
Abstract
Background
In the vertebrate spinal cord, motor neurons (MN) are generated in stereotypical numbers from a pool of dedicated progenitors (pMN) whose number depends on signals that control their specification but also their proliferation and differentiation rates. Although the initial steps of pMN specification have been extensively studied, how pMN numbers are regulated over time is less well characterized.
Results
Here, we show that ephrinB2 and ephrinB3 are differentially expressed in progenitor domains in the ventral spinal cord with several Eph receptors more broadly expressed. Genetic loss-of-function analyses show that ephrinB2 and ephrinB3 inversely control pMN numbers and that these changes in progenitor numbers correlate with changes in motor neuron numbers. Detailed phenotypic analyses by immunostaining and genetic interaction studies between ephrinB2 and Shh indicate that changes in pMN numbers in ephrin mutants are due to alteration in progenitor identity at late stages of development.
Conclusions
Altogether our data reveal that Eph:ephrin signaling is required to control progenitor identities in the ventral spinal cord.
Similar content being viewed by others


Background
In vertebrates, motor neurons (MN) innervating skeletal muscles are born in the ventral neural tube, the future spinal cord, from a pool of progenitors located in the ventricular zone. As for other neuronal subtypes, the production of stereotyped numbers of MN requires the integration of different processes such as specification and proliferation of progenitors, followed by cell cycle exit and differentiation [1]. While these processes are common to the genesis of all neuronal subtypes throughout the central nervous system, one distinguishing feature of MN development is the fact that progenitor specification is dependent on their spatial organization within the neural tube. Indeed, the vertebrate neural tube is organized along its dorsoventral axis in different progenitor domains which first give rise to distinct neuronal subtypes and later on to subtypes of glial cells. Combinatorial positional information provided by graded Sonic Hedgehog (Shh), Wnt, BMP and FGF signaling induces the regionalized expression of homeodomain and helix-loop-helix identity transcription factors (iTFs) in different progenitor domains [1]. For instance, progenitors of motor neurons (pMNs) express the iTF Olig2 while adjacent progenitors (p3) which give rise to v3 interneurons express the iTF Nkx2.2. Ventral patterning of the spinal cord, including specification of pMN and p3 progenitors, is controlled by the morphogen Sonic Hedgehog (Shh) produced by the notochord and the floor plate [2]. In addition to doses, different exposure times to Shh modulates the expression of these iTFs in progenitors, thus specifying the distinct progenitor identities [3, 4]. Specifically, the progressive emergence of a gene regulatory network (GRN) composed of three transcription factors- Pax6, Olig2 and Nkx2.2 whose expression is refined by cross-repressive interactions interprets graded Shh signaling, to control the size of the p3 and pMN progenitor domains [5, 6]. Although early steps of progenitor specification are fairly well characterized, mechanisms that ensure stereotypy in the size of progenitor domains as the tissue grows are less well understood. These mechanisms include maintenance of progenitor identities [4, 7, 8] as well as control of proliferation and differentiation rates that vary between progenitor types and over time [9]. In addition, mechanisms such as cell sorting have been shown to participate in defining and/or maintaining domain boundaries thus indirectly contributing to pattern progenitor domains [10,11,12,13].
Eph:ephrin signaling is a cell-to-cell communication pathway that has been implicated in numerous developmental processes [14, 15]. A distinctive feature of Eph:ephrin signaling is its ability to trigger forward signaling downstream of Eph receptors and reverse signaling downstream of ephrins. One of its major biological functions is to control cell adhesion and repulsion events in developing and adult tissues thus leading to the establishment and/or maintenance of axon tracts, tissue organization and patterning [16, 17]. In addition, Eph:ephrin signaling has been shown to control various aspects of neural progenitor development and homeostasis in the developing and adult mammalian cortex including self-renewal, proliferation, quiescence and differentiation [18]. In the developing spinal cord, the role of Eph:ephrin signaling has been prominently studied in post-mitotic neurons, specifically in axon guidance and fasciculation of MNs [19,20,21], as a consequence, virtually nothing is known on the function of this pathway in spinal progenitors. Here, we show that two B-class ephrins, ephrinB2 and ephrinB3 are differentially expressed in pMN and p3 progenitors. Loss of function analyses indicate that expression of ephrinB2 and ephrinB3 is not required for initial specification of these progenitors. However, at later developmental stages, expression of ephrinB2 and ephrinB3 is essential to maintain appropriate numbers of pMN and p3 progenitors. Interestingly, ephrinB2 and ephrinB3 mutants exhibit opposite phenotypes, matching their opposite differential expression patterns. Detailed analyses of ephinB2 mutants indicate that the change in pMN number is not due to a change in proliferation or differentiation rates. Rather, our data shows that Efnb2 interacts with Shh to control the ratio between pMN and p3 progenitors. Lastly, loss of ephrinB3 -but not ephrinB2- leads to pMN and p3 progenitor intermingling. Altogether our data suggests that Eph:ephrin signaling plays a role in controlling progenitor identity.
Methods
Mice
Ephrin mutant mice were maintained in a mixed background and genotyped by PCR. The mouse lines Shh ko, Efnb3 ko, Efnb2 lox and Efnb2 GFP have been described previously [22,23,24]. The Olig2-Cre mouse line [3] was maintained in a pure C57Bl6/J genetic background. For Efnb2 cKO, control genotypes used in the study include Efnb2 lox/lox, Efnb2 lox/GFP, Efnb2 +/GFP and Efnb2 +/GFP ; Olig2-Cre. For Efnb3 KO, control genotypes are always Efnb3 +/-. E0.5 is defined as the day on which a vaginal plug was detected.
In Situ Hybridization
In situ hybridization was performed using standard protocols on 70μm vibratome sections at brachial level. Antisense RNA probes labeled with digoxigenin were used to detect in vivo gene expression with a 72 h incubation time.
Immunostaining
All analyses for Efnb2 cKO were performed on control and mutant littermates collected from at least two different litters. On the other hand, control and Efnb3 mutant embryos were collected from independent litters. The number of embryo analyzed for each immunostaining and each developmental stage is indicated in the figure legends. To avoid bias in rostro-caudal axis, data was collected on thick vibratome sections covering the entire brachial region (600 μm). Antibody staining was performed following standard protocol on 70μm vibratome sections of mouse embryos at brachial level. For BrdU incorporation, pregnant dams were injected with BrdU (10mg/ml; 100mg/kg) with intraperitoneal injection. After 1 h, embryos were dissected in cold PBS and processed for subsequent immunostaining.
Antibodies used were: goat anti-Nkx2.2 (1/100, Santa Cruz Biotechnology); rabbit anti-Olig2 (1/1000, Sigma); mouse anti-Islet1/2, 39-4D5 (1/50, DSHB); rabbit anti-Foxp1 (1/200, Abcam), rabbit anti-P-H3 (1/1000, Millipore), rabbit anti-EphA4 (1/100, Santa Cruz Biotechnology), goat anti-EphB2 (1/50, R&D Systems), Tuj1 (1/1000, Covance). All secondary antibodies were from Jackson ImmunoResearch (1/1000).
Image processing and quantification
Images were collected on a Leica SP5 confocal microscope or Nikon eclipse 80i microscope for in situ hybridization data. Cell numbers were collected blindly on 5 vibratome sections (n=25 confocal Z-sections) per embryo and at least 2000 nuclei were recorded per embryo. The number of embryo analyzed for each immunostaining and each developmental stage is indicated in the figure legends. Acquisitions of nuclei 2D positions and semi quantitative analyses of fluorescence intensity were performed using Fiji [25]. Spatial distribution of progenitor subtypes was quantified using the R Project (http://www.r-project.org/), see Additional file 1: (Sup Code) for details on the code.
Statistical Analysis
For all analyses sample size was estimated empirically. Sample sizes are indicated in Figure legends and further details are provided in Additional file 2: Table S1. Statistical analyses were performed with GraphPad, using Mann-Whitney-Wilcoxon test or ANOVA, depending on the data set. P<0.05 was considered statistically significant.
Results
EphrinB2 and ephrinB3 exhibit restricted expression in progenitors of the ventral spinal cord.
A survey of members of the B-type Eph receptor family in the mouse ventral spinal cord (Fig. 1a) indicated that spinal progenitors co-express several EphB receptors, as well as EphA4, as shown by in situ hybridization (Fig. 1b-d) and immunofluorescence (Fig. 1e-g). Concerning B-type ephrin ligands, in situ hybridization at different developmental stages reveals that while Efnb1 is not expressed at significant levels in progenitors of the ventral spinal cord (Fig. 1h-j), both Efnb2 and Efnb3 are expressed in subsets of these cells. More precisely, at all stages analyzed, Efnb2 is expressed by progenitors located at an intermediate dorso-ventral position within the spinal cord, its expression never extending to the ventral-most region (Fig. 1k-m). Conversely, expression of Efnb3 is highest in the ventral-most region of the spinal cord at all stages analyzed, with a lower expression extending more dorsally (Fig. 1n-p). Because Efnb2 and Efnb3 were expressed in distinct progenitor domains of the spinal cord, we asked whether these corresponded to progenitors with distinct identities, namely pMN progenitors expressing Olig2 and p3 progenitors expressing Nkx2.2. Since the expression of Efnb2 in progenitors of the ventral neural tube detected by in situ hybridization was low, we took advantage of a reporter mouse line that expresses H2BGFP under the control of the Efnb2 endogenous promoter [22]. The benefit of this reporter strategy is that H2BGFP accumulates in the nucleus thus highlighting low domains of expression and facilitating co-expression analyses. In accordance with in situ hybridization data, H2BGFP expression was detected in a restricted population of neural progenitors from E9.5 to E11.5 (Fig. 2a-d). Co-staining with Olig2 showed that the expression domain of Efnb2 overlapped with the Olig2+ (pMN) domain (Fig. 2a-l). Co-staining with Olig2 and Nkx2.2, the iTFs for pMN and p3 respectively, showed that the ventral boundary of Efnb2 expression strictly corresponds to the p3/pMN boundary (Fig. 2m-p). Conversely, in situ hybridization for Efnb3 followed by Olig2 immunostaining showed that the highest domain of Efnb3 expression corresponds to Olig2- floor plate and p3 progenitors (Fig. 2q-s). Altogether, these expression analyses indicate that all progenitors of the ventral spinal cord co-express several Eph receptors and reveal that ephrinB2 and ephrinB3 are differentially expressed in pMN and p3 progenitors (Fig. 2t).

Eph receptors and ephrins are expressed in progenitors of the ventral spinal cord. a. Schematic representation of the ventral spinal cord at E10.5. Progenitors are located in the ventricular zone, three progenitor domains are shown: p2, pMN and p3. Differentiated motor neurons (MN) are located laterally in the mantle zone. b-d. Expression of EphB1 (b), EphB2 (c) and EphB3 (d) was monitored by in situ hybridization on transverse sections of E10.5 embryos. Scale bars: 50 μm. e-g. Transverse sections of E10.5 embryos were immunostained to detect EphA4 ((e), red), EphB2 ((f), blue) and differentiated neurons (Tuj1, green in (g)). Scale bars: 40 μm. (h-p). Expression of Efnb1 (h-j), Efnb2 (k-m) and Efnb3 (n-p) was monitored by in situ hybridization on transverse sections of E9.5, E10.5 and E11.5 embryos, as indicated. Scale bars: 50 μm. Brackets indicate domains of Efnb2 expression in progenitors. FP: floor plate, NC: notochord.

EphrinB2 and ephrinB3 are expressed in complementary domains in progenitors of the ventral spinal cord. a-l. Transverse sections of Efnb2 +/GFP embryos at E8.5, E9.5, E10.5 and E11.5 (as indicated) were immunostained to detect Olig2 (e-h). Epifluorescence is shown on (a-d) and merged images are shown on (i-l). Scale bars: 50 μm. Dashed lines highlight the ventricular zone and brackets indicate domains of Efnb2:H2BGFP expression in progenitors. m-p. Transverse sections of Efnb2 +/GFP E11.5 embryos were immunostained to detect Olig2 (m) and Nkx2.2 (n). Epifluorescence is shown in (o) and a merged image is shown in (p). The dashed line marks the p3/pMN boundary. Scale bars: 50 μm. q-s. Transverse sections of wild type E10.5 embryos were processed for Olig2 immunostaining (q) and for Efnb3 in situ hybridization (r). A merged image is shown in (s). The dashed line marks the p3/pMN boundary. Scale bars: 25 μm. T. Schematic representation of Efnb2 and Efnb3 expression in relation to pMN and p3 progenitors domains at E11.5.
EphrinB2 controls the number of pMN and their progeny
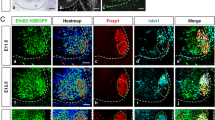
Based on the expression of ephrinB2 in pMN but not p3 progenitors we hypothesized that it may be required for pMN development. We thus generated Efnb2 loss-of-function mutant embryos and quantified the number of Olig2+ progenitors at three different developmental stages. Since Efnb2 -/- (KO) embryos exhibit precocious lethality (E10.5) due to cardiovascular defects [26, 27] to analyze later stages of development we generated Efnb2 conditional mutant embryos, using the Olig2-Cre mouse line (Efnb2 lox/lox ; Olig2-Cre thereafter called cKO). At E9.5 and E10.5, no difference was observed in the number of Olig2+ progenitors when comparing wild type and Efnb2 KO or Efnb2 cKO (Fig. 3a-d, g). On the contrary, at E11.5, the number of Olig2+ progenitors was significantly decreased in Efnb2 cKO (Fig. 3e-g), indicating that ephrinB2 is not required for initial pMN specification but is necessary at later stages to control the number of pMN progenitors. The decrease in the number of Olig2+ progenitors between E10.5 and E11.5 which is observed in wild type embryos is partly driven by differentiation of these cells into MN [9], raising the possibility that the decrease in the number of pMN progenitors in absence of ephrinB2 could be due to increased differentiation. We thus assessed the number of pMN progeny in wild type and Efnb2 mutant embryos. In the spinal cord, pMN first give rise to MN which settle in specific motor columns in the mantle zone and second, after the neuroglial transition, pMN give rise to oligodendrocyte precursors (OLP) that maintain Olig2 expression and migrate in the mantle zone. Immunostaining for Islet1/2 and Foxp1 to label MN showed that the reduction in pMN numbers correlates with a reduction in the total number of MN in E12.5 Efnb2 cKO (Fig. 3h-j), which is not consistent with increased differentiation. To assess whether one subtype of MN was preferentially affected, we used different combination of Foxp1 and Islet1/2 to discriminate different motor columns. These quantifications showed that the reduction of total MN number does not correlate with reduction of one specific motor column, however, we observed that within this reduced pool of MN, slightly more LMCm MN were present in Efnb2 cKO (Fig. 3k). Next we assessed the number of OLP by quantifying Olig2+ nuclei in the mantle zone of E13.5 Efnb2 cKO. Similar to what was observed for MN, loss of Efnb2 in pMN correlates with a decrease in OLP numbers (Fig. 3l-p). Altogether, this data reveal that ephrinB2 is required to produce a stereotyped number of pMN and of their progeny by mechanisms likely independent of differentiation.

EphrinB2 controls the number of pMN progenitors and their progeny.
a, b. Transverse sections of wild type (a) and Efnb2 -/- (b) E9.5 embryos were immunostained to detect Olig2. c, d. Transverse sections of Efnb2 lox/GFP (c) and Efnb2 lox/GFP ;Olig2-Cre (d) E10.5 embryos were immunostained to detect Olig2. e, f. Transverse sections of Efnb2 lox/GFP (e) and Efnb2 lox/GFP ;Olig2-Cre (f) E11.5 embryos were immunostained to detect Olig2. g. The number of Olig2+ progenitors was quantified for all genotypes. Error bars indicate s.e.m. (n=3 embryos per genotype at E9.5; n=4 embryos per genotype at E10.5; n=5 embryos per genotype at E11.5); **P<0.01, ns= non significant (Mann-Whitney-Wilcoxon test). h, i. Transverse sections of E12.5 Efnb2 lox/GFP (h) and Efnb2 lox/GFP ; Olig2-Cre (i) embryos were immunostained to detect Foxp1 (green) and Islet 1/2 (red). j. Quantification of the total number of motor neurons (Foxp1+ and Islet 1/2+) in both genotypes. k. Repartition of motor neurons in motor columns in both genotypes. Error bars indicate s.e.m. (n=6 embryos per group); *P<0.05; **P<0.01; ns= non significant (Mann-Whitney-Wilcoxon test). l-o. Transverse sections of E13.5 Efnb2 lox/GFP (l, n) and Efnb2 lox/GFP ; Olig2-Cre (m, o) embryos were immunostained to detect Olig2 (red) and NeuN (green). n, o are zoomed areas indicated by a box in m, n respectively. p. Quantification of the total number of Olig2+ cells in the mantle zone in both genotypes. Error bars indicate s.e.m. (n=4 embryos per group); **P<0.01 (Mann-Whitney-Wilcoxon test). Scale bars A-I: 50 μm; L-O: 200 μm.
Efnb2 interacts with Shh to control the ratio between pMN and p3 progenitors
To characterize the underlying causes of the decreased pMN number in ephrinB2 mutants, we first tested whether this reduction was due to altered rates of proliferation of pMN. We performed BrdU incorporation and immunostaining for Olig2 and quantified BrdU+/Olig2+ progenitors at two different developmental stages to monitor proliferation. At both stages, no difference in pMN proliferation was observed in Efnb2 cKO compared to control embryos (Fig. 4a-f). We also assessed apoptosis and observed no change in the number of cleaved caspase positive pMN at E10.5 and E11.5 (data not shown). In addition, to confirm that the rate of pMN differentiation was unchanged in Efnb2 cKO, we performed co-immunostaining for Olig2 and for the motor neuron (MN) marker Islet1/2 and quantified the fraction of Islet1/2+ nuclei at the basal side of the ventricular zone (intermediate zone) which represent newborn MN. As expected, no difference was observed between control and Efnb2 cKO embryos (Fig. 4d, e, g). These results show that the reduction in pMN number in absence of ephrinB2 is not due to alteration in their rate of proliferation or differentiation.

No change in pMN proliferation or differentiation rates in absence of ephrinB2. a, b. Transverse sections of E10.5 Efnb2 lox/GFP (a) and Efnb2 lox/GFP ; Olig2-Cre (b) embryos were immunostained to detect Olig2 (red) and BrdU (green). c. Quantification of BrdU+ nuclei in the Olig2+ population in both genotypes. d, e. Transverse sections of E11.5 Efnb2 lox/GFP (d) and Efnb2 lox/GFP ; Olig2-Cre (e) embryos were immunostained to detect Olig2 (red), Islet 1/2 (blue) and BrdU (green). f. Quantification of BrdU+ nuclei in the Olig2+ population in both genotypes. g. Quantification of Islet 1/2+ nuclei in the intermediate zone relative to the Olig2+ population in both genotypes. Error bars indicate s.e.m. (n=4 embryos per group); ns= non significant (Mann-Whitney-Wilcoxon test). Scale bars: 50 μm.
An alternate possible cause for the decreased number of Olig2+ progenitors at later stages of development could be that a fraction of progenitors wrongly acquire a non-pMN identity after E9.5. To assess this possibility we quantified the number of Nkx2.2+ progenitors in E11.5 Efnb2 cKO embryos and observed that the decreased number of Olig2+ progenitors was matched by an increased number of Nkx2.2+ progenitors (Fig. 5a-c). Importantly, this was not due to an increase in the number of progenitors of mixed identity (expressing both iTFs) which was similar in control and Efnb2 cKO embryos (Fig. 5c). Remarkably, the total number of pMN+p3 progenitors was also similar in control and Efnb2 cKO embryos (Fig. 5a-c), strongly suggesting that in absence of ephrinB2, a fraction of progenitors wrongly acquire the p3 identity (Nkx2.2+) at the expense of the pMN identity (Olig2+). To challenge this interpretation, we tested for a potential genetic interaction between Efnb2 and Shh, a key player in the specification of p3 and pMN identities [2]. First, we verified that Shh expression pattern was not changed in Efnb2 mutants (Additional file 3: Figure S1). Next, we quantified the number of pMN and p3 progenitors in single Efnb2 and Shh heterozygotes or in compound heterozygote embryos. While the number of Olig2+ and Nkx2.2+ progenitors in Efnb2 +/- and Shh +/- heterozygous embryos was equivalent to wild type embryos, Efnb2 +/- ; Shh +/- double heterozygous embryos exhibited a phenotype similar to Efnb2 cKO embryos, with a decrease in Olig2+ balanced by an increase in Nkx2.2+ progenitors (Fig. 5d-h). These results establish that ephrinB2 and Shh interact genetically to control the ratio between pMN and p3 progenitors in the ventral spinal cord.

Efnb2 interacts with Shh to control the ratio between pMN and p3 progenitors. a, b. Transverse sections of Efnb2 lox/GFP (a) and Efnb2 lox/GFP ;Olig2-Cre (b) E11.5 embryos were immunostained to detect Olig2 (red) and Nkx2.2 (blue). c. The number of Olig2+, Nkx2.2+ and Olig2+/ Nkx2.2+ (double) progenitors was quantified (n=5 embryos per genotype). Total refers to the sum of Olig2+ and Nkx2.2+ progenitors. Error bars indicate s.e.m.; **P<0.01 ns= non significant (Mann-Whitney-Wilcoxon test). d-g. Transverse sections of E11.5 embryos of different genotypes (as indicated) were immunostained for Olig2 (red) and Nkx2.2 (blue). h. Quantification of the number of Olig2+, Nkx2.2+ and Olig2+/ Nkx2.2+ (double) progenitors was quantified for each genotype (n=5 embryos per genotype). Total refers to the sum of Olig2+ and Nkx2.2+ progenitors. Error bars indicate s.e.m.; *P<0.05; **P<0.01; (Mann-Whitney-Wilcoxon test). Scale bars: 50 μm.
EphrinB3 inversely controls the ratio between pMN and p3 identities
The above data suggests that expression of ephrinB2 in pMN is required to impose the pMN identity (Olig2+). As shown in Fig. 2, ephrinB3 is highly expressed in p3 progenitors (Fig. 2q-s). To test whether ephrinB3 plays a similar role in controlling p3 progenitor identity, we performed immunostaining for Nkx2.2 and Olig2 in Efnb3 -/- (KO) E11.5 embryos. Quantification of the number of Olig2+ and Nkx2.2+ progenitors showed that loss of Efnb3 led to a decrease in the number of Nkx2.2+ progenitors which was balanced by an increase in Olig2+ progenitors (Fig. 6a-c). Importantly, the number of progenitors with a mixed identity and the total number of p3+pMN progenitors was unchanged in Efnb3 KO compared to Efnb3 +/- embryos (Fig. 6c), suggesting that a fraction of progenitors wrongly acquired the pMN at the expense of p3 identity in absence of ephrinB3. No change in progenitor numbers was observed at E9.5 (data not shown) and the increase in pMN number in Efnb3 KO correlated with an increase in MN numbers (Additional file 3: Figure S2A-D). Interestingly, in addition to a change in the number of pMN and p3 progenitors, intermingling between these progenitors was observed in an increased fraction of sections from Efnb3 KO compared to sections from Efnb3 +/- embryos (Fig. 6b, d). To further quantify this phenotype, we measured surfaces encompassing all Olig2+ or Nkx2.2+ nuclei on multiple transverse sections of Efnb3 +/- and Efnb3 KO embryos and deduced their region of overlap (Fig. 6e, f). The surface of overlap between Olig2+ and Nkx2.2+ domains was increased in Efnb3 KO compared to control embryos (Fig. 6e, f), confirming intermingling between Olig2+ and Nkx2.2+ progenitors in absence of ephrinB3. On the contrary, no overlap between Olig2+ and Nkx2.2+ domains was detected in Efnb2 cKO (Additional file 3: Figure S3). Altogether, these results indicate that similar to ephrinB2, ephrinB3 is required to control the ratio between p3 and pMN progenitors and that in addition, ephrinB3 is required to maintain a sharp boundary between pMN and p3 progenitor domains.

EphrinB3 inversely controls the ratio between pMN and p3 progenitors. a, b. Transverse sections of Efnb3 +/- (a) and Efnb3 -/- (b) E11.5 embryos were immunostained to detect Olig2 (red) and Nkx2.2 (blue). c. The number of Olig2+, Nkx2.2+ and Olig2+/ Nkx2.2+ (double) progenitors was quantified. Total refers to the sum of Olig2+ and Nkx2.2+ progenitors. d. Quantification of the proportion of sections showing an overlap in Efnb3 +/- and Efnb3 -/- embryos. e. Example of spatial positioning of Olig2+and Nkx2.2+ progenitors in an overlap situation in an Efnb3 -/- embryo. f. Quantification of the surface of overlap between Olig2+ and Nkx2.2+ domains in Efnb3 +/- and Efnb3 -/- embryos. Error bars indicate s.e.m. (n=5 embryos per genotype); *P<0.05; **P<0.01; (Mann-Whitney-Wilcoxon test), ns: non significant. Scale bars: 50 μm.
Discussion
While early steps of ventral pMN specification have been extensively studied, highlighting the critical role of Shh, mechanisms that control the number of ventral progenitors over time are less well characterized. Here we show that Eph:ephrin signaling is required to precisely control the number of pMN (and p3) progenitors at late stages of development (after E9.5). It has been proposed previously that the modulation of p3 and pMN numbers after E9.5 is driven mainly by differentiation and/or proliferation, which vary over time and according to progenitor types [9]. Despite the fact that a number of studies in the developing and adult cortex have shown a role for Eph:ephrinB signaling in controling the balance between proliferation and differentiation of neural progenitors in the cerebral cortex [18], our data unexpectedly show that changes in pMN progenitor numbers in ephrin mutants are not due to alterations of proliferation or differentiation rates.
Instead, our data indicate that ephrinB2 and ephrinB3 are respectively required to impose pMN and p3 identities at later stages of development (Fig. 7). What could be the underlying mechanisms? It has been shown previously that progenitor identity has to be actively maintained after initial specification. Indeed, mouse mutants in which Shh signaling is altered after ventral identities have been assigned exhibit a progressive loss of Olig2+ progenitors (and to a lesser extend Nkx2.2+ progenitors) indicating that continuous Shh signaling is required to maintain pMN (and p3) identity [4, 7, 8]. Maintenance of identity could thus be one mechanism requiring cooperation between ephrins and Shh. However, we observed that the change in progenitor number in absence of ephrinB2 and ephrinB3 concerns only a small fraction of p3 and pMN progenitors, the majority of which maintain a correct identity in both ephrin mutants. An alternative possibility is thus that after E9.5, a fraction of progenitors requires cooperation between Shh and ephrins to commit to a specific fate. It is interesting to speculate that cells of mixed identity, which represent a small fraction of p3+pMN progenitors at all stages analyzed and are located close to the p3/pMN boundary, could be such a population susceptible to adopt one or the other identity depending on external signals. In cooperation with Shh signaling, EphrinB2 or ephrinB3 may tilt the balance in Olig2 and Nkx2.2 expression and due to the repressive regulatory loop between these iTFs, a shift in expression of one iTF would be amplified and result in commitment to a specific fate. In support to this, lineage tracing studies have shown that the majority of pMN progenitors derive from cells that transiently activate an enhancer for Nkx2.2 supporting the notion that pMN and p3 progenitors share a common origin [12]. In addition, it has been shown that Olig2 -/- embryos exhibit only a mild increase in the size of the Nkx2.2+ domain [5], suggesting that only a small fraction of progenitors are competent to adopt a p3 fate even in complete absence of Olig2. Further, a similar shift in the ratio between Nkx2.2+ and Olig2+ progenitors has been described in Tcf3 -/- ;Tcf4 -/- double mutants and this was linked to the role of Tcf3/4 in inhibiting Nkx2.2 expression in progenitors fated to become pMN [12]. Because Eph:ephrin signaling is a cell-to-cell signaling pathway, this function would be akin to the resolution of binary fates that has been described for Notch signaling in other neural contexts [28]. Of note, this role of ephrinB2 and ephrinB3 in progenitor identities is consistent with their expression patterns in pMN and p3 progenitors, respectively. However, ventral progenitors are in contact with newly generated neurons which also express ephrinB2 and ephrinB3, it is thus possible that the neuronal expression of these ligands may also contribute to maintain a correct ratio between p3 and pMN identities.

Schematized representation of phenotypes. Schematized representation of transverse sections of the spinal cord showing the dorso-ventral position of p3 (blue), pMN (red) and mixed identity (blue/red) progenitors at different developmental stages and in different genetic backgrounds. Cartoons show 1) the evolution of p3 and pMN numbers over time in wild type embryos and 2) the phenotypes observed in Efnb2 and Efnb3 mutants compared to WT at E11.5. Shh gradient (grey) is represented on the left hand side, while domains of ephrinB2 and ephrinB3 expression in progenitors are represented in green and orange, respectively. In Efnb2 cKO and in Efnb2;Shh trans-heterzygotes (not shown), pMN progenitors are fewer while the number of p3 progenitors is increased. Conversely, in Efnb3 KO, less Nkx2.2+ and more Olig2+ progenitors are present. In addition, pMN and p3 progenitors are intermingled in Efnb3 KO.
As a consequence of these changes in progenitor identities, Efnb2 and Efnb3 mutants exhibit opposite alterations in MN numbers at E12.5. It would be interesting to assess whether these changes are still present postnataly, however, postnatal changes in MN number may be due to distinct mechanisms, for instance a decrease in MN number associated with cell death at later stages than those analyzed here has been described in EphA4 -/- mutants [29].
Traditionally, the role of Eph:ephrin signaling in specification processes has been linked to its function in boundary maintenance. For instance, a recent study has shown that loss of ephrinB2 in the developing cochlea leads to a switch in cell identity from supporting cell to hair cell fate and this was attributed to the mis-positioning of supporting cells into the hair cell layer [30]. No mis-positioning of p3 and pMN progenitors was observed in the Efnb2 cKO mutants analyzed here. Whether excision of Efnb2 in all neural tube progenitors would lead to a similar phenotype remains an open question. Here, we observed mis-positioning of progenitors only in ephrinB3 mutants although both ephrinB2 and ephrinB3 mutants exhibited changes in p3 and pMN progenitor ratio, indicating that resolution of identity is independent of mis-positioning. This is consistent with a growing number of published studies reporting a role for Eph:ephrin signaling in lineage commitment or cell fate maintenance via the modulation of intracellular signal transduction pathways and gene expression, independently of cell sorting at boundaries [31,32,33,34,35,36]. Another possibility, consistent with the genetic interaction between Efnb2 and Shh, could be that ephrins impact on Shh signal transduction cascade as was recently described for Notch [37, 38]. In fact, genetic interaction between Shh and cell surface proteins has been reported previously and such studies identified Gas1, Cdo and Boc as components of the Shh signaling pathway [7, 39, 40]. In this context, it would be interesting to test for a genetic interaction between Efnb3 and Shh in the control of p3 and pMN progenitor identity and positioning.
Conclusions
In conclusion, our study shows that ephrinB2 and ephrinB3 are required to control progenitor identities in the ventral spinal cord and suggests a role for Eph:ephrin signaling in refining morphogen-dependent tissue patterning.
Abbreviations
- cKO:
-
Conditional knock-out
- h:
-
Hour
- kg:
-
Kilogram
- KO:
-
Knock-out
- mg:
-
Milligram
- ml:
-
Milliliter
- MN:
-
Motor neurons
- OLP:
-
Oligodendrocyte precursors
- Shh:
-
Sonic hedgehog
- WT:
-
Wild type
- μm:
-
Micrometer
References
Briscoe J, Novitch BG. Regulatory pathways linking progenitor patterning, cell fates and neurogenesis in the ventral neural tube. Philos Trans R Soc Lond B Biol Sci. 2008;363:57–70.
Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development. 2008;135:2489–503.
Dessaud E, et al. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature. 2007;450(7170):717–20.
Dessaud E, et al. Dynamic assignment and maintenance of positional identity in the ventral neural tube by the morphogen sonic hedgehog. PLoS Biol. 2010;8(6):e1000382.
Balaskas N, et al. Gene regulatory logic for reading the Sonic Hedgehog signaling gradient in the vertebrate neural tube. Cell. 2012;148:273–84.
Cohen M, Briscoe J, Blassberg R. Morphogen interpretation: the transcriptional logic of neural tube patterning. Curr Opin Genet Dev. 2013;23(4):423–8.
Allen BL, Tenzen T, McMahon AP. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007;21(10):1244–57.
Allen BL, et al. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell. 2011;20(6):775–87.
Kicheva A, et al. Coordination of progenitor specification and growth in mouse and chick spinal cord. Science. 2014;345(6204):1254927.
Wijgerde M, et al. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002;16:2849–64.
Lei Q, et al. Transduction of graded Hedgehog signaling by a combination of Gli2 and Gli3 activator functions in the developing spinal cord. Development. 2004;131:3593–604.
Wang H, et al. Tcf/Lef repressors differentially regulate Shh-Gli target gene activation thresholds to generate progenitor patterning in the developing CNS. Development. 2011;138:3711–21.
Xiong F, et al. Specified neural progenitors sort to form sharp domains after noisy Shh signaling. Cell. 2013;153:550–61.
Lisabeth EM, Falivelli G, Pasquale EB. Eph Receptor Signaling and Ephrins. Cold Spring Harb Perspect Biol. 2013;5:a009159.
Kania, A. and R. Klein, Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat Rev Mol Cell Biol. 2016;17:240–56.
Fagotto F, Winklbauer R, Rohani N. Ephrin-Eph signaling in embryonic tissue separation. Cell Adh Migr. 2014;8(4):308–26.
Cayuso, J., Q. Xu, and D.G. Wilkinson, Mechanisms of boundary formation by Eph receptor and ephrin signaling. Dev Biol. 2015;401:122–31.
Laussu J, et al. Beyond boundaries: Eph/ephrin signaling in neurogenesis. Cell Adh Migr. 2014;8:349–59.
Kao TJ, Kania A. Ephrin-mediated cis-attenuation of Eph receptor signaling is essential for spinal motor axon guidance. Neuron. 2011;71:76–91.
Luxey M, et al. Eph/ephrin-B1 forward signaling controls fasciculation of motor and sensory axons. Dev. Biol. 2013;383:264–74.
Luxey M, Laussu J, Davy A. EphrinB2 sharpens lateral motor column division in the developing spinal cord. Neural Dev. 2015;10:25.
Davy A, Soriano P. Ephrin-B2 forward signaling regulates somite patterning and neural crest cell development. Dev. Biol. 2007;304:182–93.
Grunwald IC, et al. Hippocampal plasticity requires postsynaptic ephrinBs. Nat. Neurosci. 2004;7:33–40.
Yokoyama N, et al. Forward signaling mediated by ephrin-B3 prevents contralateral corticospinal axons from recrossing the spinal cord midline. Neuron. 2001;29:85–97.
Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82.
Adams RH, et al. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1998;13:295–306.
Wang HU, Chen Z-F, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor eph-B4. Cell. 1998;93:741–53.
Cau E, Blader P. Notch activity in the nervous system: to switch or not to switch? Neural Dev. 2009;4:36.
Helmbacher F, et al. Targeting of the EphA4 tyrosine kinase receptor affects dorsal/ventral pathfinding of limb motor axons. Development. 2000;127:3313–24.
Defourny J, et al. Cochlear supporting cell transdifferentiation and integration into hair cell layers by inhibition of ephrin-B2 signalling. Nat Commun. 2015;6:7017.
Picco V, Hudson C, Yasuo H. Ephrin-Eph signaliing drives the asymmetric division of notochord/neural precursors in Ciona embryos. Development. 2007;134:1491–7.
Stolfi A, et al. Neural tube patterning by Ephrin, FGF and Notch signaling relays. Development. 2011;138:5429–39.
Ashton RS, et al. Astrocytes regulate adult hippocampal neurogenesis through ephrin-B signaling. Nat. Neurosci. 2012;15:1399–407.
Haupaix N, et al. p120RasGAP mediates ephrin/Eph-dependent attenuation of FGF/ERK signals during cell fate specification in ascidian embryos. Development. 2013;140:4347–52.
Ottone C, et al. Direct cell-cell contact with the vascular niche maintains quiescent neural stem cells. Nat Cell Biol. 2014;16(11):1045–56.
Chen S, et al. Interrogating cellular fate decisions with high-throughput arrays of multiplexed cellular communities. Nat Commun. 2016;7:10309.
Stasiulewicz, M., et al., A conserved role for Notch in priming the cellular response to Shh through ciliary localisation of the key Shh transducer, Smoothened. Development, 2015.
Kong JH, et al. Notch activity modulates the responsiveness of neural progenitors to sonic hedgehog signaling. Dev Cell. 2015;33(4):373–87.
Martinelli DC, Fan CM. Gas1 extends the range of Hedgehog action by facilitating its signaling. Genes Dev. 2007;21:1231–43.
Tenzen T, et al. The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Dev Cell. 2006;10:647–56.
Acknowledgements
The Islet 1/2 antibody was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Iowa City, IA 52242. We thank Dr Novitch for sharing the Olig2-Cre mice. Dr Kania and Dr Henkemeyer provided some molecular reagents used in this study. We are grateful to Brice Ronsin for his help with confocal microscopy (TRI Imaging Core Facility) and to Marion Aguirrebengoa for her help with statistical analyses. We thank the ABC facility and ANEXPLO for housing mice. We are grateful for Sylvain Touret’s help for writing the R code. We thank Eric Agius, Serge Plaza and Alain Vincent for critical reading of the manuscript.
Funding
Research in the Davy team is financed by the CNRS, by the Fondation ARC and by ANR (ANR-15-CE13-0010-01). JL received support from the French Ministère de l’Enseignement Supérieur et de la Recherche and from the Fondation pour la Recherche Médicale (FDT20140931010). DJL is funded by the Miami Project to Cure Paralysis and PAN by NIH/NINDS (NS089325).
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Authors’ contributions
JL planned, performed and analyzed experiments, and he participated in writing the manuscript; AK, CA and NE performed experiments; PA and DL collected and provided mutant embryos and revised the manuscript; CS provided scientific input on the project and revised the manuscript; AD supervised the project, planned the experiments, analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare no conflict of interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All animal procedures were approved by the institution ethical committee (protocol number: APAFIS#1289-2015110609133558 v5).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Supplemental Code. (PDF 42 kb)
Additional file 2: Table S1.
Figure-by-figure details on sample size (DOCX 13 kb)
Additional file 3: Figure S1–3.
Expression of Shh is not changed in ephrin mutants. (PDF 330 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Laussu, J., Audouard, C., Kischel, A. et al. Eph/Ephrin Signaling Controls Progenitor Identities In The Ventral Spinal Cord. Neural Dev 12, 10 (2017). https://doi.org/10.1186/s13064-017-0087-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13064-017-0087-0