this methodology is to avoid toxic reagent and intermediate. These easily prepared salts were successfully employed for stereoselective synthesis of enol ethers as well as for carbon homologation in aldehydes. The new methodology will be useful for organic synthetic chemists as well as others working in associated fields.
Experimental
All experiments were carried out under inert atmosphere using standard Schlenk technique with oven dried glassware and magnetic stirring. All solvents were freshly dried and distilled before use. All chemicals were purchased from Sigma Aldrich, Alfa Aesar and Merck. IR spectra were measured on a Perkin–Elmer Paragon 1000 (thin film) or on a Perkin–Elmer BXII spectrometer (neat). Bruker Avance NMR spectrometer of 300, 400 and 500 MHz were used for NMR spectral studies. Optical rotation was measured on Polarimeter P-2000. Crystal structure was confirmed by single crystal X-ray diffractometer Bruker Enrauf–Nonius Apex smart and Siemens P4. Mass spectra were measured on GC–MS 5977A, MAT312-EI, JEOL-600H-2, and JEOL MS-600H-1. Reactions were monitored by TLC plates from Merck (silica gel 60 F254, aluminum oxide 60 F254). TLCs were visualized by UV fluorescence and phosphomolybdic acid spraying reagent.
General procedure for synthesis of α-alkoxymethyltriphenylphosphonium iodides (2a–j)
In a seal tube triphenylphosphine (20 mmol) and iodine (1.1 equiv) were taken in toluene (4 mL) and mixture was allowed to stir for 5 min. Solution of bis-alkoxymethane (1, 10 mmol in 1 mL toluene) was added to the reaction mixture and allowed to stir for 5 h at room temperature (28 °C). After completion of reaction, solvent was removed under reduced pressure and residue was washed with hexane to obtain required salt.
Butoxymethyltriphenylphosphonium iodide (2a)
Lemon yellow thick oil, yield = 80%, IR: υ (cm−1) = 689, 730, 1115, 1302, 1412, 2835. 1H-NMR (300 MHz, MeOD): δ ppm. 7.93–7.91 (3H, m, CH aromatic), 7.90–7.89 (3H, m, CH aromatic), 7.88–7.79 (2H, m, CH aromatic), 7.78–7.76 (3H, m, CH aromatic), 7.76–7.75 (3H, m, CH aromatic), 7.74–7.72 (1H, m, CH aromatic), 5.40 (2H, d, J = 4.8, CH2), 3.71 (2H, t, J = 6.4, CH2), 1.56–1.51 (2H, m, CH2), 1.28–1.22 (2H, m, CH2), 0.84 (3H, t, J = 7.6, CH3). 13C-NMR (75 MHz, MeOD): δ ppm. 136.62, 136.60 (2 carbons), 135.25, 135.15, 133.74 (3 carbons), 133.08, 132.97 (3 carbons), 131.55, 131.42, 130.01, 129.89 (2 carbons), 118.60, 117.74, 75.88, 35.76, 20.07, 13.99. 31P (202 MHz, CDCl3): δ ppm 18.83. EIMS = 349 (M+-I), 277.2 (48.4%), 262.2 (100%), 183.1 (59.6%), 108.0 (57.2%), 56 (36.3%).
Methoxymethyltriphenylphosphonium iodide (2b) [25]
Lemon yellow thick oil, yield = 73%, IR: υ (cm−1) = 691, 724, 1112, 1437, 2877, 2958. 1H-NMR (300 MHz, CDCl3): δ ppm 7.69–7.66 (3H, m, C–H aromatic), 7.65–7.61 (5H, m, C–H aromatic), 7.59–7.57 (2H, m, C–H aromatic), 7.56–7.51 (5H, m, C–H aromatic), 5.56 (2H, d, J = 3.9, CH2), 3.51 (3H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ ppm. 135.77, 135.39, 135.35, 134.34 (3 carbons), 133.97, 133.84, 133.62, 133.49 (2 carbons), 130.78, 130.47, 130.30, 130.05, 129.89 (3 carbons), 116.85, 66.19. 31P (202 MHz, CDCl3): δ ppm 17.53. EIMS = 307 (M+-I), 277.2 (100%), 262.2 (67.6%), 183.1 (54.9%), 108.0 (10.9%), 77.0 (9.8%), 50.9 (5.6%).
Ethoxymethyltriphenylphosphanium iodide (2c)
Colorless oil: yield = 82%, IR: υ (cm−1) = 2846, 2794, 1946, 1586, 1484, 1437, 1317, 1112, 1092; 1H NMR (400 MHz, CDCl3) δ = 7.77–7.70 (9H, m), 7.65–7.60 (6H, m), 5.72 (2H, d, J = 3.96), 3.85 (2H, q, J = 7.0), 1.09 (3H, t, J = 7.0); 13C NMR (100 MHz, CDCl3) δ = 135.3, 135.3, 134.0 (3C), 133.9, 132.0 (3C), 131.9, 130.4, 130.3 (3C), 128.5, 128.4 (3C), 116.5, 64.21, 14.93; 31P-NMR (CDCl3): δ 25.77; HRMS +ESI calculated for C21H22OP: 321.1403; found 321.1404.
Benzoxymethyltriphenylphosphonium iodide (2d)
Yellow thick oil, yield = 87%, IR υ (cm−1) = 681, 734, 1103, 1305, 1425, 2767. 1H-NMR (300 MHz, MeOD): δ ppm. 7.82–7.76 (3H, m, CH aromatic), 7.75–7.71 (3H, m, CH aromatic), 7.70–7.67 (3H, m, CH aromatic), 7.66–7.62 (2H, m, CH aromatic), 7.58–7.55 (3H, m, CH aromatic), 7.48–7.45 (3H, m, CH aromatic), 7.37–7.29 (3H, m, CH aromatic), 5.72 (2H, d, J = 3, CH2), 4.97 (2H, s, CH2). 13C-NMR (75 MHz, MeOD): δ ppm. 134.57 (3 carbons), 133.60 (4 carbons), 133.19 (3 carbons), 132.33, 131.91 (4 carbons), 130.49 (4 carbons), 129.78, 129.41 (4 carbons), 97.76, 78.39. 31P (202 MHz, MeOD): δ ppm 17.55. EIMS = 383 (M+-I), 277.2 (59.6%), 262.2 (100%), 183.1 (48.4%), 108.0 (10.9%), 50.9 (9.8%).
Phenethoxymethyltriphenylphosphonium iodide (2e)
Yellowish powder, m.p = 171–173 °C, yield = 81%, IR υ (cm−1) = 690, 730, 1124, 1317, 2917. 1H-NMR (300 MHz, CDCl3): δ ppm. 7.78–7.36 (20H, m, CH aromatic), 5.45 (2H, d, J = 1.2 Hz, CH2), 4.21 (2H, t, J = 6.4, CH2), 2.75 (2H, t, J = 7.2, CH2). 13C-NMR (75 MHz, CDCl3): δ ppm. 138.43 (4 carbons), 137.98, 137.81 (2 carbons), 137.23, 136.31 (4 carbons), 136.06, 135.78, 135.23, 134.94 (3 carbons), 134.24, 129.81 (2 carbons), 129.12 (2 carbons), 117.89, 94.67, 77.78, 37.54. 31P (202 MHz, CDCl3): δ ppm 17.74. EIMS = 397 (M+-I), 277.2 (100%), 262.2 (67.6%), 183.1 (59.6%), 108.0 (13.4%), 91 (43%).
(S)-sec-Butoxymethyltriphenylphosphonium iodide (2f)
Yellowish white crystals, m.p = 58 °C, yield = 89%, \(\left[ \alpha \right]_{D}^{25}\) = − 11 (c = 0.0018, MeOH), IR: υ (cm−1) = 682, 709, 1107, 1311, 1444, 2863. 1H-NMR (300 MHz, MeOD): δ ppm. 7.93–7.88 (3H, m, CH aromatic), 7.85–7.83 (1H, m, CH aromatic), 7.82–7.78 (3H, m, CH aromatic), 7.77–7.67 (3H, m, CH aromatic), 7.64–7.63 (3H, m, CH aromatic), 7.63–7.60 (1H, m, CH aromatic), 7.56–7.54 (1H, m, CH aromatic), 5.51 (1H, dd, J = 13.5, 4.8, CH2), 5.39 (1H, dd, J = 13.5, 5.7, CH2), 3.70–3.64 (1H, m, CH), 1.60–1.43 (2H, m, CH2), 1.18 (3H, d, J = 6.0 Hz, CH3), 0.75 (3H, t, J = 7.5, CH3). 13C-NMR (75 MHz, MeOD): δ ppm. 139.32, 135.11, 134.98, 134.72, 134.46, 133.95, 133.60, 133.32, 133.00, 132.93, 132.60, 131.82, 131.27, 130.87, 130.04, 129.90, 129.83, 128.60, 94.89, 79.51, 30.51, 20.10, 10.09. 31P (202 MHz, CDCl3): δ ppm 19.01. EIMS = 349 (M+-I), 277.2 (7.7%), 262.2 (55.9%), 183.1 (100%), 167.1 (49.8%), 152.1 (14.8%), 108.0 (13.4%), 91.0 (43.9%).
Triphenyl((((2R)-1,3,3-trimethylbicyclo[2.2.1]heptan-2-yl)oxy)methyl) phosphonium iodide (2g)
Lemon yellow thick oil, yield = 91%, \(\left[ \alpha \right]_{D}^{25}\) = + 55 (c = 0.004, MeOH), IR: υ (cm−1) = 684, 968, 1112, 2948. 1H-NMR (300 MHz, MeOD): δ ppm. 7.89–7.83 (4H, m, CH aromatic), 7.82–7.80 (1H, m, CH aromatic), 7.80–7.63 (4H, m, CH aromatic), 7.61–7.55 (3H, m, CH aromatic), 7.54–7.51 (3H, m, CH aromatic), 5.53 (2H, dd, J = 1.2, 4.8 Hz, CH2), 3.10 (1H, d, J = 14.1, CH), 1.67–1.53 (2H, m, CH2), 1.49–1.37 (2H, m, CH2), 1.06–1.01 (1H, m, CH), 1.06–0.96 (2H, m, CH2), 0.91 (3H, s, CH3), 0.83 (3H, s, CH3), 0.73 (3H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ ppm. 135.48, 135.45 (2 carbons), 134.26, 134.18, 132.06 (3 carbons), 132.01, 131.99, 130.53 (3 carbons), 130.43, 128.56, 128.47, 116.88, 116.20, 98.49, 66.63, 49.50, 48.38, 41.18, 40.01, 31.10, 26.10, 25.80, 20.72, 19.93. 31P (202 MHz, CDCl3): δ ppm 19.46. EIMS = 429 (M+-I), 277.2 (48.4%), 262.2 (100%), 183.1 (59.6%), 108.0 (57.2%), 56 (36.3%).
((((1S,2R)-2-isopropyl-5-methylcyclohexyl)oxy)methyl)triphenylphosphonium iodide (2h)
Light yellow semisolid, yield = 80%, \(\left[ \alpha \right]_{D}^{25}\) = + 8 (c = 0.027, MeOH), IR: υ (cm−1) = 687, 963, 1112, 2914. 1H-NMR (300 MHz, MeOD): δ ppm. 7.91–7.90 (2H, m, CH aromatic), 7.89–7.88 (1H, m, CH aromatic), 7.88–7.87 (2H, m, CH aromatic), 7.86–7.83 (4H, m, CH aromatic), 7.80–7.75 (1H, m, CH aromatic), 7.73–7.32 (1H, m, CH aromatic), 7.31–7.30 (1H, m, CH aromatic), 7.28–7.27 (1H, m, CH aromatic), 7.30–7.25 (1H, m, CH aromatic), 7.23–7.22 (1H, m, CH aromatic), 5.62 (1H, dd, J = 6.7, 3.3, CH2), 5.24 (1H, dd, J = 6.9, 2.9, CH2), 3.44 (1H, td, J = 5.7, 9.6, CH), 2.32–2.23 (1H, m, CH), 1.69–1.57 (2H, m, CH2), 1.41–1.33 (2H, m, CH2), 1.23–1.19 (2H, m, CH2), 0.95–0.91 (1H, m, CH), 0.79 (3H, d, J = 6.9, CH3), 0.75 (3H, d, J = 6.9, CH3), 0.56 (3H, d, J = 6.9, CH3). 13C-NMR (75 MHz, CDCl3): δ ppm. 135.32, 135.28, 134.32, 134.18, 134.02, 133.87, 133.61, 132.20, 132.07, 130.48, 130.32, 128.86, 128.66, 128.59, 128.50, 117.41, 116.27, 83.81, 74.16, 48.28, 46.95, 40.90, 39.42, 34.21, 31.19, 25.57, 23.41, 22.27, 16.18. 31P (202 MHz, MeOH): δ ppm 19.19. EIMS = 431 (M+-I), 277.2 (100%), 262.2 (67.6%), 183.1 (54.9%), 108.0 (10.9%), 77 (9.8%), 56 (36.3%).
Triphenyl((((2R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)oxy)methyl) phosphonium iodide (2i)
Light brown semi solid, yield = 70%, \(\left[ \alpha \right]_{D}^{25}\) = + 2.13 (c = 5 mg/15 mL MeOH), IR: υ (cm−1) = 683, 981, 1114, 2914. 1H-NMR (300 MHz, CDCl3): δ ppm. 7.89–7.83 (4H, m, CH aromatic), 7.82–7.80 (2H, m, CH aromatic), 7.77–7.71 (4H, m, CH aromatic), 7.67–7.61 (3H, m, CH aromatic), 7.57–7.51 (2H, m, CH aromatic), 5.69 (2H, dd, J = 6, 12, CH2), 3.03 (1H, dt, J = 3.9, 6.91, CH), 1.85–1.74 (2H, m, CH2), 1.65–1.64 (2H, m, CH2), 1.63–1.57 (1H, m, CH), 1.53–1.38 (2H, m), 0.90 (3H, s, CH3), 0.72 (3H, s, CH3), 0.51 (3H, s, CH3) 13C-NMR (75 MHz, CDCl3): δ ppm. 135.3, 135.3, 134.0 (3C), 133.9, 132.0, 131.9, 130.4, 130.3 (3C), 128.5 (3C), 128.4 (3C), 116.5 (d, J = 85), 76.3, 49.0, 48.6, 41.5, 41.4, 39.2, 26.2, 21.0, 20.2, 19.8; 31P (202 MHz, CDCl3): δ ppm 19.00. EIMS = 430 (M+-I), 277.2 (7.7%), 262.2 (55.9%), 183.1 (100%), 167.1 (49.8%), 152.1 (14.8%), 108.0 (13.4%), 91.0 (43.9%).
tert-Butoxymethyltriphenylphosphonium iodide (2j)
Yellowish thick oil, yield = 77%, IR: υ (cm−1) = 690, 713, 1127, 1295, 1405, 2799. 1H-NMR (400 MHz, MeOD): δ ppm. 7.91–7.90 (2H, m, CH aromatic), 7.89–7.86 (4H, m, CH aromatic), 7.83–7.75 (3H, m, CH aromatic), 7.34–7.31 (4H, m, CH aromatic), 7.25–7.23 (2H, m, CH aromatic), 5.45 (2H, dd, J = 1.6, 16.8, CH2), 0.047 (9H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ ppm. 136.69 (3 carbons), 136.63, 135.39, 135.27, 134.81, 134.76 (3 carbons), 133.66, 133.13, 132.67, 131.13, 131.09, 129.79 (3 carbons), 117.69, 89.54, 28.76 (3 carbons). 31P (202 MHz, CDCl3): δ ppm 18.98. EIMS = 349 (M+-I), 277.2 (100%), 262.2 (67.6%), 201.1 (24.5%), 183.1 (54.9%), 152.1 (11.4%), 108.0 (10.9%), 77.0 (9.8%).
General method for synthesis of vinyl ethers 3a–e
In a two neck round bottom flask n-BuLi (1.5 eq) was added to stirred solution of phosphonium iodide 2 (1 eq) in THF at − 78 °C and mixture was allowed to stir under argon. After 20 min solution of aldehyde (1 eq) in THF was added drop wise at the same temperature and reaction mixture was allowed to stir for further 4 h allowing the temperature to come to room temperature slowly. Reaction was monitored on TLC, after completion reaction was quenched with methanol and solvent was evaporated under reduced pressure. Products were purified on silica gel column by combinations of ethyl acetate and pet ether as eluent.
2-Ethoxyethenyl benzene (3a–a′, mixture of cis and trans isomers) [38]
1H NMR (400 MHz, CDCl3) δ ppm. 8.00–7.97 (1H, m), 7.62–7.56 (1H, m), 7.50–7.46 (1 H, m), 7.32–7.25 (5H, m), 7.17–7.13 (1H, m), 7.01 (0.76H, d, J = 12.9), 6.23 (0.26H, d, J = 7.0), 5.86 (0.73H, d, J = 12.9), 5.24 (0.27H, d, J = 8.0), 4.01 (0.56H, q, J = 7.2), 3.92 (1.5 H, q, J = 7.3), 1.46–1.35 (6H, m); HRMS GC/MS calculated for C10H12O: 148.0883; found 148.0879.
1-Chloro-4[2-ethoxyethenyl]benzene (3b–b′, mixture of cis and trans isomers) [39]
1H NMR (400 MHz, CDCl3) δ ppm. 7.51–7.15 (4H, m), 6.94 (0.69H, d, J = 12.0), 6.37 (0.31H, d, J = 8.0), 5.83 (0.71H, d, J = 12.0), 5.69 (0.29H, d, J = 7.4), 3.95 (0.62H, q, J = 7.4), 3.86 (1.43H, q, J = 7.2), 1.34–1.26 (6H, m); HRMS GC/MS calculated for C10H11OCl: 182.0493; found 182.0501.
1-Bromo-4[2-ethoxyethenyl]benzene (3c–c′, mixture of cis and trans isomers) [42, 62]
1H NMR (400 MHz, CDCl3) δ = 7.31–7.21 (4H, m), 7.01 (0.73H, d, J = 12.8), 6.51 (0.29H, d, J = 7.1), 5.83 (0.70H, d, J = 12.8), 5.69 (0.31H, d, J = 7.3), 4.12 (1.42H, q, J = 7.2), 3.93 (0.63H, q, J = 7.5), 1.45–1.37 (6H, m); HRMS GC/MS calculated for C10H11OBr: 225.9988; found 225.9988.
1-[(1E & Z)-2-ethoxyethenyl]-4-methoxybenzene (3d–d′) [42, 62]
(Mixture of cis and trans isomers) 1H NMR (400 MHz, CDCl3) δ ppm 7.57–7.15 (4H, m), 6.79 (0.63H, d, J = 13.0), 6.13 (0.37H, d, J = 8.0), 6.10 (0.64H, d, J = 12.9), 5.65 (0.38H, d, J = 7.8), 3.89 (4H, q, J = 7.5), 1.43 (6H, m). HRMS GC/MS calculated for C11H14O2: 178.0988; found 178.0991.
(E)-(2-((2-isopropyl-5-methylcyclohexyl)oxy)vinyl)benzene (3e′) [40]
Colorless oil; yield = 43%, 1H NMR (CDCl3, 400 MHz): δ ppm 7.28–7.22 (4H, m), 7.15–7.11 (1H, m), 6.92 (1H, d, J = 12.6), 5.93 (1H, d, J = 12.6), 3.62 (1H, td, J = 4.3), 2.21–2.10 (2H, m), 1.72–1.71 (1H, m), 1.69–1.68 (1H, m), 1.58–1.52 (1H, m), 1.49–1.39 (2H, m), 1.11–1.01 (2H, m), 0.95 (3H, d, J = 6.16), 0.94 (3H, d, J = 6.6), 0.82 (3H, d, J = 6.9); 13C NMR (CDCl3, 100 MHz): δ ppm 147.5, 136.7, 128.6 (2C), 125.4 (2C), 124.9, 107.0, 81.6, 47.8, 41.4, 34.3, 31.5, 25.8, 23.4, 22.1, 20.7, 16.4. HRMS GC/MS calculated for C18H26O; 258.1984, found; 258.1987.
General method for carbon homologation in aldehydes
In a two neck round bottom flask containing phosphonim iodide 2a (1 eq) in dry THF (5 mL), n-BuLi (1.5 eq) was added dropwise at − 78 °C and mixture was allowed to stir for 30 min. Solution of aldehyde (1 eq) in THF was added dropwise to the phosphorene reaction mixture and further allowed to stir for 5 h. After acidic hydrolysis, crude product was extracted with EtOAc (10 mL × 2). Combined extract was dried over Na2SO4, concentrated and purified on preparative TLC (silica gel) to obtain higher analogue of aldehydes (see Additional file 1).
General procedure for asymmetric reduction reaction
In a two-neck round bottom flask, acetophenone (1.5 mmol), NaBH4 (2.25 mmol) along with iodide salt 2g (10 mol%) was taken in methanol (5 mL). Reaction mixture was stirred for 2 h at room temperature. The reaction progress was monitored by TLC and after completion; the mixture was quenched with water and extracted EtOAc (2 × 3 mL). Combined organic layer was dried over MgSO4 and the solvent was evaporated under reduced pressure to afford the corresponding (R)-1-phenylethanol (92% yield, 4% ee). Enantiomeric excess (ee) was calculated on HPLC using chiral cellulose OD-H column, hexane/i-PrOH, 95:5, flow rate 1 mL/min (see Additional file 1).
References
Bahadori L, Manan NS, Chakrabarti MH, Hashim MA, Mjalli FS, AlNashef IM, Hussain MA, Low CT (2013) The electrochemical behaviour of ferrocene in deep eutectic solvents based on quaternary ammonium and phosphonium salts. Phys Chem Chem Phys 15(5):1707–1714
Hayyan M, Hashim MA, Al-Saadi MA, Hayyan A, AlNashef IM, Mirghani ME (2013) Assessment of cytotoxicity and toxicity for phosphonium-based deep eutectic solvents. Chemosphere 93(2):455–459
Marsden SP (2009) Organic synthesis: the Wittig reaction cleans up. Nat Chem 1(9):685
Zaragoza F (2002) One-step conversion of alcohols into nitriles with simultaneous two-carbon chain elongation. (Cyanomethyl) trimethylphosphonium iodide as a reagent with a dual mode of action. J Org Chem 67(14):4963–4964
Hayyan A, Hashim MA, Mjalli FS, Hayyan M, AlNashef IM (2013) A novel phosphonium-based deep eutectic catalyst for biodiesel production from industrial low grade crude palm oil. Chem Eng Sci 5(92):81–88
Wang P, Liao S, Zhu JB, Tang Y (2014) Double γ-alkylation of allylic phosphorus ylides: a unique access to oxa-bicyclic [3.3.0] diene skeletons. Chem Comm 50(7):808–810
Okada H, Mori T, Saikawa Y, Nakata M (2009) Formation of α-hydroxyketones via irregular Wittig reaction. Tetrahedron Lett 50(12):1276–1278
Gentile G, Di Fabio R, Pavone F, Sabbatini FM, St-Denis Y, Zampori MG, Vitulli G, Worby A (2007) Novel substituted tetrahydrotriazaacenaphthylene derivatives as potent CRF 1 receptor antagonists. Bioorg Med Chem Lett 17(18):5218–5221
Fröhlich J, Sauter F, Hametner C, Pfalz M (2009) Synthesis of novel 3-heterospiro [5.5] undecanes. ARKIVOC 1(6):298–308
Wiebe DA, Burton DJ (2012) Chemoselective halogenation of 2-hydroperfluoroalkyl aldehydes. J Fluorine Chem 1(139):4–11
Ko KY, Wagner S, Yang SH, Furkert DP, Brimble MA (2015) Improved synthesis of the unnatural amino acids AHMOD and AMD, components of the anticancer peptaibol culicinin D. J Org Chem 80(17):8631–8636
Pindur U, Lutz G, Rogge M (1995) First synthesis of chiral 3-vinylindoles as 4π-components for Diels–Alder reactions. J Heterocyclic Chem 32(1):201–206
Lambert WT, Hanson GH, Benayoud F, Burke SD (2005) Halichondrin B: synthesis of the C1–C22 subunit. J Org Chem 70(23):9382–9398
Assefa H, Nimrod A, Walker L, Sindelar R (2001) Enantioselective synthesis and complement inhibitory assay of A/B-ring partial analogues of oleanolic acid. Bioorg Med Chem Lett 11(13):1619–1623
Treu M, Jordis U (2002) 4a,5,9,10,11,12-Hexahydro-6H-benzo[a]cyclohepta[hi] benzofuran-synthesis of unnatural galanthamine analogs. Molecules 7(4):374–381
Poschalko A, Welzig S, Treu M, Nerdinger S, Mereiter K, Jordis U (2002) Synthesis of (±)-6H-benzofuro[3a,3,2, ef][3]benzazepine: an unnatural analog of (−)-galanthamine. Tetrahedron 58(8):1513–1518
Mangold SL, Carpenter RT, Kiessling LL (2008) Synthesis of fluorogenic polymers for visualizing cellular internalization. Org Lett 10(14):2997–3000
Fujioka H, Goto A, Otake K, Kubo O, Yahata K, Sawama Y, Maegawa T (2010) Remarkable effect of phosphine on the reactivity of O,P-acetal—efficient substitution reaction of O,P-acetal. Chem Comm 46(22):3976–3978
Fujioka H, Goto A, Otake K, Kubo O, Sawama Y, Maegawa T (2011) An unusual reaction of α-alkoxyphosphonium salts with Grignard reagents under an O2 atmosphere. Chem Comm 47(35):9894–9896
Goto A, Otake K, Kubo O, Sawama Y, Maegawa T, Fujioka H (2012) Effects of phosphorus substituents on reactions of α-alkoxyphosphonium salts with nucleophiles. Chem Eur J 18(36):11423–11432
Deng Z, Lin JH, Xiao JC (2016) Nucleophilic arylation with tetraarylphosphonium salts. Nat Commun 29(7):10337
Szymczyk M (2017) Unexpected course of Wittig reaction when using cinnamylaldehyde as a substrate. Phosphorus Sulfur 192(3):264–266
Wittig G, Schlosser M (1961) Über die Herstellung von Vinyläthern, Vinylthioäthern und Vinylhalogeniden auf der Phosphylen-Basis; IV. Mitteil. über Phosphin-alkylene als olefinbildende Reagenzien. Eur J Inorg Chem 94(5):1373–1383
Mazurek MA (1978) A new efficient synthesis of iodomethyl methyl ether. Synthesis 08:588–589
Morcillo SP, Alvarez de Cienfuegos L, Mota AJ, Justicia J, Robles R (2011) Mild method for the selective esterification of carboxylic acids based on the Garegg–Samuelsson reaction. J Org Chem 76(7):2277–2281
Xu F, Wang NG, Tian YP, Chen YM, Liu WC (2012) Ph3P/I2-catalyzed beckmann rearrangement of ketoximes into amides. Synth Commun 42(23):3532–3539
Lockman JW, Klimova Y, Anderson MB, Willardsen JA (2012) Synthesis of substituted quinazolines: application to the synthesis of verubulin. Synth Commun 42(12):1715–1723
Kumar A, Akula HK, Lakshman MK (2010) Simple synthesis of amides and Weinreb amides using PPh3 or polymer-supported PPh3 and iodine. Eur J Org Chem 2010(14):2709–2715
Samimi HA, Kiyani H, Shams Z (2013) Stereo-controlled deamination of ketoaziridines using Ph3P/I2. J Chem Res 37(5):282–284
Duangkamol C, Wangngae S, Pattarawarapan M, Phakhodee W (2014) Acyloxyphosphonium versus aminophosphonium intermediates: application to the synthesis of N-acylbenzotriazoles. Eur J Org Chem 32:7109–7112
Wangngae S, Duangkamol C, Pattarawarapan M, Phakhodee W (2015) Significance of reagent addition sequence in the amidation of carboxylic acids mediated by PPh3 and I2. RSC Adv 5(33):25789–25793
Sun G, Lv X, Zhang Y, Lei M, Hu L (2017) Palladium-catalyzed formylation of Aryl iodides with HCOOH as CO source. Org let 19(16):4235–4238
Mumtaz S, Wali KS, Zaidi J, Iqbal A, Maqsood CZ, Mohammed KK, Perveen S (2013) Synthesis of chiral menthoxymethyl ether of phenol and substituted phenol and their use in directed ortho metalation. Lett Org Chem 10(8):578–583
Pruess DL, Scannell JP, Kellett M, Ax HA, Janecek J, Williams TH, Berger J (1947) Antimetabolites produced by microorganisms. X. J Antibio 27(4):229–233
González-Coloma A, Escoubas P, Mizutani J, Lajide L (1994) Insect growth inhibitors from Machilus japonica. Phytochemistry 35(3):607–610
Nebois P, Greene AE (1999) Novel enantioselective approach to γ-lactams from chiral enol ethers: synthesis of (−)-statine. J Org Chem 61(16):5210–5211
Corey EJ (2002) Catalytic enantioselective Diels–Alder reactions: methods, mechanistic fundamentals, pathways, and applications. Angew Chem Int Ed 41:1650–1667
Tian J, Moeller Kevin D (2005) Electrochemically assisted Heck reactions. Org Lett 24:5381–5383
Miyaura N, Maeda K, Suginome H (1982) Palladium-catalyzed cross-coupling of (2-ethoxyvinyl) boranes with aryl and benzyl halides. A new method for conversion of organic halides into aldehydes with two more carbon atoms. J Org Chem 47(11):2117–2120
Fuwa H, Sasaki M (2007) An efficient method for the synthesis of enol ethers and enecarbamates. Total syntheses of isoindolobenzazepine alkaloids, lennoxamine and chilenine. Org Biomol Chem 5(12):1849–1853
Satoh M, Miyaura N, Suzuki A (1987) Palladium-catalyzed cross-coupling reaction of (1-ethoxy-1-alken-2-yl) boranes with ortho-functionalized iodoarenes. A novel and convenient synthesis of benzo-fused heteroaromatic compounds. Synthesis 04:373–377
Sakamoto T, Kondo Y, Yasuhara A, Yamanaka H (1991) Condensed heteroaromatic ring systems. XVIII. Palladium-catalyzed cross-coupling reaction of aryl bromides with (Z)-1-ethoxy-2-tributylstannylethene and its utilization for construction of condensed heteroaromatics. Tetrahedron 47:1877–1886
Beletskaya IP, Cheprakov AV (2000) The Heck reaction as a sharpening stone of palladium catalysis. Chem Rev 100:3009–3066
Fürstner A (2000) Olefin metathesis and beyond. Angew Chem Int Ed 39:3012–3043
Rehbein J, Hiersemann M (2013) Claisen rearrangement of aliphatic allyl vinyl ethers from 1912 to 2012: 100 years of electrophilic catalysis. Synthesis 45(09):1121–1159
Hansen HJ, Schmid H (1974) Stereochemie von [3.3]-und [5.5]-sigmatropischen umlagerungen. Tetrahedron 30(13):1959–1969
He W, Herrick IR, Atesin TA, Caruana PA, Kellenberger CA, Frontier AJ (2008) Polarizing the Nazarov cyclization: the impact of dienone substitution pattern on reactivity and selectivity. J Am Chem Soc 130(3):1003–1011
Malona JA, Cariou K, Frontier AJ (2009) Nazarov cyclization initiated by peracid oxidation: the total synthesis of (±)-rocaglamide. J Am Chem Soc 131(22):7560–7561
Aoshima S, Kanaoka S (2009) A renaissance in living cationic polymerization. Chem Rev 109(11):5245–5287
Friesen RW (2001) Generation and reactivity of α-metalated vinyl ethers. J Chem Soc Perkin Trans 17:1969–2001
Dehli JR, Legros J, Bolm C (2005) Synthesis of enamines, enol ethers and related compounds by cross-coupling reactions. Chem Commun 8:973–986
Winternheimer DJ, Shade RE, Merlic CA (2010) Methods for vinyl ether synthesis. Synthesis 15:2497–2511
Wan Z, Jones CD, Koenig TM, Pu YJ, Mitchell D (2003) Vinyl aryl ethers from copper-catalyzed coupling of vinyl halides and phenols. Tetrahedron Lett 44(45):8257–8259
Shade RE, Hyde AM, Olsen JC, Merlic CA (2010) Copper-promoted coupling of vinyl boronates and alcohols: a mild synthesis of allyl vinyl ethers. J Am Chem Soc 132(4):1202–1203
Kondo M, Kochi T, Kakiuchi F (2010) Rhodium-catalyzed anti-Markovnikov intermolecular hydroalkoxylation of terminal acetylenes. J Am Chem Soc 133(1):32–34
Moyano A, Charbonnier F, Greene AE (1987) Simple preparation of chiral acetylenic ethers. J Org Chem 52(13):2919–2922
Keegstra MA (1992) Copper catalysed preparation of vinyl ethers from unactivated vinylic halides. Tetrahedron 48(13):2681–2690
Ronson TO, Voelkel MH, Taylor RJ, Fairlamb IJ (2015) Macrocyclic polyenynes: a stereoselective route to vinyl-ether-containing skipped diene systems. Chem Commun 51(38):8034–8036
Lam PY, Vincent G, Bonne D, Clark CG (2003) Copper-promoted/catalyzed C–N and C–O bond cross-coupling with vinylboronic acid and its utilities. Tetrahedron Lett 44(26):4927–4931
Dussault PH, Sloss DG, Symonsbergen DJ (1998) Application of the Sonogashira coupling reaction to the stereoselective synthesis of chiral 1,3-dienol ethers. Synlett 12:1387–1389
Maeda K, Shinokubo H, Oshima K, Utimoto K (1996) Stereoselective synthesis of allyl vinyl ethers from silyl enol ethers. J Org Chem 61(7):2262–2263
Engesser T, Brückner R (2017) Synthesis of trans configured enol ethers by a sequence of syn selective glycolate aldol addition, hydrolysis, and grob fragmentation. Eur J Org Chem 38:5789–5794
Kulkarni MG, Rasne RM, Davawala SI, Doke AK (2002) Allyl vinyl ethers via Wittig olefination: a short and efficient synthesis of (±)-mesembrine. Tetrahedron Lett 43(12):2297–2298
Balti M, Efrit ML, Leadbeater NE (2016) Preparation of vinyl ethers using a Wittig approach, and their subsequent hydrogenation employing continuous-flow processing. Tetrahedron Lett 57(16):1804–1806
Authors’ contributions
HYG designed and supervised the project and wrote the paper. ZMC performed experiments and assist in manuscript preparation. JHZ guided in data interpretation and reaction mechanism. SY solved X-ray structure. MIC provided instrumental facilities. All authors read and approved the final manuscript.
Acknowledgements
Authors are obliged to Pakistan Science Foundation (PSF) Islamabad for support of this research project (P-US/Chem 427). We are also grateful to HEJ Research institute of Chemistry, ICCBS Karachi for providing analytical facilities.
Availability of data
CCDC. 1537362 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; or deposit@ccdc.cam.ac.uk). General procedure and spectral data of substrates bis-alkoxy methanes (1) and specimen NMR spectra of α-alkoxymethyl phosphonium iodides (2) and vinyl ethers (3) are given in Additional file 1.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1.
General method for synthesis of Bis-alkoxy methanes.
Additional file 2.
Carbon Homologation in aldehydes.
Additional file 3.
Asymmetric reduction of acetophenone.
Additional file 4.
Crystallography data for(S)-sec-Butoxymethyltriphenylphosphonium iodide.
Additional file 5.
Specimen NMR Spectra of alkoxymethyltriphenylphosphonium iodides.
Additional file 6.
Specimen NMR Spectrum of vinyl ether.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gondal, H.Y., Cheema, Z.M., Zaidi, J.H. et al. Facile synthesis of α-alkoxymethyltriphenylphosphonium iodides: new application of PPh3/I2. Chemistry Central Journal 12, 62 (2018). https://doi.org/10.1186/s13065-018-0421-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0421-6