
Abstract
Background
Many stroke survivors remain with residual cognitive and motor impairments despite receiving timely acute and sub-acute rehabilitation. This indicates that rehabilitation following stroke should be continuous to meet the needs of individual stroke patients. Both cognitive and motor functions are essential for mastering daily life and, therefore, should be aimed at with rehabilitation. Exergames, motor-cognitive exercises performed using video games, are an auspicious method to train both motor and cognitive functions and at the same time may foster the long-term motivation for training. This study aims to assess the effect of concept-guided, personalised, motor-cognitive exergame training on cognitive and motor functions in chronic stroke survivors.
Methods
This study is a single-blinded, randomised controlled trial. Assessments are performed at baseline, after a 12-week intervention, and at a 24-weeks follow-up. Chronic stroke patients (≥ 18 years old, ≥ 6 months post-stroke) able to stand for 3 min, independently walk 10 m, follow a two-stage command, and without other neurological diseases apart from cognitive deficits or dementia are included. Participants in the intervention group perform the exergame training twice per week for 30 (beginning) up to 40 (end) minutes additionally to their usual care programme. Participants in the control group receive usual care without additional intervention(s). Global cognitive functioning (total Montreal Cognitive Assessment (MoCA) score) is the primary outcome. Secondary outcomes include health-related quality of life, specific cognitive functions, single- and dual-task mobility, and spatiotemporal gait parameters. The target sample size for this trial is 38 participants. Linear mixed models with the post-outcome scores as dependent variables and group and time as fixed effects will be performed for analysis.
Discussion
Superior improvements in global cognitive functioning and in the abovementioned secondary outcomes in the intervention group compared to the control group are hypothesised. The results of this study may guide future design of long-term rehabilitation interventions after stroke.
Trial registration
ClinicalTrials.gov (NCT05524727). Registered on September 1, 2022.
Similar content being viewed by others


Administrative Information
Note: the numbers in curly brackets in this protocol refer to SPIRIT checklist item numbers. The order of the items has been modified to group similar items (see http://www.equator-network.org/reporting-guidelines/spirit-2013-statement-defining-standard-protocol-items-for-clinical-trials/).
Title {1} | PEMOCS: Evaluating the effects of a concept-guided, PErsonalized, MOtor-Cognitive exergame training in chronic Stroke – study protocol for a randomized controlled trial |
Trial registration {2a and 2b} | clinicaltrials.gov (NCT05524727) |
Protocol version {3} | #5, 20/10/2023 |
Funding {4} | This study is supported by grants of the USZ Innovation Pool (https://usz-foundation.com/en/usz-innovation-pool/) and the Swiss Association of Physiotherapy (physioswiss). |
Author details {5a} | Huber S.K., Knols R.H., Held J.P.O., Betschart M., de Bruin, E.D. |
Name and contact information for the trial sponsor {5b} | Rudolf H. Knols, PhD, University Hospital Zurich, Rämistrasse 100, CH-8091 Zurich, ruud.knols@usz.ch |
Role of sponsor {5c} | Oversight of Good Clinical Practice regulatory requirements |
Introduction
Background and rationale {6a}
Stroke is a dominant global health burden and a major cause of long-term disability in adults [1,2,3,4]. In Switzerland, approximately 20,000 persons suffer a stroke each year [5]. A stroke can cause motor and cognitive impairments [6, 7]. The most common motoric consequence of stroke is hemiparesis, a unilateral paralysis or weakness of either one or both extremities [8]. It occurs in up to 80% of patients with stroke [9]. Hemiparesis typically leads to gait and balance impairments [10, 11], which can restrict mobility and independent ambulation [12]. Common cognitive impairments after stroke include deficits in executive functions, attention, spatial perception, and psychomotor processing speed [13, 14]. These cognitive impairments occur in a comparable frequency as motor impairments; depending on the specific cognitive function, 30 to 90% of stroke survivors suffer from cognitive impairment [7, 15, 16].
Residual impairments, consequences from the stroke lasting after the acute and sub-acute rehabilitation phase, are common after stroke [17, 18]. This is especially true for cognitive impairments. It is striking in this context that cognitive impairments have so far gained much less attention in research compared with motor impairments notwithstanding the fact patients mentioning ‘What are the best ways to improve cognition after stroke?’ being one of their foremost research priorities in relation to life after stroke [19]. Remaining cognitive deficits are often responsible for limited independence and quality of life in patients, who have regained good motor functioning and activities-of-daily-living ability [20]. The optimal treatment of cognitive deficits in chronic stroke patients is a clearly identifiable research gap [17, 21].
Motor-cognitive training is a comprehensive rehabilitation method that combines motor and cognitive training [22]. Combined motor-cognitive trainings may be an auspicious method to tackle this research gap. Motor-cognitive trainings can either be performed sequentially (first motor training component, then cognitive training component, or vice versa) or simultaneously (both task components executed at the same time) [22, 23]. Simultaneous motor-cognitive trainings, where both tasks are executed concurrently, may have the highest relevance for daily life, because daily life almost exclusively sets combined challenges [22]. Motor and cognitive functions have been shown to share structural and functional roots [24]. Fittingly, the ‘guided plasticity facilitation’ model suggests that motor-cognitive training can lead to additional benefits due to interaction effects of the two components [25, 26]. Confirming the theory, multiple systematic reviews in healthy older adults found that combined motor-cognitive trainings were superior in improving motor, cognitive, and dual-task functions [27,28,29,30,31,32,33,34,35]. In (chronic) stroke, however, there is less and yet unclear evidence. While first systematic reviews report beneficial effects of motor-cognitive over single interventions on balance and gait [36, 37], the effects on cognitive functions remain unclear to date [37,38,39].
The most promising type of simultaneous motor-cognitive trainings may be exergames [37], cognitively demanding video games, which require the player to be physically active to complete the gaming tasks [40, 41]. Gamification of training and the use of virtual reality can increase the motivation for and adherence to exergame training [42,43,44], while also giving the training ecological validity [45]. In healthy older adults, exergames have been shown to improve motor and dual-task functions [46,47,48,49,50,51], while potential has been reported for improving cognitive functions [52,53,54,55,56]. In other neurological populations, exergames showed beneficial effects on balance, mobility, and walking capacity [57,58,59,60,61,62], while the evidence for improving cognitive functions with exergames is yet scarce and inconsistent [63,64,65,66]. In (chronic) stroke, exergames have been found a suitable adjunct to conventional rehabilitation for improving motor functions [67,68,69]. However, little is yet known about the effect of motor-cognitive exergames on cognitive functions in chronic stroke, so further studies are needed [37]. Moreover, in most previous studies including stroke survivors, motor-cognitive exergame interventions were applied without systematically considering training principles, especially personalised progression [37]. It is known, however, that applying training principles and personally tailoring the training to the individual is important for the success of any training intervention [70, 71]. Therefore, we developed a training concept considering the FITT-VP (frequency, intensity, time, type, volume, progression) training principles in combination with neuroplasticity and motor learning principles [70, 72]. Applying this concept, it is the aim of this study to investigate if adding motor-cognitive exergame training to usual care has a beneficial effect on the long-term rehabilitation of chronic stroke survivors and if global cognitive functioning and secondary outcomes can be improved compared to a control group who does not receive the exergame training.
Objectives {7}
To address the gaps of knowledge regarding rehabilitation of cognitive functions and effects of motor-cognitive exergames in chronic stroke, the primary objective of this study is to evaluate the effect of a 12-week concept-guided, personalised, motor-cognitive exergame training when added to usual care, in comparison to the effect of usual care alone on global cognitive functioning in chronic stroke survivors.
The secondary objectives of this study are to explore (1) the acute and (2) the persistent effect up to a 12-week follow-up of the exergame intervention on health-related quality of life, specific cognitive functions (sub-functions of attentional, executive, and visuospatial functions), single- and dual-task mobility, and spatiotemporal parameters in chronic stroke patients.
Trial design {8}
This study is a randomised, controlled trial (RCT) with two parallel arms, investigating the superiority of a concept-guided, personalised, motor-cognitive exergame training added to usual care over usual care alone. The trial employs a single-blinded approach, with outcome assessors being unaware of group assignments. The randomisation is performed with a 1:1 ratio using stratification by sex (female or male [73]) and cognitive impairment (MoCA ≥ 24 or MoCA < 24 [74]).
Methods: participants, interventions, and outcomes
Study setting {9}
This study takes place in different academic and rehabilitation hospitals in the Canton of Zurich, Switzerland.
Eligibility criteria {10}
Inclusion criteria
Participants fulfilling all the following inclusion criteria are eligible for the study:
-
Adults (≥ 18 years) with chronic stroke (≥ 6 months post-stroke, ischemic or haemorrhagic [75])
-
Able to stand for 3 min and walk 10 m, Functional Ambulation Category (FAC) ≥ 3
-
Able to follow a two-stage command (no MoCA threshold for inclusion was set to prevent exclusion of persons with subtle or specific cognitive deficits [76])
-
Able to give informed consent as documented by signature
Exclusion criteria
Participants fulfilling one or more of the following exclusion criteria are not eligible for the study:
-
Unable or not willing to give informed consent
-
Having been diagnosed with other neurological diseases (e.g. Parkinson’s Disease, multiple sclerosis), except cognitive deficits or dementia
-
Clinical contra-indications for the study intervention
-
Unable to follow the study intervention or the test for the primary endpoint (MoCA), e.g. due to a neglect, aphasia, or other language problems
-
Overlapping enrolment in another clinical trial
Who will take informed consent? {26a}
Movement scientists and therapists who are part of the study team and have been trained for this task obtain written informed consent from participants, after they have been written and orally informed about the study, its benefits and risks, and their rights and had at least 24 h for consideration.
Additional consent provision for collection and use of participant data and biological specimens {26b}
The consent includes collection and use of individually identifiable participant data for the study procedures (through members of the study team only) and for audit trials by the cantonal ethical committee and other local authorities. Moreover, participants consent that their de-identified data is used for the analysis and publication of the study results. This study does not involve collecting biological specimens for storage.
Interventions
Explanation for choice of comparators {6b}
Participants in the control group continue with their usual care and receive no additional intervention. They are called once a week to align for contact to the study team and to gather their physical and cognitive activity data (see the ‘ Intervention and further activity outcomes’ section). This comparator was chosen as the aim of this study is to determine the effect of additional motor-cognitive exergame training. This aim was based on systematic reviews, which recommend the application of exergame training in addition to usual care, aiming at increasing the amount of rehabilitation offered to patients [63, 67, 77].
Intervention description {11a}
The intervention group receives concept-guided, personalised, motor-cognitive exergame training additionally to usual care. The personalised motor-cognitive exergame training for stroke (PEMOCS) concept, which guides the intervention, was developed specifically for this study and, in accordance with the Modified Consensus on Exercise Reporting Template (CERT) for Therapeutic Exercise Interventions [78, 79], published elsewhere [80]. It determines the training dosage with regards to frequency, intensity, time, type, and volume of the exercises, which are based on recommendations from scientific literature for motor-cognitive and exergame trainings in chronic stroke and healthy older adults. Participants train twice a week for 12 weeks (intervention period, see Fig. 1). Training sessions last between 30 (beginning) and 40 (end) minutes, progressing in duration for 2 min every second week and resulting in 840 min total planned training time (see Table 1). Additionally, the PEMOCS concept is designed to provide personalised progression and variability in training considering principles for neuroplasticity, motor learning, and training [80,81,82,83]. It was developed based on Gentile’s Taxonomy for motor learning [84] and tested for its feasibility in the target population [85]. In short, motor and cognitive tasks of the exergame training are allocated to various difficulty levels along a skill-progression scheme with three dimensions. Based on the participants’ subjective ratings of their perceived motor-cognitive task difficulty and their perceived performance (see ‘Intervention outcomes’), progression through the difficulty levels is determined individually for each participant. Therefore, the training is personalised within a standardised progression scheme. Additionally, variability rules ensure variation in training tasks.

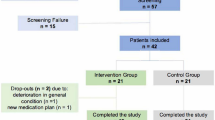
Study flow. Overview of the study procedures. ADH, adherence; CPL, compliance; PCA, physical and cognitive activities; (S)AE, (serious) adverse event; UC, usual care
The training is performed using the exergame device Dividat Senso (Dividat AG, Schindellegi, Switzerland, for a detailed description of the device see [85]). The Dividat Senso consists of a TV screen and a pressure-sensitive plate as well as a handrail on three sides to provide security to trainees (Fig. 2). Participants perform stepping movements on the plate to control the games. The system provides real-time feedback on the gamer’s performance, including visual, auditory, and tactile cues facilitating the interaction of the participant with the video games. These video games target different cognitive functions (incl. attentional, executive, memory, and visuospatial functions). Via an online platform, personalised training programmes can be created within the Dividat training system.

Study device: Dividat Senso in action. Pressure-sensitive plate with handrails on three sides and screen on head-height of the participant, showing the video game. The participant performs a stepping movement to play the game
Criteria for discontinuing or modifying allocated interventions {11b}
The intervention is discontinued in case of withdrawal of the informed consent, participant request, or if the health status of the participant or any harm does not allow the continuation of the intervention. Minor individual modifications of the planned intervention are possible (e.g. using a training aid for additional stability) if following the training concept is possible.
Strategies to improve adherence to interventions {11c}
All sessions are supervised one-to-one by a trained movement scientist, who engages the participant in the study procedures and encourages complying with the set appointments. Additionally, considering each individual’s game preferences is part of the PEMOCS concept’s variability rules to foster motivation and fun during the gaming sessions. Compliance with training sessions and adherence to scheduled training time are recorded.
Relevant concomitant care permitted or prohibited during the trial {11d}
Concomitant care (usual care) including other therapies is allowed and recorded in both groups during the whole study.
Provisions for post-trial care {30}
N/A, as there are no disadvantages likely to arise from the intervention.
Outcomes {12}
Primary and secondary outcomes are collected at three time points (T0–week 0, T1–week 12, T2–week 24; see Table 2, Figs. 1 and 3) by cognitive and motor assessments as well as a health-related-quality-of-life questionnaire. At the baseline measurement (T0), participant characteristics and baseline factors are recorded. Intervention and activity outcomes are collected during the intervention (T0–T1) and the follow-up (T1–T2) periods, respectively. All variables will be aggregated as means or medians, depending on the distribution of the data, and the analysis metric for all will be final values (see the ‘ Statistical methods’ section). An overview of all outcome variables is presented in Tables 3 and 4.

SPIRIT figure. ADH, adherence; BBS, Berg Balance Scale; BQ, baseline questionnaire, incl. see the ‘ Baseline factors’ section; TUG(-Cogn), (cognitive dual-task) Timed Up and Go test; CPL, compliance; FAC, Functional Ambulation Category; FM-LE, lower-extremity component of Fugl-Meyer assessment; MoCA, Montreal Cognitive Assessment; mRS, modified Rankin scale; MRT, mental rotation test; NBT, N-back test; NIHSS, National Institute of Health Stroke Scale; OWA, outdoor walking assessment; PCA, physical and cognitive activities; PP, perceived performance; PTD, perceived task difficulty; SIS 3.0, Stroke Impact Scale; SRT, simple reaction test; Stroop, Stroop Interference test; TMT, Trial Making test; UC, usual care; 10MWT, 10-m walk test
Baseline factors
At baseline, the following information is recorded to describe the study population (see Table 2):
-
Demographics (collected via baseline questionnaire): age, sex, years of education, marital status
-
Other characteristics (collected via baseline questionnaire): weight, height, handedness
-
Stroke diagnostic details (collected via baseline questionnaire): number of strokes, time point/type (ischemic, haemorrhagic)/lesion site/side and location of initial paresis of the (most recent) stroke, initial (if available), and current (at baseline) National Institutes of Health Stroke Scale (NIHSS [ 88])
-
Clinical characteristics A (collected via baseline questionnaire): comorbidities (Charlson Comorbidity Index, CCI [ 89]), modified Rankin scale (mRS[ 90])
-
Clinical characteristics B (collected by the assessor at the baseline measurement): Functional Ambulation Category (FAC [ 91]), Lower-Extremity component of the Fugl-Meyer assessment (FM-LE), Berg Balance Scale (BBS [ 92])
Primary outcome
The primary outcome is global cognitive functioning measured by the total score of the Montreal Cognitive Assessment (MoCA) [93]. The MoCA is composed of several tests assessing different cognitive domains including attention, executive functions, working memory, short-term memory recall, visuospatial skills, and orientation [74]. The MoCA has been successful in detecting cognitive decline [94] and showed good reliability in stroke patients and healthy older adults [95,96,97] as well as fair to good responsiveness and validity [98] in chronic stroke patients [99]. The maximum achievable score is 30 points, where more points represent better cognitive functioning. For individuals with 12 or less years of education and a total score < 30, an additional point is added [93]. A MoCA score below 24 points indicates mild cognitive impairment in individuals after stroke [74, 100, 101], and an improvement of 1.22 points was found to be a clinically relevant change [99].
Global cognitive functioning, measured by the total score of the MoCA, covers the cognitive domains typically impaired after stroke. Further cognitive tests examining specific cognitive domains/functions will be implemented as secondary outcomes [102].
Secondary outcomes
Health-related quality of life and perceived recovery are assessed using the total score and the single domain scores of the Stroke Impact Scale [SIS 3.0 [103]]. The SIS 3.0 is a stroke-specific questionnaire assessing the self-reported health status on 5-point Likert scales [98]. It encompasses eight domains (strength, memory/thinking, emotion, communication, ADL/IADL, mobility, hand function, and participation) and a visual analogue scale, where the perceived state of recovery is rated (0 to 100%) [98]. The final score lies between 0 and 100, where a higher score indicates better health-related quality of life. The German SIS (DE-SIS, translated and cross-culturally adapted) was found reliable and valid for the use in German-speaking stroke survivors [104].
Secondary cognitive outcomes include the following computer-based cognitive assessments:
-
To assess alertness, a simple reaction test (SRT) (‘WAFA’ within the Vienna Test System, VTS, see the ‘Plans for assessment and collection of outcomes’) is applied. The SRT is a reliable and valid neuropsychological assessment for alertness [105,106,107]. It composes of six tests with visual and auditory stimuli, of which two evaluate intrinsic alertness (participant has to react to an appearing stimulus as fast as possible), two evaluate crossmodal-phasic alertness (a crossmodal warning stimulus precedes the actual stimulus and the participant has to only react to the actual and not the warning stimulus), and two evaluate unimodal-phasic alertness (a unimodal warning stimulus precedes the actual stimulus and the participant has to only react to the actual and not the warning stimulus) [107].
-
Processing speed and cognitive flexibility, a sub-domain of executive functions, are assessed using the Trail Making test (TMT) (‘TMT—Langensteinbacher Version’ within the VTS), which is a widely used, reliable, and valid neuropsychological assessment [108,109,110]. The TMT consists of two parts: TMT-A assesses general information-processing speed; it asks to connect rising numbers (1–25) as fast as possible. TMT-B is used to test cognitive flexibility; the task is to connect rising numbers and letters alternatingly [111].
-
To assess interference inhibition, another sub-domain of executive functions, the Stroop Interference test (‘STROOP’ within the VTS) is used [112]. The Stroop test is a widely used, reliable, and valid neuropsychological assessment testing the ability to inhibit the reaction to a more dominant stimulus in favour of the inquired reaction to a less dominant stimulus [113]. This assessment contains four sub-tests: two baseline and two interference conditions. In the first baseline condition, colour words are presented in grey font and the participant must select the correct colour (baseline reading). In the second baseline condition, coloured bars are presented, and the participant must select the correct colour (baseline naming). In the interference conditions, the colour words are shown in coloured fonts and the participant has to either select the correct colour of the word (interference reading) or of the font (interference naming) [114].
-
To assess working memory and related cognitive functions, the N-back test (NBT) (‘NBV’ within the VTS) is used, which is a widely used, reliable, and valid neuropsychological test [115,116,117]. The participant is presented a row of letters and has to decide upon every letter whether it corresponds to the one shown N letters earlier [118]. In this study, the test condition N = 2 is used.
-
Mental rotation ability, a sub-domain of visuospatial functions, is assessed using the mental rotation test (MRT) (‘3D’ within the VTS), which is based on the paradigm by Shepard and Metzler [119]. It determines the ability to mentally rotate abstract objects and has been used in stroke patients before [85, 120, 121]. Each item consists of a figure composed of a number of blocks. The participant has to imagine how the arrangement of the blocks looks when viewed from another perspective and choose the correct 2D-view from a selection of four possible solutions [122].
Secondary mobility and dual-task outcomes included the following assessments:
-
The Timed Up and Go test (TUG), a reliable and valid assessment in stroke patients [123, 124], is conducted to analyse changes in mobility and dynamic balance. Participants are instructed to perform the TUG ‘as fast and safely as possible’. Time is measured from the moment the participant’s back leaves the backrest of the chair until it touches the backrest of the chair again [125].
-
To assess mobility under dual-task conditions and dual-task effects, the TUG-Cognitive is also performed [126]. The TUG-Cognitive (TUG-Cog) is a reliable and valid assessment of dual-task mobility in stroke patients and healthy adults [125, 126]. A cognitive task is first executed in single-task mode during 60 s while being seated on a chair. After this, both tasks, the TUG and the cognitive task, are performed simultaneously, while participants are instructed to not prioritise one over the other. As cognitive task, serial subtraction of 3 from a random number between 50 and 100 (not in the row of three) will be used in participants who are able to complete this task [126, 127]. Participants, who are not able to accomplish serial subtraction, perform a verbal fluency task instead, naming nouns from categories (e.g. fruits, animals, cloths) starting with a specific letter [126, 127]. For all single- and dual-task trials, a familiarisation trial is performed before executing three test trials each. The TUG and TUG-Cog are conducted under laboratory conditions at the participating study centres.
Secondary gait outcomes included a 10-m walk test (10MWT) and an outdoor walking assessment (OWA) using inertial gait sensors to analyse temporal and spatial gait parameters.
-
The 10MWT has been found reliable and valid in stroke patients [128]. The 10MWT is performed according to the protocol by Cheng et al. [128], where participants walk 14 m, where only the middle 10 m are timed. The participant starts walking at the 0-m mark, and the stopwatch is started as soon as the first foot crosses the 2-m mark and stopped again when the first foot crosses the 12-m mark, while the participant continues walking to the 14-m mark. At first, a familiarisation trial is performed followed by three trials at comfortable walking and three trials at fast walking speed. Participants are instructed to ‘walk at a comfortable speed’ and ‘walk as fast but safely as possible’, respectively. Participants use their usual walking aid if needed.
-
The OWA is a 400-m walk following an outdoor route without stairs [129]. At each study centre, a suitable route nearby was pre-defined and all participants follow this same route. Participants use their usual walking aid if needed and wear proper footwear for an outdoor walk. They are instructed to ‘walk at a comfortable speed, as if they were on a stroll’. To ensure safety, participants are accompanied by two investigators.
Intervention and further activity outcomes
Compliance and adherence to the trainings and the reasons for not attending or aborting a training session are recorded during the intervention period (T0–T1, see Table 2, Figs. 1 and 3). Additionally, participants in the intervention group are asked to rate the motor-cognitive task difficulty of the training tasks (perceived task difficulty, PTD) and their motor-cognitive performance (perceived performance, PP) in every training session (during T0–T1, see Table 2, Figs. 1 and 3). Visual analogue scales (VAS) in the eCRF based on the cognitive load theory [130] and the NASA-TLX [National Aeronautics and Space Administration Task Load Index [131]] are used to collect these ratings (Additional file 1). PTD and PP are expressed as percentage values where higher percentages stand for more difficult tasks and better performance, respectively. The intervention outcomes are summarised in Table 4.
All participants are interviewed weekly throughout the whole study (T0–T2, see Table 2, Figs. 1 and 3) regarding the dose (frequency and time) and content (intensity and type) of moderate to intense physical and cognitive activities, which they perform as part of their usual care or in their leisure time. Definitions for moderate to intense activities are based on the World Health Organisation’s (WHO) 2020 Guidelines on Physical Activity and Sedentary Behaviour [132]. The interviews are done using a structured questionnaire (Additional file 1), which implies the FITT-VP principles [70] and the TIDieR checklist [Template for Intervention Description and Replication [133]]. The further activity outcomes are summarised in Table 4.
Participant timeline {13}
Each participant is involved for approximately 24 weeks (12 weeks intervention period, 12 weeks follow-up period, Figs. 1 and 3). Participants are contacted and screened for eligibility by their therapist, physician, and/or the study team. Eligible, potential participants are provided with detailed study information in oral and written form. Interested potential participants are invited to a first study appointment, where they are first again provided with the study information, especially outlining the benefits, risks, and their rights associated with the study, and can clarify remaining questions. Trained movement scientists and therapists then obtain written informed consent from those willing and able to participate in the study, before any study-related procedures start. After that, participants first attend the baseline measurement (T0, Figs. 1 and 3). Subsequently, the participant is allocated randomly to one of the two study arms. Randomisation for each participant is run in Research Electronic Data Capture (REDCap) by an investigator other than the blinded assessor. Participants allocated to the intervention group thereafter attend concept-guided, personalised motor-cognitive training, twice a week for 12 weeks (T0–T1). Participants allocated to the control group receive no additional intervention during the same 12 weeks (T0–T1). After completion of the intervention period, all participants attend the post-intervention measurement (T1, Figs. 1 and 3). During the subsequent 12-week follow-up period (T1-T2), participants in both groups receive no additional intervention. At the end of this period, all participants attend the follow-up measurement (T2).
Sample size {14}
A sample size of 38 participants (approx. 19 per group) was estimated for this study. The sample size estimation was based on systematic reviews investigating the effects of motor-cognitive training and exergames on cognitive functions. Stanmore et al. included studies with any population (including five out of seventeen studies with stroke or other neurological patients) and found a small to medium effect for global cognitive functioning (SMD = 0.44, p = 0.001) and several small to large effects for cognitive domains including executive functions, processing speed and visuospatial skills (0.26 ≤ SMD ≤ 0.90, p < 0.05) (132). Five further reviews included studies with older adults and found small to large effect sizes for global or overall cognitive functioning and cognitive domains including attention, executive functions, learning and memory, and processing speed (0.30 ≤ SMD ≤ 1.37, p < 0.05) [31, 35, 54, 56, 134]. Based on this evidence, a small to medium effect on global cognitive functioning is anticipated for the planned study (f = 0.21). The sample size was estimated using G*Power, entering this effect size (requiring no expected difference between groups and standard deviations) and the following parameters into the mask for a two-way mixed ANOVA; α-level = 0.05, power = 0.80, number of groups = 2, number of measurements = 3, correlation among rep measures = 0.5, nonsphericity correction = 1. Dropouts will be replaced by post-recruitment until the planned sample size is achieved. Based on the recent feasibility study [85] and comparable literature [135,136,137], a dropout rate of 10–20% can be expected.
Recruitment {15}
The PEMOCS study is a single-blind randomised control trial (RCT) with chronic stroke survivors recruited from hospitals and rehabilitation centres in the Canton of Zurich, Switzerland. Participants are recruited by therapists and physicians during therapy sessions and stroke follow-up appointments, by flyers on the ward, and by contact of the study team in case of provided general consent. Regular contact between the recruiters and study team meetings should help to maintain an acceptable recruitment rate.
Assignment of interventions: allocation
Sequence generation {16a}, concealment mechanism {16b}, and implementation {16c}
The randomisation is stratified by cognitive status (cognitive impairment absent or present, determined by a MoCA score ≥ 24 or < 24, respectively [74, 100, 101]) and by sex (female or male [138,139,140]). Both stratifying variables are allocated 1:1 to both groups. Participants are randomised using REDCap, the same tool as utilised for eCRF keeping (see the ‘Data management’ section [141, 142]). To perform the randomisation in REDCap, a pre-defined randomisation list in the form of an excel document needs to be uploaded onto the platform, which is then being used by the software. Instructions by REDCap show how this list must be structured to provide the desired allocation ratio and stratification [141, 142]. These instructions were followed by the randomisation-list creator, a person otherwise not involved in the study. This way it was ensured that no member of the study team would know the allocation sequence. After creating the list, the randomisation-list creator encrypted the excel document containing the list with a password. Both, the list and the password, are stored in a secure place, where the investigators of the study team have no access. Moreover, the REDCap user rights to access and view the randomisation setup feature were removed from all investigators of the study team before the final randomisation list was uploaded onto the REDCap platform [141, 142].
Study investigators other than the blinded assessor enrol participants and allocate them to groups. A separate instrument in REDCap, which is invisible for the assessor through user privileges, is used for randomisation.
Assignment of interventions: blinding
Who will be blinded {17a}
Outcome assessors are blinded to group allocation. They cannot access the randomisation tool in REDCap and are not involved in intervention procedures. To blind the data analyst, a de-identified dataset will be used for analysis, which will not contain unique identifiers such as the study ID.
Procedure for unblinding if needed {17b}
N/A, as assessors, who are the only blinded members of the study team, are always accompanied by a training supervisor, who knows the participants and their group allocation. Hence, in case any situation during a measurement session would require knowledge of group allocation, the training supervisor can handle it and the assessor does not need to be unblinded.
Data collection and management
Plans for assessment and collection of outcomes {18a}
All outcome variables are gathered in an eCRF (see the ‘Data management’ section). All study-team members gathering data receive specific training for the relevant study procedures. Primary and secondary outcomes are collected at baseline (T0), post-intervention (T1), and follow-up (T2) measurements. The MoCA (primary outcome) is executed on paper following the instructions of the providers (mocacognition.com [93]), and the data is transferred into the eCRF. All assessors obtain a MoCA certificate (mocacognition.com) for trained execution of the test before performing the assessment in the study. The SIS 3.0 is collected via an online questionnaire filled out by the participants electronically in the eCRF or, in case not able to do so, on paper and transferred to the eCRF by the investigators. Secondary computer-based cognitive assessments are conducted within the Vienna Test System (VTS, Schuhfried GmbH, Mödling, Austria), a valid and reliable software for neuropsychological testing. All assessments include written instructions and practice sets and are only started if the participant received clarifying responses on any questions regarding the test functionality. The tests are performed on a touch-screen computer, using either one button on the keyboard or the touch screen to answer the stimuli. All cognitive outcome variables are obtained from the VTS result sheets and transferred into the eCRF. Outcome variables for the TUG and TUG-Cog are collected using a stopwatch and by noting correct answers on paper and directly entered into the eCRF. For the TUG, TUG-Cog, and the 10MWT, practice trials are performed before the actual assessment to ensure clarity of the procedure. Outcome variables of the gait assessments are gathered using the Gait Up system (Gait Up SA, Lausanne, Switzerland) with Physilog® sensors (wearable standalone movement inertial sensors, 50 × 37 × 9.2 mm, 19 g). The Gait Up system provides quantitative, objective, and valid assessment of gait movement [143] presented on output sheets, from where the data are transferred into the eCRF. The first and last two gait cycles are excluded from the analysis to eliminate acceleration and deceleration [86]. Baseline characteristics (see the ‘ Baseline factors’ section) are collected via a questionnaire filled out by the participants electronically in the eCRF or, in case not able to do so, on paper and transferred to the eCRF by the investigators. The results of FMA-LE, FAC, and BBS (see the ‘ Baseline factors’ section) are directly entered into the eCRF. ‘ Intervention and further activity outcomes’ are collected within the eCRF throughout the intervention (T0–T1) and follow-up (T1–T2) periods, respectively. In all phases, deviations from the protocol are recorded in the eCRF to ensure traceability and the ability to exactly repeat the assessment procedures at T0, T1, and T2 for each individual participant.
Plans to promote participant retention and complete follow-up {18b}
During the follow-up period, participants in both groups are contacted once a week to inquire their usual care, general physical, and cognitive activities (see ‘Intervention and activity outcomes’). This keeps them engaged in the study procedures and, therefore, promotes successful retention.
Data management {19}
An electronic case report form (eCRF) is kept for each enrolled participant using REDCap electronic data capture tools hosted at ETH Zurich [141, 142]. REDCap (Research Electronic Data Capture) is a secure, web-based software platform designed to support data capture for research studies, providing (1) an intuitive interface for validated data capture, (2) audit trails for tracking data manipulation and export procedures, (3) automated export procedures for seamless data downloads to common statistical packages, and (4) procedures for data integration and interoperability with external sources [141, 142]. This eCRF has been validated before enrolment of the first participant. Study team members who are authorised to enter or edit data in the eCRFs, receive a login to the REDCap study platform, and are listed with signatures in the trial master file (TMF) and the investigator site file (ISF). To assure that any authorised person, who may perform data entries and changes in the eCRF, can be identified, all entries/edits are recorded with name, date, and time. Data entry of the primary and secondary outcomes in REDCap is performed by one and double-checked by another investigator (verification, four-eyes-principle). Should any previously entered data need to be changed (e.g. because a mistake was identified during the verification), a reason must be given to proceed. eCRFs are kept current to reflect participant status at each phase during the study.
Study and participant data will be handled with uttermost discretion and are only accessible to authorised personnel who require the data to fulfil their duties within the scope of the study. Participants are coded and not identifiable in the eCRF or on any other study-specific documents. Appropriate coded identification (study ID) is used. Each study ID composes of four random letters or numbers, which are not related to any characteristics, or the time point of inclusion of the participants. The sponsor will store the participant identification list in a secured and locked location. All study data are archived for ten years after study termination or premature termination of the study.
Confidentiality {27}
Personal information of potential and enrolled participants is kept confidential and only accessible for involved study-team members for study-related purpose. Data protection is kept according to current guidelines of the Swiss law. Participants, who have withdrawn from the study, can ask the deletion of their personal information at any time. All participants receive a study ID (a random sequence of four letters and numbers) not associable with their personal data. All study data is stored only with this ID and never related to any personal data. The key to decode study data is kept locked and only accessible for involved study-team members for study-related purpose(s). After completion of the study procedures, study data are archived according to Good Clinical Practice (GCP) guidelines for at least 10 years.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in the trial/future use {33}
N/A as no biological specimens are collected in this study.
Statistical methods
Microsoft Excel (Microsoft Corporation, 2016) will be used to aggregate and tabulate the data. All statistical analyses will be performed using RStudio open-source software (Bosten, USA [144]) or SPSS Statistics (version 26 for windows; IBM, Chicago, IL, USA).
Statistical methods for primary and secondary outcomes {20a}
Distributions of all baseline factors and primary and secondary outcome variables will be checked with the Shapiro–Wilk test [145]. Appropriate descriptive statistics will be obtained for all baseline factors and outcome variables (means and standard deviations for normally distributed data, medians and inter-quartile ranges for non-normally distributed data, frequencies for categorical data). Differences between groups in baseline factors will be evaluated using an independent t-test if assumptions for parametric testing are met or a non-parametric alternative otherwise. For categorical data, a chi-square test or a Fisher’s exact test will be used as appropriate [146].
Assumptions on the residuals of primary and secondary outcomes will be checked using the DHARMa package in R [147]. Appropriate actions will be taken if one or more assumptions are not met. All primary and secondary outcomes will be analysed following the standard intention-to-treat (ITT [148, 149]) principle using linear mixed-effects models (LMEM, lme4 package in R). Subject-specific random intercepts will account for within-subject correlations between time points. Follow-up scores (T2) of the outcomes will be the dependent variables of the models, while group (intervention vs. control, control being the reference), time (T0, T1, T2), and group x time interactions at T1 and at T2, respectively, will be included as independent variables (fixed effects). Baseline factors such as age, sex, and time since stroke will be considered as covariates. Missing data of whole measurement time points (i.e. of dropouts) will be accounted for with the ‘last observation carried forward’ method. For single missing data points (e.g. if a participant did not perform an assessment at one time point or a technical issue produced missing data at one time point), however, no data imputation will be performed as LMEMs can be fitted even if some outcome data are missing [145, 150]. For the outdoor walking assessment (OWA), high occurrence of missing data is expected, as ability to walk 400 m is not covered by the eligibility criteria and varying weather conditions on the measurement days may interfere with the assessment. Therefore, for the OWA outcome parameters, datasets of participants who did not perform the OWA at one or several time points will be excluded from the analysis. Significance will be set to p < 0.05. Effect sizes will be calculated as r (Bravais-Person correlation coefficient) and interpreted as small (r < 0.3), medium (r < 0.5), and large (r ≥ 0.5) [145].
Methods for analysis of intervention and further activity outcomes
Mean/median compliance and adherence rates with standard deviations/inter-quartile ranges will be reported overall and for each week of the intervention period. Additionally, reasons for not attending or aborting a training session will be summarised. Mean/median ratings of perceived motor-cognitive task difficulty and perceived performance with standard deviations/inter-quartile ranges will be reported overall and for each week of the intervention period. These will be compared to the targeted ranges for task difficulty and perceived performance to establish if an optimal training load was achieved. The volume of usual care (physical, cognitive, and other therapies) and general physical as well as cognitive activities will be descriptively summarised and considered as covariates in the LMEMs of the primary and secondary analyses.
Interim analysis {21b}
N/A. No interim analyses are planned, as preliminary analysis of the effect will most probably be underpowered and, therefore, not informative for the decision of an early study termination.
Methods for additional analysis (e.g. sub-group analyses) {20b}
As the ITT analyses may underestimate a present treatment effect [148], the analyses of the primary and secondary outcomes will be repeated with only those participants, who did not withdraw from the study within the intervention period (T0–T1), and with adherence rates of ≥ 85% (per protocol analysis). This cut-off was chosen based on systematic reviews covering comparable interventions, outcomes, and populations [27, 151], where a minimum of 720 min in at least 12 weeks or an intervention duration of at least 8 weeks were recommended. Moreover, 85% of 24 training sessions results in an ‘acceptable’ absence of 3.6 sessions, which seems practical to account for sickness and conflicting schedules. The results of the ITT and per-protocol analyses will be compared in the discussion of the study report.
Furthermore, to assess clinical meaningfulness of possible treatment effects, the following analyses will be performed on outcomes that (a) revealed a significant between group effect at either T1 or T2 [152, 153] and (b) a clinically important difference is reported in literature. On the one hand, the difference in change score between the two groups will be compared to clinical meaningful change scores (e.g. 1.22 points in the MoCA [99]). On the other hand, frequencies of ‘responders’ (individual change score above clinically meaningful change) and ‘non-responders’ (individual change score below clinically meaningful change) between the two groups will be compared [152, 153].
Plans to give access to the full protocol, participant-level data, and statistical code {31c}
The full protocol of this study was published on https://clinicaltrials.gov (NCT05524727). De-identified participant-level data and the statistical code will be available from the corresponding author on reasonable request.
Oversight and monitoring
Composition of the coordinating centre and trial steering committee {5d}
Recruitment and screening are performed at all study cites by the (local) principal investigators or their delegated staff. All other study-related procedures are performed by trained members of the sponsor/principal investigator team, who are in daily contact regarding the organisation of the trial.
Composition of the data monitoring committee, its role and reporting structure {21a}
Data monitoring in this study is performed by a senior researcher not otherwise involved in the study procedures. On at least three monitoring visits, the monitor reviews the study (team) documents and regulatory aspects, the enrolment process, the participant data, safety aspects, and protocol deviations. Upon monitoring visits, the monitor generates a report including any findings that must be resolved. Resolution of these findings is performed by the principal investigator. Additionally, the clinical trial centre (CTC) of the University Hospital Zurich performs a quality visit on the protocol and monitoring reports. Both the monitor and the clinical trial centre are independent of the sponsor.
Adverse event reporting and harms {22}
Adverse events are recorded in the eCRF throughout the study and managed according to GCP guidelines.
Frequency and plans for auditing trial conduct {23}
The project management group (including study staff from all sites) meets at least weekly to track correct trial conducted. As this is a low-risk study, no data monitoring committee exists; however, according to the approved study protocol by the cantonal ethical committee, an independent monitor performs at monitoring visits on the study documentation and procedures, including but not limited to the following actions:
-
1.
An initiation visit before the study start, where is evaluated whether ethical approval is granted, training of study staff was performed and documented, study and safety documentation as well as case report forms were appropriately prepared, and insurance is valid;
-
2.
At least one routine monitoring visit (further visits are planned if indicated due to inconveniences), where it is checked whether protocol and study documentation is up-to-date, informed consent procedures and enrolment are correctly performed, source data is correctly filed and data transferred to case report forms, safety is correctly reported and treated, and blinding has been maintained; and
-
3.
A closure visit upon the end of the study, where the monitor repeats the checks from the routine monitoring visit, and additionally assesses whether documentation is complete and archiving appropriately prepared.
Plans for communicating important protocol amendments to relevant parties (e.g. trial participants, ethical committee) {25}
Necessary protocol amendments are submitted to the ethical committee before implementation. All study-team members are informed about changes to the protocol on the weekly project management group meetings. Updated versions of the protocol are added to the investigator site files upon approval of the ethical committee. In case of an amendment, which changes study procedures or conditions for participants, participants are informed immediately and the clinical trial registry is updated upon approval of the ethical committee.
Dissemination plans {31a}
The findings of this study will be published in scientific journal articles and scientific presentations. All publications will be authored by the study team, following established authorship guidelines. Participants will receive a copy of their individual study data upon request.
Discussion
The PEMOCS study evaluates the effect of a 12-week concept-guided, personalised, motor-cognitive exergame training added to usual care compared to usual care alone on global cognitive functions and explores effects on specific cognitive functions, health-related quality of life, and gait in chronic stroke survivors. The results will give insight into under-investigated research topics in chronic stroke: the effect of a motor-cognitive exergame integrating whole-body movements on cognitive functions, its effect on spatiotemporal gait parameters relevant in stroke, and the benefits of personally-tailored progression and variability in exergame interventions [37]. The feasibility trial preceding this RCT showed that chronic stroke survivors were continuously motivated for and satisfied with the personalised exergame training and adherence was high [85]. Therefore, satisfactory adherence in the training group is expected in this study. Based on results from studies with exergame trainings in healthy older adults or other neurological patients, superior effects on cognitive functions of the additional exergame training compared to usual care alone are hypothesised [34, 63, 154]. We expect most participants with chronic stroke to exhibit cognitive decline, as cognitive impairment is widely reported in chronic stroke, even in patients with seemingly good clinical outcome [155, 156]. We found significantly lower MoCA values in a high-functioning stroke sample compared to healthy adults with comparable age in a previous study [129]. We did not, however, include a MoCA-based threshold for cognitive impairment for inclusion because such a screening instrument seems unsuitable to identify subtle or specific cognitive deficits [76]. Furthermore, previous research has shown that exergame training may have positive effects on gait and mobility [37, 67], including gait speed, spatiotemporal gait parameters, and mobility in chronic stroke. Therefore, superior improvements in various spatiotemporal gait parameters and mobility in the intervention compared to the control group are expected in this study.
The findings of this study could be considered for the design and prescription of future long-term rehabilitation interventions for chronic stroke survivors that focus on cognitive functioning next to restoring motor functions. This is an identified need since cognitive dysfunction after stroke has a persistently high prevalence [157,158,159]. Exergames have the potential to increase motivation for training [42, 85], to produce additional benefits on physical functions as compared with conventional care modes [60, 67], as well as on cognitive functions [55, 63, 154]. Exergames that contain aspects of virtual reality have shown to be able to significantly effect on Body Structure/Function and Activity level outcomes, including improvements in cognitive function, and there is, therefore, evidence supporting the use of such interventions as an adjunct for stroke rehabilitation [160, 161]. Therefore, exergames could help expand existing rehabilitation services for chronic stroke survivors, which may lead to further improvements of cognitive and motor functioning and, therefore, contribute to increased quality of life after stroke [162].
Due to its design, this study will have two major limitations. (1) Study participants will train 840 min with 100% adherence rate, which is at the lower border of recommended training volume for improving cognitive functions in chronic stroke and older adults [23, 27, 163, 164]. Similarly, some authors recommend more than two and longer sessions per week [55, 164]. However, practical reasons including that participants needed to come into the study centre twice a week limited more training time in this study. Moreover, the feasibility study preceding this RCT showed that participants preferred two sessions per week, lasting 30 to 40 min and no longer [85]. To prevent over-request of time expenditure of the participants and, thereby, harm recruitment, the total training volume was kept at this lower border of recommended time. (2) The black box of usual care and leisure time physical and cognitive activities may influence the measured outcomes as well. Usual care is a wide term and has been reported to include a wide range of interventions, doses, intensities, and implementations [165, 166]. This limitation is addressed by recording the participants’ usual care and general activities besides the study intervention and by considering this in the analysis. (3) This study’s sample size was not based on a standard RCT sample size calculation, as at the time of planning, no expected values for difference between groups and standard deviations for the MoCA in chronic stroke were available due to the novelty of the topic in research. Therefore, the sample size was estimated based on effect sizes from systematic reviews in related literature (see the ‘Sample size’ section).
Trial status
The protocol version 2 of this study was approved by the local Ethics Committee in Switzerland (Ethics Committee of the Canton Zurich, project-ID: 2022–01211) in August 2022. Since then, three amendments were submitted to the ethics committee: (1) protocol version 3 (October 2022) enclosed a change in the patient information regarding travelling costs to study centres, (2) protocol version 4 (March 2023) reported a new principal investigator at one of the study sites, and (3) protocol version 5 (October 2023) included a more detailed description of the data management, which was suggested by the monitor. None of the amendments changed any of the study procedures. The current version 5 of the protocol was approved by the Ethics Committee on October 20, 2023. Study procedures began in September 2022 and are expected to be completed by July 2024. At the time point of submission of this manuscript, the study is running. At submission of this manuscript, 37 participants were enrolled and planned to completed the study or had already completed it. Nine additional participants had been enrolled but withdrew before study completion. The study was registered on https://clinicaltrials.gov (NCT05524727) on September 1, 2022, as well as on https://.kofam.ch, the portal for human research in Switzerland, which ensures that none of the study procedures have been changed since the start of the study.
Availability of data and materials {29}
No preliminary clinical results will be published before the end of the trial. The final trial dataset with anonymous data will be available on a repository or included with the publication as additional file.
Abbreviations
- ADH:
-
Adherence
- ANOVA:
-
Analysis of variance
- BBS:
-
Berg Balance Scale
- BQ:
-
Baseline questionnaire, incl. see the ‘Baseline factors’ section
- CA:
-
Cognitive activity
- CCI:
-
Charlson comorbidity index
- CPL:
-
Compliance
- CT:
-
Cognitive therapy
- eCRF:
-
Electronic case report form
- FAC:
-
Functional Ambulation Category
- FITT-VP:
-
Frequency, intensity, time, type, volume, progression
- FM-LE:
-
Lower-extremity component of the Fugl-Meyer assessment
- GCP:
-
Good clinical practice
- ISF:
-
Investigator side file
- LMEM:
-
Linear mixed-effects model
- MoCA:
-
Montreal Cognitive Assessment
- mRS:
-
Modified Rankin scale
- MRT:
-
Mental rotation test
- NASA-TLX:
-
National Aeronautics and Space Administration Task Load Index
- NBT:
-
N-back test
- NIHSS:
-
National institutes of health stroke scale
- OWA:
-
Outdoor walking assessment
- PA:
-
Physical activity
- PCA:
-
Physical and cognitive activities
- PI:
-
Principal investigator
- PP:
-
Perceived performance
- PT:
-
Physical therapy
- PTD:
-
Perceived task difficulty
- RCT:
-
Randomised, controlled trial
- REDCap:
-
Research Electronic data capture
- (S)AE:
-
(Serious) adverse event
- SIS 3.0:
-
Stroke Impact Scale
- SMD:
-
Standardised mean difference
- SRT:
-
Simple reaction test
- Stroop:
-
Stroop Interference test
- T0:
-
Baseline
- T0-T1:
-
Intervention period
- T1:
-
Post-intervention
- T1-T2:
-
Follow-up period
- T2:
-
Follow-up
- TIDieR:
-
Template for intervention description and replication
- TMF:
-
Trial master file
- TMT:
-
Trail Making test
- TUG:
-
Timed Up and Go test
- TUG-Cogn:
-
Cognitive dual-task Timed Up and Go test
- UC:
-
Usual care
- VAS:
-
Visual analogue scale
- VTS:
-
Vienna Test System
- 10MWT:
-
10-M walk test
References
Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–528. https://doi.org/10.1161/cir.0000000000000659.
Johnson CO, Nguyen M, Roth GA, Nichols E, Alam T, Abate D, et al. Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):439–58. https://doi.org/10.1016/S1474-4422(19)30034-1.
Feigin VL, Roth GA, Naghavi M, Parmar P, Krishnamurthi R, Chugh S, et al. Global burden of stroke and risk factors in 188 countries, during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. The Lancet Neurology. 2016;15(9):913–24. https://doi.org/10.1016/s1474-4422(16)30073-4.
Katan M, Luft A. Global burden of stroke. Semin Neurol. 2018;38(2):208–11. https://doi.org/10.1055/s-0038-1649503.
Schweizerisches_Gesundheitsobservatorium. Hirnschlag: Inzidenz und Letalität in der Schweiz 2019 [accessed 2022 9th May]. Available from: https://ind.obsan.admin.ch/de/indicator/obsan/hirnschlag.
Ursin MH, Bergland A, Fure B, Thommessen B, Hagberg G, Oksengard AR, Ihle-Hansen H. Gait and balance one year after stroke; relationships with lesion side, subtypes of cognitive impairment and neuroimaging findings-a longitudinal, cohort study. Physiotherapy. 2019;105(2):254–61. https://doi.org/10.1016/j.physio.2018.07.007.
Sun JH, Tan L, Yu JT. Post-stroke cognitive impairment: epidemiology, mechanisms and management. Ann Transl Med. 2014;2(8):80. https://doi.org/10.3978/j.issn.2305-5839.2014.08.05.
Patten C, Lexell J, Brown HE. Weakness and strength training in persons with poststroke hemiplegia: rationale, method, and efficacy. J Rehabil Res Dev. 2004;41(3A):293–312. https://doi.org/10.1682/jrrd.2004.03.0293.
Barker WH, Mullooly JP. Stroke in a defined elderly population, 1967–1985. A less lethal and disabling but no less common disease. Stroke. 1997;28(2):284–90. https://doi.org/10.1161/01.STR.28.2.284.
Tyson SF, Hanley M, Chillala J, Selley A, Tallis RC. Balance disability after stroke. Phys Ther. 2006;86(1):30–8. https://doi.org/10.1093/ptj/86.1.30.
Balaban B, Tok F. Gait disturbances in patients with stroke. PM R. 2014;6(7):635–42. https://doi.org/10.1016/j.pmrj.2013.12.017.
Durcan S, Flavin E, Horgan F. Factors associated with community ambulation in chronic stroke. Disabil Rehabil. 2016;38(3):245–9. https://doi.org/10.3109/09638288.2015.1035460.
Cumming TB, Marshall RS, Lazar RM. Stroke, cognitive deficits, and rehabilitation: still an incomplete picture. Int J Stroke. 2013;8(1):38–45. https://doi.org/10.1111/j.1747-4949.2012.00972.x.
Middleton LE, Lam B, Fahmi H, Black SE, McIlroy WE, Stuss DT, et al. Frequency of domain-specific cognitive impairment in sub-acute and chronic stroke. NeuroRehabilitation. 2014;34(2):305–12. https://doi.org/10.3233/NRE-131030.
Mellon L, Brewer L, Hall P, Horgan F, Williams D, Hickey A, group A-Ss. Cognitive impairment six months after ischaemic stroke: a profile from the ASPIRE-S study. BMC Neurol. 2015;15(1):31. https://doi.org/10.1186/s12883-015-0288-2.
Lamb F, Anderson J, Saling M, Dewey H. Predictors of subjective cognitive complaint in postacute older adult stroke patients. Arch Phys Med Rehabil. 2013;94(9):1747–52. https://doi.org/10.1016/j.apmr.2013.02.026.
Hotter B, Padberg I, Liebenau A, Knispel P, Heel S, Steube D, et al. Identifying unmet needs in long-term stroke care using in-depth assessment and the Post-Stroke Checklist - The Managing Aftercare for Stroke (MAS-I) study. Eur Stroke J. 2018;3(3):237–45. https://doi.org/10.1177/2396987318771174.
www.swissheart.ch. Swiss Heart Foundation - Rehabilitation after Stroke [accessed 2022 9th Feb]. Available from: https://www.swissheart.ch/herzkrankheiten-hirnschlag/fuer-ihre-gesundheit/rehabilitation-nach-einem-hirnschlag.html.
Pollock A, St George B, Fenton M, Firkins L. Top ten research priorities relating to life after stroke. Lancet Neurol. 2012;11(3):209. https://doi.org/10.1016/S1474-4422(12)70029-7.
Kapoor A, Lanctot KL, Bayley M, Kiss A, Herrmann N, Murray BJ, Swartz RH. “Good outcome” isn’t good enough: cognitive impairment, depressive symptoms, and social restrictions in physically recovered stroke patients. Stroke. 2017;48(6):1688–90. https://doi.org/10.1161/STROKEAHA.117.016728.
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–596. https://doi.org/10.1161/CIR.0000000000000757.
Herold F, Hamacher D, Schega L, Muller NG. Thinking while moving or moving while thinking - concepts of motor-cognitive training for cognitive performance enhancement. Front Aging Neurosci. 2018;10:228. https://doi.org/10.3389/fnagi.2018.00228.
Tait JL, Duckham RL, Milte CM, Main LC, Daly RM. Influence of sequential vs. simultaneous dual-task exercise training on cognitive function in older adults. Front Aging Neurosci. 2017;9(368):368. https://doi.org/10.3389/fnagi.2017.00368.
Verstraeten S, Mark R, Sitskoorn M. Motor and cognitive impairment after stroke: a common bond or a simultaneous deficit. Stroke Res Ther. 2016;1(1). not available. URL: https://stroke.imedpub.com/motor-and-cognitive-impairment-after-strokea-common-bond-or-a-simultaneous-deficit.php?aid=9074.
Kraft E. Cognitive function, physical activity, and aging: possible biological links and implications for multimodal interventions. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2012;19(1–2):248–63. https://doi.org/10.1080/13825585.2011.645010.
Fissler P, Kuster O, Schlee W, Kolassa IT. Novelty interventions to enhance broad cognitive abilities and prevent dementia: synergistic approaches for the facilitation of positive plastic change. Prog Brain Res. 2013;207:403–34. https://doi.org/10.1016/B978-0-444-63327-9.00017-5.
Lauenroth A, Ioannidis AE, Teichmann B. Influence of combined physical and cognitive training on cognition: a systematic review. BMC Geriatr. 2016;16(1):141. https://doi.org/10.1186/s12877-016-0315-1.
Bamidis PD, Vivas AB, Styliadis C, Frantzidis C, Klados M, Schlee W, et al. A review of physical and cognitive interventions in aging. Neurosci Biobehav Rev. 2014;44:206–20. https://doi.org/10.1016/j.neubiorev.2014.03.019.
Levin O, Netz Y, Ziv G. The beneficial effects of different types of exercise interventions on motor and cognitive functions in older age: a systematic review. European review of aging and physical activity : official journal of the European Group for Research into Elderly and Physical Activity. 2017;14(1):20. https://doi.org/10.1186/s11556-017-0189-z.
Dhir S, Teo WP, Chamberlain SR, Tyler K, Yucel M, Segrave RA. The effects of combined physical and cognitive training on inhibitory control: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2021;128:735–48. https://doi.org/10.1016/j.neubiorev.2021.07.008.
Gavelin HM, Dong C, Minkov R, Bahar-Fuchs A, Ellis KA, Lautenschlager NT, et al. Combined physical and cognitive training for older adults with and without cognitive impairment: a systematic review and network meta-analysis of randomized controlled trials. Ageing Res Rev. 2020;66:101232. https://doi.org/10.1016/j.arr.2020.101232.
Rieker JA, Reales JM, Muinos M, Ballesteros S. The effects of combined cognitive-physical interventions on cognitive functioning in healthy older adults: a systematic review and multilevel meta-analysis. Front Hum Neurosci. 2022;16:838968. https://doi.org/10.3389/fnhum.2022.838968.
Teraz K, Slosar L, Paravlic AH, de Bruin ED, Marusic U. Impact of motor-cognitive interventions on selected gait and balance outcomes in older adults: a systematic review and meta-analysis of randomized controlled trials. Front Psychol. 2022;13:837710. https://doi.org/10.3389/fpsyg.2022.837710.
Wollesen B, Wildbredt A, van Schooten KS, Lim ML, Delbaere K. The effects of cognitive-motor training interventions on executive functions in older people: a systematic review and meta-analysis. European review of aging and physical activity : official journal of the European Group for Research into Elderly and Physical Activity. 2020;17:9. https://doi.org/10.1186/s11556-020-00240-y.
Zhu X, Yin S, Lang M, He R, Li J. The more the better? A meta-analysis on effects of combined cognitive and physical intervention on cognition in healthy older adults. Ageing Res Rev. 2016;31:67–79. https://doi.org/10.1016/j.arr.2016.07.003.
He Y, Yang L, Zhou J, Yao LQ, Pang MYC. Dual-task training effects on motor and cognitive functional abilities in individuals with stroke: a systematic review. Clin Rehabil. 2018;32(7):865–77. https://doi.org/10.1177/0269215518758482.
Huber SK, Knols RH, Arnet P, de Bruin ED. Motor-cognitive intervention concepts can improve gait in chronic stroke, but their effect on cognitive functions is unclear: A systematic review with meta-analyses. Neurosci Biobehav Rev. 2022;132:818–37. https://doi.org/10.1016/j.neubiorev.2021.11.013.
Amoros-Aguilar L, Rodriguez-Quiroga E, Sanchez-Santolaya S, Coll-Andreu M. Effects of combined interventions with aerobic physical exercise and cognitive training on cognitive function in stroke patients: a systematic review. Brain sciences. 2021;11(4):473. https://doi.org/10.3390/brainsci11040473.
Embrechts E, McGuckian TB, Rogers JM, Dijkerman CH, Steenbergen B, Wilson PH, Nijboer TCW. Cognitive and motor therapy after stroke is not superior to motor and cognitive therapy alone to improve cognitive and motor outcomes: new insights from a meta-analysis. Arch Phys Med Rehabil. 2023;104(10):1720–34. https://doi.org/10.1016/j.apmr.2023.05.010.
Adams MA, Marshall SJ, Dillon L, Caparosa S, Ramirez E, Phillips J, Norman GJ. A theory-based framework for evaluating exergames as persuasive technology. Proceedings of the 4th International Conference on Persuasive Technology; Claremont, California, USA: Association for Computing Machinery; 2009. p. 1–8. https://doi.org/10.1145/1541948.1542006.
de Boissieu P, Denormandie P, Armaingaud D, Sanchez S, Jeandel C. Exergames and elderly: a non-systematic review of the literature. European geriatric medicine. 2017;8(2):111–6. https://doi.org/10.1016/j.eurger.2017.02.003.
Swanson LR, Whittinghill DM. Intrinsic or extrinsic? Using videogames to motivate stroke survivors: a systematic review. Games Health J. 2015;4(3):253–8. https://doi.org/10.1089/g4h.2014.0074.
Johnson D, Deterding S, Kuhn K-A, Staneva A, Stoyanov S, Hides L. Gamification for health and wellbeing: a systematic review of the literature. Internet Interv. 2016;6:89–106. https://doi.org/10.1016/j.invent.2016.10.002.
Widmer M, Held JPO, Wittmann F, Valladares B, Lambercy O, Sturzenegger C, et al. Reward during arm training improves impairment and activity after stroke: a randomized controlled trial. Neurorehabil Neural Repair. 2022;36(2):140–50. https://doi.org/10.1177/15459683211062898.
Chang M, Buchel D, Reinecke K, Lehmann T, Baumeister J. Ecological validity in exercise neuroscience research: a systematic investigation. Eur J Neurosci. 2022;55(2):487–509. https://doi.org/10.1111/ejn.15595.
Fang Q, Ghanouni P, Anderson SE, Touchett H, Shirley R, Fang F, Fang C. Effects of exergaming on balance of healthy older adults: a systematic review and meta-analysis of randomized controlled trials. Games Health J. 2020;9(1):11–23. https://doi.org/10.1089/g4h.2019.0016.
Gallou-Guyot M, Mandigout S, Bherer L, Perrochon A. Effects of exergames and cognitive-motor dual-task training on cognitive, physical and dual-task functions in cognitively healthy older adults: an overview. Ageing Res Rev. 2020;63:101135. https://doi.org/10.1016/j.arr.2020.101135.
Hai L, Hou HY, Zhou C, Li HJ. The effect of exergame training on physical functioning of healthy older adults: a meta-analysis. Games Health J. 2022;11(4):207–24. https://doi.org/10.1089/g4h.2021.0173.
Janhunen M, Karner V, Katajapuu N, Niiranen O, Immonen J, Karvanen J, et al. Effectiveness of exergame intervention on walking in older adults: a systematic review and meta-analysis of randomized controlled trials. Phys Ther. 2021;101(9):pzab152. https://doi.org/10.1093/ptj/pzab152.
Pacheco TBF, de Medeiros CSP, de Oliveira VHB, Vieira ER, de Cavalcanti FAC. Effectiveness of exergames for improving mobility and balance in older adults: a systematic review and meta-analysis. Syst Rev. 2020;9(1):163. https://doi.org/10.1186/s13643-020-01421-7.
Taylor LM, Kerse N, Frakking T, Maddison R. Active video games for improving physical performance measures in older people: a meta-analysis. Journal of geriatric physical therapy (2001). 2018;41(2):108–23. https://doi.org/10.1519/JPT.0000000000000078.
Buyle M, Jung Y, Pavlou M, Gonzalez SC, Bamiou DE. The role of motivation factors in exergame interventions for fall prevention in older adults: a systematic review and meta-analysis. Front Neurol. 2022;13:903673. https://doi.org/10.3389/fneur.2022.903673.
Jiang J, Guo W, Wang B. Effects of exergaming on executive function of older adults: a systematic review and meta-analysis. PeerJ. 2022;10:e13194. https://doi.org/10.7717/peerj.13194.
Soares VN, Yoshida HM, Magna TS, Sampaio RAC, Fernandes PT. Comparison of exergames versus conventional exercises on the cognitive skills of older adults: a systematic review with meta-analysis. Arch Gerontol Geriatr. 2021;97:104485. https://doi.org/10.1016/j.archger.2021.104485.
Stojan R, Voelcker-Rehage C. A systematic review on the cognitive benefits and neurophysiological correlates of exergaming in healthy older adults. J Clin Med. 2019;8(5):734. https://doi.org/10.3390/jcm8050734.
Yen H-Y, Chiu H-L. Virtual reality exergames for improving older adults’ cognition and depression: a systematic review and meta-analysis of randomized control trials. J Am Med Dir Assoc. 2021;22(5):995–1002. https://doi.org/10.1016/j.jamda.2021.03.009.
Calafiore D, Invernizzi M, Ammendolia A, Marotta N, Fortunato F, Paolucci T, et al. Efficacy of virtual reality and exergaming in improving balance in patients with multiple sclerosis: a systematic review and meta-analysis. Front Neurol. 2021;12:773459. https://doi.org/10.3389/fneur.2021.773459.
Elena P, Demetris S, Christina M, Marios P. Differences between exergaming rehabilitation and conventional physiotherapy on quality of life in Parkinson’s disease: a systematic review and meta-analysis. Front Neurol. 2021;12:683385. https://doi.org/10.3389/fneur.2021.683385.
Malone LA, Mendonca CJ, Kim Y. Active videogaming interventions in adults with neuromuscular conditions: a scoping review. Games Health J. 2022;11(3):141–56. https://doi.org/10.1089/g4h.2021.0096.
Prosperini L, Tomassini V, Castelli L, Tacchino A, Brichetto G, Cattaneo D, Solaro CM. Exergames for balance dysfunction in neurological disability: a meta-analysis with meta-regression. J Neurol. 2021;268(9):3223–37. https://doi.org/10.1007/s00415-020-09918-w.
Santos P, Scaldaferri G, Santos L, Ribeiro N, Neto M, Melo A. Effects of the Nintendo Wii training on balance rehabilitation and quality of life of patients with Parkinson’s disease: a systematic review and meta-analysis. NeuroRehabilitation. 2019;44(4):569–77. https://doi.org/10.3233/NRE-192700.
Wang D, Cui WJ, Hou ZH, Gao Y. Effectiveness of different exercises in improving postural balance among Parkinson’s disease patients: a systematic review and network meta-analysis. Front Aging Neurosci. 2023;15:1215495. https://doi.org/10.3389/fnagi.2023.1215495.
Mura G, Carta MG, Sancassiani F, Machado S, Prosperini L. Active exergames to improve cognitive functioning in neurological disabilities: a systematic review and meta-analysis. Eur J Phys Rehabil Med. 2018;54(3):450–62. https://doi.org/10.23736/S1973-9087.17.04680-9.
Cai Z, Ma Y, Li L, Lu GZ. Effects of exergaming in older individuals with mild cognitive impairment and dementia: a systematic review and meta-analysis. Geriatr Nurs. 2023;51:351–9. https://doi.org/10.1016/j.gerinurse.2023.03.028.
Elhusein AM, Fadlalmola HA, Awadalkareem EM, Alhusain EYM, Alnassry SM, Alshammari M, et al. Exercise-based gaming in patients with multiple sclerosis: a systematic review and meta-analysis. Belitung Nurs J. 2024;10(1):1–14. https://doi.org/10.33546/bnj.3006.
Li K, Wang Y, Wu Z, Yao X, Fan Y. Effectiveness of active exergames for improving cognitive function in patients with neurological disabilities: a systematic review and meta-analysis. Games Health J. 2023;12(3):198–210. https://doi.org/10.1089/g4h.2022.0134.
Chan KGF, Jiang Y, Choo WT, Ramachandran HJ, Lin Y, Wang W. Effects of exergaming on functional outcomes in people with chronic stroke: a systematic review and meta-analysis. J Adv Nurs. 2022;78(4):929–46. https://doi.org/10.1111/jan.15125.
Cheok G, Tan D, Low A, Hewitt J. Is Nintendo Wii an effective intervention for individuals with stroke? A systematic review and meta-analysis. J Am Med Dir Assoc. 2015;16(11):923–32. https://doi.org/10.1016/j.jamda.2015.06.010.
Ghazavi Dozin SM, Mohammad Rahimi N, Aminzadeh R. Wii Fit-based biofeedback rehabilitation among post-stroke patients: a systematic review and meta-analysis of randomized controlled trial. Biol Res Nurs. 2024;26(1):5–20. https://doi.org/10.1177/10998004231180316.
Zaleski AL, Taylor BA, Panza GA, Wu Y, Pescatello LS, Thompson PD, Fernandez AB. Coming of age: considerations in the prescription of exercise for older adults. Methodist Debakey Cardiovasc J. 2016;12(2):98–104. https://doi.org/10.14797/mdcj-12-2-98.
Billinger SA, Arena R, Bernhardt J, Eng JJ, Franklin BA, Johnson CM, et al. Physical activity and exercise recommendations for stroke survivors: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(8):2532–53. https://doi.org/10.1161/STR.0000000000000022.
Kitago T, Krakauer JW. Motor learning principles for neurorehabilitation. In: Barnes MP, Good DC, editors. Handbook of Clinical Neurology. 110: Elsevier; 2013. p. 93–103.
Appelros P, Åsberg S. Sex differences in stroke. Handb Clin Neurol. 2020;175:299–312. https://doi.org/10.1016/b978-0-444-64123-6.00021-7.
Chiti G, Pantoni L. Use of Montreal Cognitive Assessment in patients with stroke. Stroke. 2014;45(10):3135–40. https://doi.org/10.1161/STROKEAHA.114.004590.
Bernhardt J, Hayward KS, Kwakkel G, Ward NS, Wolf SL, Borschmann K, et al. Agreed definitions and a shared vision for new standards in stroke recovery research: the Stroke Recovery and Rehabilitation Roundtable taskforce. Neurorehabil Neural Repair. 2017;31(9):793–9. https://doi.org/10.1177/1545968317732668.
Nakling AE, Aarsland D, Naess H, Wollschlaeger D, Fladby T, Hofstad H, Wehling E. Cognitive deficits in chronic stroke patients: neuropsychological assessment, depression, and self-reports. Dement Geriatr Cogn Dis Extra. 2017;7(2):283–96. https://doi.org/10.1159/000478851.
Wiley E, Khattab S, Tang A. Examining the effect of virtual reality therapy on cognition post-stroke: a systematic review and meta-analysis. Disabil Rehabil Assist Technol. 2020;17(1):50–60. https://doi.org/10.1080/17483107.2020.1755376.
Slade SC, Finnegan S, Dionne CE, Underwood M, Buchbinder R. The Consensus on Exercise Reporting Template (CERT) applied to exercise interventions in musculoskeletal trials demonstrated good rater agreement and incomplete reporting. J Clin Epidemiol. 2018;103:120–30. https://doi.org/10.1016/j.jclinepi.2018.07.009.
Page P, Hoogenboom B, Voight M. Improving the reporting of therapeutic exercise interventions in rehabilitation research. Int J Sports Phys Ther. 2017;12(2):297–304.
Huber SK, Manser P, de Bruin ED. PEMOCS: theory derivation of a concept for PErsonalized MOtor-Cognitive exergame training in chronic Stroke—a methodological paper with an application example. Frontiers in sports and active living. 2024;6. https://doi.org/10.3389/fspor.2024.1397949.
Kleim JA, Jones TA. Principles of experience-dependent neural plasticity: implications for rehabilitation after brain damage. J Speech Lang Hear Res. 2008;51(1):S225–39. https://doi.org/10.1044/1092-4388(2008/018).
Bayles MP. ACSM’s exercise testing and prescription. Indianapolis, USA: Lippincott Williams & Wilkins; 2023.
Maier M, Ballester BR, Verschure P. Principles of neurorehabilitation after stroke based on motor learning and brain plasticity mechanisms. Front Syst Neurosci. 2019;13:74. https://doi.org/10.3389/fnsys.2019.00074.
Gentile AM. A working model of skill acquisition with application to teaching. Quest. 1972;17(1):3–23. https://doi.org/10.1080/00336297.1972.10519717.
Huber SK, Held JPO, de Bruin ED, Knols RH. Personalized motor-cognitive exergame training in chronic stroke patients-a feasibility study. Front Aging Neurosci. 2021;13(663):730801. https://doi.org/10.3389/fnagi.2021.730801.
Anker R. PhysiGait Lab: user manual and outcome parameters.2019. Available from: https://clinical.gaitup.com/wp-content/uploads/2019/10/PhysiGaitLab_User_Manual_v131.pdf.
Sekiya N, Nagasaki H. Reproducibility of the walking patterns of normal young adults: test-retest reliability of the walk ratio(step-length/step-rate). Gait Posture. 1998;7(3):225–7. https://doi.org/10.1016/s0966-6362(98)00009-5.
Kwah LK, Diong J. National Institutes of Health Stroke Scale (NIHSS). J Physiother. 2014;60(1):61. https://doi.org/10.1016/j.jphys.2013.12.012.
Charlson M, Szatrowski TP, Peterson J, Gold J. Validation of a combined comorbidity index. J Clin Epidemiol. 1994;47(11):1245–51. https://doi.org/10.1016/0895-4356(94)90129-5.
Banks JL, Marotta CA. Outcomes validity and reliability of the modified Rankin scale: implications for stroke clinical trials: a literature review and synthesis. Stroke. 2007;38(3):1091–6. https://doi.org/10.1161/01.STR.0000258355.23810.c6.
Mehrholz J, Wagner K, Rutte K, Meissner D, Pohl M. Predictive validity and responsiveness of the functional ambulation category in hemiparetic patients after stroke. Arch Phys Med Rehabil. 2007;88(10):1314–9. https://doi.org/10.1016/j.apmr.2007.06.764.
Blum L, Korner-Bitensky N. Usefulness of the Berg Balance Scale in stroke rehabilitation: a systematic review. Phys Ther. 2008;88(5):559–66. https://doi.org/10.2522/ptj.20070205.
Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695–9. https://doi.org/10.1111/j.1532-5415.2005.53221.x.
Tan HH, Xu J, Teoh HL, Chan BP, Seet RC, Venketasubramanian N, et al. Decline in changing Montreal Cognitive Assessment (MoCA) scores is associated with post-stroke cognitive decline determined by a formal neuropsychological evaluation. PLoS ONE. 2017;12(3):e0173291. https://doi.org/10.1371/journal.pone.0173291.
Shen YJ, Wang WA, Huang FD, Chen J, Liu HY, Xia YL, et al. The use of MMSE and MoCA in patients with acute ischemic stroke in clinical. Int J Neurosci. 2016;126(5):442–7. https://doi.org/10.3109/00207454.2015.1031749.
Cumming TB, Lowe D, Linden T, Bernhardt J. The AVERT MoCA data: scoring reliability in a large multicenter trial. Assessment. 2020;27(5):976–81. https://doi.org/10.1177/1073191118771516.
Feeney J, Savva GM, O’Regan C, King-Kallimanis B, Cronin H, Kenny RA. Measurement error, reliability, and minimum detectable change in the Mini-Mental State Examination, Montreal Cognitive Assessment, and Color Trails Test among community living middle-aged and older adults. Journal of Alzheimers Disease. 2016;53(3):1107–14. https://doi.org/10.3233/Jad-160248.
Duncan PW, Wallace D, Lai SM, Johnson D, Embretson S, Laster LJ. The Stroke Impact Scale version 20: evaluation of reliability, validity, and sensitivity to change. Stroke. 1999;30(10):2131–40 (none).
Wu CY, Hung SJ, Lin KC, Chen KH, Chen P, Tsay PK. Responsiveness, minimal clinically important difference, and validity of the MoCA in stroke rehabilitation. Occup Ther Int. 2019;2019:2517658. https://doi.org/10.1155/2019/2517658.
Shi D, Chen X, Li Z. Diagnostic test accuracy of the Montreal Cognitive Assessment in the detection of post-stroke cognitive impairment under different stages and cutoffs: a systematic review and meta-analysis. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2018;39(4):705–16. https://doi.org/10.1007/s10072-018-3254-0.
Potocnik J, Ovcar Stante K, Rakusa M. The validity of the Montreal Cognitive Assessment (MoCA) for the screening of vascular cognitive impairment after ischemic stroke. Acta Neurol Belg. 2020;120(3):681–5. https://doi.org/10.1007/s13760-020-01330-5.
McLeod C, Norman R, Litton E, Saville BR, Webb S, Snelling TL. Choosing primary endpoints for clinical trials of health care interventions. Contemporary Clinical Trials Communications. 2019;16:100486. https://doi.org/10.1016/j.conctc.2019.100486.
Vellone E, Savini S, Fida R, Dickson VV, Melkus GDE, Carod-Artal FJ, et al. Psychometric evaluation of the Stroke Impact Scale 3.0. Journal of Cardiovascular Nursing. 2015;30(3):229–41. https://doi.org/10.1097/JCN.0000000000000145.
Petersen C, Morfeld M, Bullinger M. Testing and validation of the German version of the Stroke Impact Scale. Fortschr Neurol Psychiatr. 2001;69(6):284–90. https://doi.org/10.1055/s-2001-14465.
Spikman J, van Zomeren E. Assessment of Attention. In: Gurd J, Kischka U, Marshall J, editors. The Handbook of Clinical Neuropsychology. 2nd ed: Oxford University Press; 2010. 81–96.
Zimmermann P, Fimm B. Testbatterie zur Aufmerksamkeitsprüfung TAP, Handbuch. Psychologische Testsysteme PsyTest, Freiburg. 1992.
Sturm W. Vienna test system - Perceptional and attentional functions: Alertness (WAFA) - Manual. 2006. Available from: https://marketplace.schuhfried.com/en/WAFA.
Bowie CR, Harvey PD. Administration and interpretation of the Trail Making Test. Nat Protoc. 2006;1(5):2277–81. https://doi.org/10.1038/nprot.2006.390.
Tombaugh TN. Trail Making Test A and B: normative data stratified by age and education. Arch Clin Neuropsychol. 2004;19(2):203–14. https://doi.org/10.1016/S0887-6177(03)00039-8.
Reitan RM. Validity of the Trail Making Test as an indicator of organic brain damage. 1958;8(3):271–6. https://doi.org/10.2466/pms.1958.8.3.271.
Rodewald K, Weisbrod M, S. A. Vienna test system - Trail making test - Langensteinbacher version (TMT-L) - Manual.2012. Available from: https://marketplace.schuhfried.com/en/TMT.
Faria CA, Alves HVD, Charchat-Fichman H. The most frequently used tests for assessing executive functions in aging. Dement Neuropsychol. 2015;9(2):149–55. https://doi.org/10.1590/1980-57642015DN92000009.
Scarpina F, Tagini S. The Stroop Color and Word Test. Front Psychol. 2017;8(557):557. https://doi.org/10.3389/fpsyg.2017.00557.
Schuhfried G. Vienna test system - Stroop Interence test (Stroop) - Manual.1999. Available from: https://marketplace.schuhfried.com/en/Stroop.
Owen AM, McMillan KM, Laird AR, Bullmore E. N-back working memory paradigm: a meta-analysis of normative functional neuroimaging studies. Hum Brain Mapp. 2005;25(1):46–59. https://doi.org/10.1002/hbm.20131.
León-Domínguez U, Martín-Rodríguez JF, León-Carrión J. Executive n-back tasks for the neuropsychological assessment of working memory. Behav Brain Res. 2015;292:167–73. https://doi.org/10.1016/j.bbr.2015.06.002.
Gajewski PD, Hanisch E, Falkenstein M, Thönes S, Wascher E. What does the n-Back task measure as we get older? Relations between working-memory measures and other cognitive functions across the lifespan. Front Psychol. 2018;9:2208. https://doi.org/10.3389/fpsyg.2018.02208.
Schellig D, Schuri U. Vienna test system - N-Back verbal (NBV) - Manual.2009. Available from: https://marketplace.schuhfried.com/en/NBV.
Shepard RN, Metzler J. Mental rotation of three-dimensional objects. Science (New York, NY). 1971;171(3972):701–3. https://doi.org/10.1126/science.171.3972.701.
Jansen P, Schachten T. The improvement of visual-spatial performance after golf training in patients with stroke: a pilot study. Cognitie, Brain, Behavior, an Interdisciplinary Journal. 2016;20(3):159–69 (none).
Bo W, Lei M, Tao S, Jie LT, Qian L, Lin FQ, Ping WX. Effects of combined intervention of physical exercise and cognitive training on cognitive function in stroke survivors with vascular cognitive impairment: a randomized controlled trial. Clin Rehabil. 2019;33(1):54–63. https://doi.org/10.1177/0269215518791007.
Bratfisch O, Hagmann E. Vienna test system - Visual awarness (3D) - Manual.2004. Available from: https://marketplace.schuhfried.com/en/3D.
Ng MM, Hill KD, Batchelor F, Burton E. Factors predicting falls and mobility outcomes in patients with stroke returning home after rehabilitation who are at risk of falling. Arch Phys Med Rehabil. 2017;98(12):2433–41. https://doi.org/10.1016/j.apmr.2017.05.018.
Ng SS, Hui-Chan CW. The Timed Up & Go test: its reliability and association with lower-limb impairments and locomotor capacities in people with chronic stroke. Arch Phys Med Rehabil. 2005;86(8):1641–7. https://doi.org/10.1016/j.apmr.2005.01.011.
Hofheinz M, Schusterschitz C. Dual task interference in estimating the risk of falls and measuring change: a comparative, psychometric study of four measurements. Clin Rehabil. 2010;24(9):831–42. https://doi.org/10.1177/0269215510367993.
Yang L, He C, Pang MY. Reliability and validity of dual-task mobility assessments in people with chronic stroke. PLoS ONE. 2016;11(1):e0147833. https://doi.org/10.1371/journal.pone.0147833.
Pumpho A, Chaikeeree N, Saengsirisuwan V, Boonsinsukh R. Selection of the better dual-Timed Up and Go cognitive task to be used in patients with stroke characterized by subtraction operation difficulties. Front Neurol. 2020;11(262):262. https://doi.org/10.3389/fneur.2020.00262.
Cheng DK, Nelson M, Brooks D, Salbach NM. Validation of stroke-specific protocols for the 10-meter walk test and 6-minute walk test conducted using 15-meter and 30-meter walkways. Top Stroke Rehabil. 2020;27(4):251–61. https://doi.org/10.1080/10749357.2019.1691815.
Huber SK, Knols RH, Held JPO, Christen T, de Bruin ED. Agreement, reliability, and concurrent validity of an outdoor, wearable-based walk ratio assessment in healthy adults and chronic stroke survivors. Front Physiol. 2022;13:857963. https://doi.org/10.3389/fphys.2022.857963.
Paas F, Tuovinen JE, Tabbers H, Van Gerven PWM. Cognitive load measurement as a means to advance cognitive load theory. Educational Psychologist. 2003;38(1):63–71. https://doi.org/10.1207/S15326985EP3801_8.
Flagel K, Galler B, Steinhauser J, Gotz K. The, “National Aeronautics and Space Administration-Task Load Index” (NASA-TLX) - an instrument for measuring consultation workload within general practice: evaluation of psychometric properties. Z Evid Fortbild Qual Gesundhwes. 2019;147–148:90–6. https://doi.org/10.1016/j.zefq.2019.10.003.
Bull FC, Al-Ansari SS, Biddle S, Borodulin K, Buman MP, Cardon G, et al. World Health Organization 2020 guidelines on physical activity and sedentary behaviour. Br J Sports Med. 2020;54(24):1451–62. https://doi.org/10.1136/bjsports-2020-102955.
Hoffmann TC, Glasziou PP, Boutron I, Milne R, Perera R, Moher D, et al. Better reporting of interventions: template for intervention description and replication (TIDieR) checklist and guide. BMJ. 2014;348:g1687. https://doi.org/10.1136/bmj.g1687.
Yu RWL, Chan AHS. Meta-analysis of the effects of game types and devices on older adults-video game interaction: Implications for video game training on cognition. Appl Ergon. 2021;96:103477. https://doi.org/10.1016/j.apergo.2021.103477.
Nyman SR, Victor CR. Older people’s recruitment, sustained participation, and adherence to falls prevention interventions in institutional settings: a supplement to the Cochrane systematic review. Age Ageing. 2011;40(4):430–6. https://doi.org/10.1093/ageing/afr016.
Givon N, Zeilig G, Weingarden H, Rand D. Video-games used in a group setting is feasible and effective to improve indicators of physical activity in individuals with chronic stroke: a randomized controlled trial. Clin Rehabil. 2016;30(4):383–92. https://doi.org/10.1177/0269215515584382.
Hung JW, Chou CX, Chang HF, Wu WC, Hsieh YW, Chen PC, et al. Cognitive effects of weight-shifting controlled exergames in patients with chronic stroke: a pilot randomized comparison trial. Eur J Phys Rehabil Med. 2017;53(5):694–702. https://doi.org/10.23736/S1973-9087.17.04516-6.
Dong L, Briceno E, Morgenstern LB, Lisabeth LD. Poststroke cognitive outcomes: sex differences and contributing factors. J Am Heart Assoc. 2020;9(14):e016683. https://doi.org/10.1161/jaha.120.016683.
Reeves MJ, Bushnell CD, Howard G, Gargano JW, Duncan PW, Lynch G, et al. Sex differences in stroke: epidemiology, clinical presentation, medical care, and outcomes. Lancet Neurol. 2008;7(10):915–26. https://doi.org/10.1016/s1474-4422(08)70193-5.
Sohrabji F, Park MJ, Mahnke AH. Sex differences in stroke therapies. J Neurosci Res. 2017;95(1–2):681–91. https://doi.org/10.1002/jnr.23855.
Harris PA, Taylor R, Minor BL, Elliott V, Fernandez M, O’Neal L, et al. The REDCap consortium: building an international community of software platform partners. J Biomed Inform. 2019;95: 103208. https://doi.org/10.1016/j.jbi.2019.103208.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–81. https://doi.org/10.1016/j.jbi.2008.08.010.
De Keersmaecker E, Lefeber N, Geys M, Jespers E, Kerckhofs E, Swinnen E. Virtual reality during gait training: does it improve gait function in persons with central nervous system movement disorders? A systematic review and meta-analysis NeuroRehabilitation. 2019;44(1):43–66. https://doi.org/10.3233/NRE-182551.
RStudio_Team. RStudio: integrated development for F. RStudio, PBC, Boston. 2020. Available from: http://www.rstudio.com/.
Field A, Miles J, Field Z. Discovering statistics using R. London: SAGE Publications Ltd; 2012.
Kim HY. Statistical notes for clinical researchers: chi-squared test and Fisher’s exact test. Restor Dent Endod. 2017;42(2):152–5. https://doi.org/10.5395/rde.2017.42.2.152.
Harting F. DHARMa: residual diagnostics for hierarchical (multi-level/mixed) regression models. R package version 0.4.6. 2022 [Available from: http://florianhartig.github.io/DHARMa/.
McCoy CE. Understanding the intention-to-treat principle in randomized controlled trials. West J Emerg Med. 2017;18(6):1075–8. https://doi.org/10.5811/westjem.2017.8.35985.
Olijnyk L, Darsaut TE, Ohman J, Raymond J. Understanding intent to treat analyses: an important lesson from the international cooperative study on the timing of aneurysm surgery. Neurochirurgie. 2022;68(5):471–3. https://doi.org/10.1016/j.neuchi.2022.02.002.
Gabrio A, Plumpton C, Banerjee S, Leurent B. Linear mixed models to handle missing at random data in trial-based economic evaluations. Health Econ. 2022;31(6):1276–87. https://doi.org/10.1002/hec.4510.
Lee HS, Park YJ, Park SW. The effects of virtual reality training on function in chronic stroke patients: a systematic review and meta-analysis. Biomed Res Int. 2019;2019:7595639. https://doi.org/10.1155/2019/7595639.
Cates C, Karner C. Clinical importance cannot be ruled out using mean difference alone. Bmj-Brit Med J. 2015;351:h5496. https://doi.org/10.1136/bmj.h5496.
Uryniak T, Chan ISF, Fedorov VV, Jiang Q, Oppenheimer L, Snapinn SM, et al. Responder analyses-a PhRMA position paper. Statistics in Biopharmaceutical Research. 2011;3(3):476–87. https://doi.org/10.1198/sbr.2011.10070.
Stanmore E, Stubbs B, Vancampfort D, de Bruin ED, Firth J. The effect of active video games on cognitive functioning in clinical and non-clinical populations: a meta-analysis of randomized controlled trials. Neurosci Biobehav Rev. 2017;78:34–43. https://doi.org/10.1016/j.neubiorev.2017.04.011.
Jokinen H, Melkas S, Ylikoski R, Pohjasvaara T, Kaste M, Erkinjuntti T, Hietanen M. Post-stroke cognitive impairment is common even after successful clinical recovery. Eur J Neurol. 2015;22(9):1288–94. https://doi.org/10.1111/ene.12743.
van Rijsbergen MW, Mark RE, de Kort PL, Sitskoorn MM. Subjective cognitive complaints after stroke: a systematic review. J Stroke Cerebrovasc Dis. 2014;23(3):408–20. https://doi.org/10.1016/j.jstrokecerebrovasdis.2013.05.003.
Gillespie DC, Bowen A, Chung CS, Cockburn J, Knapp P, Pollock A. Rehabilitation for post-stroke cognitive impairment: an overview of recommendations arising from systematic reviews of current evidence. Clin Rehabil. 2015;29(2):120–8. https://doi.org/10.1177/0269215514538982.
Douiri A, Rudd AG, Wolfe CD. Prevalence of poststroke cognitive impairment: South London Stroke Register 1995–2010. Stroke. 2013;44(1):138–45. https://doi.org/10.1161/STROKEAHA.112.670844.
Lo JW, Crawford JD, Desmond DW, Bae HJ, Lim JS, Godefroy O, et al. Long-term cognitive decline after stroke: an individual participant data meta-analysis. Stroke. 2022;53(4):1318–27. https://doi.org/10.1161/strokeaha.121.035796.
Aminov A, Rogers JM, Middleton S, Caeyenberghs K, Wilson PH. What do randomized controlled trials say about virtual rehabilitation in stroke? A systematic literature review and meta-analysis of upper-limb and cognitive outcomes. J Neuroeng Rehabil. 2018;15(1):29. https://doi.org/10.1186/s12984-018-0370-2.
Corbetta D, Imeri F, Gatti R. Rehabilitation that incorporates virtual reality is more effective than standard rehabilitation for improving walking speed, balance and mobility after stroke: a systematic review. J Physiother. 2015;61(3):117–24. https://doi.org/10.1016/j.jphys.2015.05.017.
Cugusi L, Prosperini L, Mura G. Exergaming for quality of life in persons living with chronic diseases: a systematic review and meta-analysis. PM R. 2020;13:756–80. https://doi.org/10.1002/pmrj.12444.
Oberlin LE, Waiwood AM, Cumming TB, Marsland AL, Bernhardt J, Erickson KI. Effects of physical activity on poststroke cognitive function: a meta-analysis of randomized controlled trials. Stroke. 2017;48(11):3093–100. https://doi.org/10.1161/STROKEAHA.117.017319.
Yang B, Wang SM. Meta-analysis on cognitive benefit of exercise after stroke. Complexity. 2021;2021. Artn 5569346. https://doi.org/10.1155/2021/5569346.
Arienti C, Buraschi R, Pollet J, Gobbo M. Opening the black box of ‘usual care’ and finding a black hole: a numerical systematic review on ‘usual care’ control groups in stroke rehabilitation RCTs. BMJ Evidence-Based Medicine. 2019;24(Suppl 1):A27–8. https://doi.org/10.1136/bmjebm-2019-EBMLive.52.
Negrini S, Arienti C, Kiekens C. Usual care: the big but unmanaged problem of rehabilitation evidence. Lancet. 2020;395(10221):337. https://doi.org/10.1016/S0140-6736(19)32553-X.
Acknowledgements
We would like to thank Christoph Bauer, PhD, as an independent monitor, and Lore Legrand, PhD, for her intellectual input regarding neuropsychological testing.
Funding
Open access funding provided by Swiss Federal Institute of Technology Zurich This study is supported by grants of the Innovation Pool of the University Hospital of Zurich (https://usz-foundation.com/en/usz-innovation-pool/) and the Swiss Association of Physiotherapy (physioswiss).
Author information
Authors and Affiliations
Contributions
SH developed the methodology and study protocol under the supervision of EdB and RK as well as with the advisory help of JH and MB. SH performed the manuscript writing, while all authors were involved in the scientific content and manuscript revision. The final manuscript was read and approved by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate {24}
This study was approved by the Ethics Committee of the Canton Zurich, Switzerland (project-ID: 2022–01211). Written, informed consent will be obtained from all participants.
Consent for publication {32}
We have consent by Dividat AG to use the picture of the study device (Fig. 2). Dividat AG holds the rights for this picture.
Competing interests {28}
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huber, S., Knols, R., Held, J. et al. PEMOCS: Evaluating the effects of a concept-guided, PErsonalised, MOtor-Cognitive exergame training on cognitive functions and gait in chronic Stroke—study protocol for a randomised controlled trial. Trials 25, 451 (2024). https://doi.org/10.1186/s13063-024-08283-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-024-08283-7