
Abstract
Background
Myopia is increasing in prevalence worldwide. Combination therapy showed a better effect on myopia control than monotherapy. Repeated low-level red light therapy (RLRL) therapy and defocus-incorporated multiple segment (DIMS) spectacle lenses have been reported to retard myopia progression significantly. However, whether these two therapies are better than one is still unknown. The present study aims to report the study protocol of a trial designed to evaluate the efficacy and safety of combination therapy of RLRL and DIMS versus DIMS alone for reducing the progression of myopia among Chinese school-aged children.
Methods
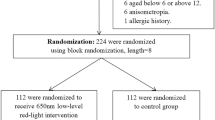
This study is a 12-month, randomized, parallel-controlled, single-center clinical trial. We will recruit children aged 8–12 years with spherical equivalence (SE) between − 0.50 D and − 6.00 D under cycloplegia in both eyes. We will recruit 66 participants with an allocation ratio of 1:1 from our hospital. Participants in the intervention group will be treated with an RLRL therapy device twice a day from Monday to Friday at home, 3 min per session, with a minimum interval of 4 h, under the supervision of their parents/guardians. They will wear DIMS spectacles for myopia correction during the day. Participants in the control group will not receive the RLRL therapy and will only wear DIMS spectacles to correct myopia. Participants from both groups will attend the hospital every 6 months.
The primary outcome is the change in axial length at 12 months. Secondary outcomes include changes in refraction under cycloplegia, optical coherence tomography (OCT), multifocal electroretinogram (mfERG), color vision, and participants’ self-reporting of adverse events at 12 months.
Discussion
This study will report the efficacy and safety outcome of the combination therapy of RLRL and DIMS versus DIMS for school-aged children with myopia in detail.
Trial registration
ChiCTR2300075398. Registered 4 September 2023. https://www.chictr.org.cn/bin/project/edit?pid=200751.
Similar content being viewed by others

Background
Myopia has become a serious worldwide public health issue [1]. The prevalence of children’s myopia has reached epidemic levels in certain areas (such as Eastern Asia) and still increasing exponentially [2]. It has been reported that the prevalence of myopia in China is as high as 90% [2]. A review predicts by 2050, approximately 50% of the world’s population will be myopic, and 10% will have high myopia [3]. High myopia is usually accompanied by the elongation of axial length, which may increase the risk of pathologic myopic retinopathy, such as macular degeneration, retinal detachment, and glaucoma, leading to blindness [4]. Therefore, it is crucial to explore treatments to control myopia.
Currently, numerous studies have focused on controlling myopia in young childhood, when it progresses rapidly [5,6,7,8]. Atropine [6, 9], orthokeratology (OK) [8], and a combination of these treatments [10, 11] have been assessed for myopia control efficacy. Topical atropine eye drops have been proven to be the most efficient in retarding myopia progression in children [6, 9]. The OK lens can reduce 40–60% of axial length (AL) elongation when compared to a single-vision spectacle [12]. The combination therapy was more effective in slowing AL elongation than the OK lens alone over a 2-year randomized clinical trial (0.29 mm vs. 0.40 mm) [13]. However, the combination treatment does not decrease the adverse effects of atropine and OK lens use. Therefore, further control treatments are still needed to evaluate myopia control combination therapies’ safety and efficacy.
Repeated low-level red light (RLRL) therapy is a novel and effective treatment for slowing the progression of children’s myopia, as demonstrated by numerous clinical trials [14,15,16,17]. Defocused incorporated multiple segment (DIMS) spectacle lenses showed significant myopia retardation through 3-year follow-up study [18]. In our clinic, we observed that combination therapy showed better control efficacy than monotherapy. However, till now, there is no RCT to prove it. The effect of the combination of these two treatments for myopia control needs to be investigated.
Therefore, we plan to conduct a randomized, parallel-controlled, and single-center trial to investigate the efficacy and safety of combination therapy of RLRL and DIMS spectacle lenses versus DIMS in slowing myopia progression in children.
Methods/design
This is a 12-month, randomized, parallel-controlled, single-center clinical trial enrolling children aged 8–12 years with myopia as subjects. The main purpose of this clinical trial is to evaluate the efficacy and safety of combination therapy of RLRL and DIMS spectacle lenses in slowing myopia progression in children. This clinical trial will be conducted at a tertiary hospital, Tianjin Medical University Eye Hospital. Ethical approval was obtained from the institutional review board of Tianjin Medical University Eye Hospital (No.2023KY-32). Written informed consent will be collected from the participants and their parents/guardians before enrollment.
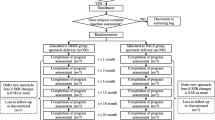
The schedule of the trial (Table 1) followed the SPIRIT (Standardized Protocol Items: Recommendations for Interventional Trials) 2013 statement (https://www.spirit-statement.org/). The study flow schedule is shown in Fig. 1.

Study flow diagram
Participants’ eligibility
Inclusion criteria
-
1.
8–12 years of age (at the time of consenting), regardless of gender;
-
2.
Myopia of a spherical equivalent (SE) between − 0.50 D and − 6.00 D under cycloplegia in both eyes, with astigmatism < 2.00 D, binocular anisometropia < 1.50 D;
-
3.
Both best-corrected distance visual acuity and best-corrected near visual acuity of at least 1.0;
-
4.
Able to actively cooperate with and adhere to treatment requirements;
-
5.
The parents/guardians and the child provide written informed consent.
Exclusion criteria
-
1.
History of photosensitivity, ocular or systematic diseases;
-
2.
Amblyopia;
-
3.
Children with the immune system and systemic diseases or mental disorders, inability to communicate normally;
-
4.
Previously received other treatments to control the development of myopia, such as atropine or orthokeratology lens within 3 months;
-
5.
Other situations that the investigators may consider unsuitable for enrolling in the study.
Recruitment
To better recruit participants, we will create a poster at the front of the optometry center of our hospital. Children diagnosed with myopia at the hospital’s outpatient clinic will be invited. The potentially eligible participants and their parents/guardians will be contacted by the clinical research coordinator (CRC). The CRC will provide a detailed introduction to the study. Interested participants and their parents/guardians will sign the informed consent. Afterward, they will be invited to undergo a screening examination. Participants will be eligible to participate in the clinical trial if they meet the inclusion criteria, and they will be excluded if they meet any of the exclusion criteria.
Randomization and masking
A statistician is responsible for the randomization of the intervention or control group. Simple randomization will be implemented. The subject file numbers (1–100) in a spreadsheet of Excel (Microsoft Office) and a column of random numbers for the group allocation will be conducted by the statistician. That means that subjects will be assigned to either group by following a random software sequence generated from Excel. The allocation sequence will be sealed in an opaque envelope. Due to the nature of the intervention, the principal investigator and participants will be aware of the study allocation. While the outcome assessors, including technicians and optometrists, will be masked to the treatment allocation.
Intervention
Eligible participants will be randomly assigned to the intervention group or control group, at a 1:1 ratio. Participants in the intervention group will receive RLRL treatment twice a day from Monday to Friday, with a treatment duration of 3 min and a minimum interval of 4 h. They will wear DIMS spectacle lenses for myopia correction. Subjects in the control group will only wear DIMS spectacles for myopia correction during the day.
The brand of the red light machine will be Eyerising [14], a semiconductor laser product in Suzhou, China. The wavelength is 650 ± 10 nm, the illuminance level is approximately 1600 lx, and the power is 2.00 ± 0.50 mW. The light power entering a 4-mm pupil is supposed to be approximately 0.29 mW. According to American National Standards Institute Z136.1 standards from 2014, this laser is classified as class 1; its upper maximum power is 0.4 mW and is safe for direct ocular exposure. Several randomized clinical trials using the Eyerising RLRL laser have reported no treatment-associated severe adverse events. The machine had a central control system. Each participant in the intervention group will have a unique corresponding personal account and password; the machine will be initiated for treatment after logging into the system.
The study will evaluate AL, cycloplegic auto-refraction, OCT, multifocal electroretinogram (mfERG), color vision, visual acuity (VA) of the two groups of subjects, and participants’ self-reporting of adverse events at 6 months and 12 months after enrollment.
Primary outcome
Figure 1 displays the timeline for data collection and visits. The assessments will be conducted following a predetermined order. Comprehensive eye exams will be conducted by an ophthalmologist.
The primary outcome is to determine whether the combination therapy of RLRL and DIMS spectacle is superior to DIMS therapy, in terms of preventing myopia progression, by evaluating the AL (mm) elongation by Lenstar 900 (Haag-streit, USA) in these two groups of participants. The measurement will be conducted three times and the mean values will be recorded. For the primary outcome analysis, the myopia progression over 1 year would be determined by the difference in AL between the baseline and 12-month visit.
Secondary outcome
The secondary outcome measurements include the spherical equivalent (D) under cycloplegia by autorefractor, the sub-fovea thickness of choroid by OCT, mfERG, and color vision as well as participants’ self-reporting of adverse events at each visit. The differences between the baseline and 6-month and12-month visits of the two groups of subjects will be recorded. The change of visual acuity, spherical equivalent after cycloplegia, choroid thickness under the macular, mfERG between the baseline, and 12-month visit will be calculated as the secondary outcome.
Visual acuity
An ETDRS chart (Precision Vision, Villa Park, Illinois, USA) with standard illumination will be used to measure visual acuity at a distance of four meters. Uncorrected and best-corrected visual acuity will be measured.
AL
Before cycloplegia, Lenstar 900 (Haag-Streit, USA) will be used to measure the AL of both eyes. Three measurements will be taken for each eye. The AL measurement will be based on the mean of these 3 values if the desired precision (< 0.05 mm) is achieved.
Cycloplegia
Cycloplegia will be induced with three drops of 1% cyclopentolate administered at the 0, 5th, and 20th minute to each eye. The light reflex and pupil dilation will be checked after an additional 15 min. Full cycloplegia will be justified if the pupil dilates to 6 mm or greater and the light reflex is absent.
Cycloplegic autorefraction
Cycloplegic autorefraction will be conducted using an autorefractor (KR-8800, Topcon, Tokyo, Japan). The same autorefractor will be used throughout the study. It will be calibrated before each examination session.
Optical coherence tomography
Swept Source-OCT (DRI OCT Triton, Topcon, Tokyo, Japan) will be used to capture the macular scan. Shooting mode: 12 mm radial scan mode.
Color vision
Farnsworth Munsell 100 Hue Test (FM-100) will be used to test the changes in color vision.
Multifocal electroretinogram (mfERG)
mf-ERG will be taped using RETI-scan (Roland Consult, Germany). We will obtain P1 response density (nV/deg2), P1 amplitude (μV), P1 implicit time (ms), N1 amplitude (μV), and N1 implicit time (ms) values from central three rings.
Sample size calculation
Previous studies have investigated the efficacy of RLRL in controlling myopia in children. Jiang et al.’s multicenter randomized controlled trial [14] reported that the AL elongation in the RLRL group was 0.13 mm, while it was 0.38 mm in the single-vision spectacle group. Carly [15] showed the AL elongation was 0.10 mm in the DIMS group, compared to 0.30 mm in the single-vision spectacle group. However, our study is the first to specifically investigate the efficacy of RLRL therapy combined with DIMS in controlling myopia in children.
The parameters used for the sample size calculation included a significance level of 0.05, 95% confidence interval, 80% power, and 1:1 allocation. Based on Jiang et al.’s [15], Carly et al.’s study [19], and other published data, the AL elongation in the DIMS group will be 0.11 ± 0.02 mm/year, to detect a 20% difference (0.02 mm with 0.02 mm of SD) in AL between two groups. The rate of participant loss to follow-up over 1 year is estimated to be 20%. Calculations will be two-sided and performed using the software, NCSS PASS 11. A total sample of 66 eligible children is required in the trial, with 33 participants planned to be assigned to each of the two groups.
Data collection, management, and monitoring
The principal investigator will provide training to the site staff on trial processes and procedures, including the completion of the clinical research forms (CRF) and data collection through investigator meetings and site initiation visits. A paper-based CRF will be used throughout examinations. All data for each participant will be collected and recorded in the CRF for the study. Instrument-printed results will be appended to the CRF when available. Participant identification on the CRF will be through their unique trial identifier, allocated at the time of recruitment.
Study data will be entered into the electronic data capture (EDC) system. During submission, the EDC will automatically check for missing values. After the examinations are completed, all study documentation, including participant medical records and data, will be stored in a research facility locker to preserve participant privacy and ensure access for data review and audit.
The primary outcome of the current study is the change in AL to assess the efficacy of the combination therapy of RLRL and DIMS. At each visit, masked investigators analyzed AL and other measures. The RLRL therapy will be suspended if any odd symptoms are detected, and the investigator will study and report the signs and symptoms as soon as possible. Each negative incident will be documented in the CRF. Participants who are unable to be tracked during the study are labeled dropouts. Participants may withdraw from the study if they experience excessive side effects or if the treatment has a poor effect. Furthermore, participants can leave the research at any moment for any reason. To ensure the participants’ safety, the investigators may potentially withdraw them from the study.
Participant medical records can only be assessed by the investigators and monitors. The participants’ data and information would not be used for any other study purposes. The identities of the participants will be masked to protect their privacy throughout data processing. There will be no interim analysis. The Department of Clinical Research in Tianjin Medical University Eye Hospital oversees the data management. Regular data monitoring will be undertaken following the sponsor’s standard operating procedures. The Tianjin Medical University Eye Hospital’s Good Clinical Practice (GCP) office will conduct the audits. Every 6 months, they will examine the adherence to the protocol and study procedures. The investigators will not be involved in the audit process.
Statistical analysis
The primary analysis will be performed under the intention-to-treat (ITT) principle. All subjects who complete the protocol and those who fail to complete the protocol but do not withdraw from the study will be included in the analysis dataset. For subjects who withdraw from the study, valid data collected before withdrawal will be included in the analysis dataset.
Baseline characteristics and follow-up visits results will be summarized as mean and standard deviation (SD), median and interquartile range (IQR), or numbers and proportions (%) as appropriate, depending on the scale of the measurement and distribution. Endpoints from both eyes will be pooled in combined analysis using generalized estimating equations (GEE) to account for the correlation between eyes within the participant. Differences in the AL and other continuous secondary outcomes between the intervention group and control group will also be tested for significance using an independent t-test. Analysis of covariance will be performed to adjust for baseline characteristics and other covariates. The chi-square test will be used to explore the distribution of categorical secondary outcomes between two groups.
An unstructured covariance matrix will be used along with a restricted maximum likelihood method to estimate treatment effects. The model will include group, visit, and group-by-visit interaction as fixed effects, along with baseline age, sex, and baseline value as covariates. Subjects will be included as a random factor. The estimated mean treatment differences, corresponding 95% confidence intervals (CIs), and two-sided p values will be calculated. To assess the impact of missing data on the primary outcome, sensitivity analyses will be conducted by imputing extreme values (lowest and highest). A detailed statistical analysis plan will be completed before the final analysis is started.
Protocol compliance
A protocol deviation is defined as an incident that deviates from the normal expectation of a particular part of the trial process. Any deviations from the protocol will be fully documented on the protocol deviation form in the CRF and reported to the steering committee and ethics committee.
The Eyerising red light machine is linked to a centralized system through the Internet, allowing treatment data and compliance to be monitored centrally. One staff member will keep track of treatment compliance and usage time information, which will be collected once a week. To improve compliance, staff will notify parents/guardians of children who use the machine less than 8 times a week. Participants will be reminded by phone or email 1 week before their clinical visit; then, appointments will be made in advance according to their availability. In the event of non-compliance, such as absence, participants will be contacted by phone or WeChat to confirm their willingness to continue or terminate the study.
For very rare cases such as eye irritation, skin itching, and eye pain, participants will be advised to use the designated eye drops. Adverse events (AE) will be continuously monitored. In the case of an AE, participants will be informed about the severity of the event, and the principal investigator (PI) will decide if participants can continue further. If participants consent and agree, they will be reminded daily regarding the administration of eye drops. Any queries regarding the study will be answered by trained clinical staff at Tianjin Medical University Eye Hospital, China.
For very rare cases such as intolerable eye glare, and VA decreased, the Tianjin Medical University Eye Hospital Ethics Board will be notified; experimental treatments will be discontinued promptly, and professional ophthalmologists will provide treatment.
Post-trial care
It is recommended that children with myopia should be re-exanimated at least half a year to determine whether the defocus spectacles need to be replaced. Visual acuity, axial length, refraction, OCT, color vision, and other examinations can continue to be conducted in our outpatient clinics at our hospital.
Protocol amendments
Any protocol changes that may have an impact on the study’s conduct, such as changes in the study objectives, study design, sample sizes, or study procedures, require a formal amendment to the protocol. These amendments must be approved by the institutional review board of our hospital before implementation. Minor protocol adjustments that do not affect the study will be agreed upon and documented in a memorandum by the primary investigator.
Discussion
The present trial is designed to investigate the efficacy and safety of combination therapy of RLRL and DIMS spectacle lenses in slowing myopia progression in children. To the best of our knowledge, the present study is the first randomized, parallel-controlled, and single-center trial that obtains detailed data on the combination of RLRL therapy and defocus spectacle lenses.
The underlying mechanism of retarding the myopia progression of RLRL therapy was improving the hypoxia environment by increasing the blood flow to the choroid, which influenced sclera remodeling additionally [20,21,22]. Additionally, red light can also improve human photoreceptor function by changing thresholds for Tritan and protein function, which may modulate the metabolism of the fundus [23]. The DIMS spectacle lenses slow myopia progression by providing a relative positive power (+ 3.50 D) which imposes myopic defocus and inhibits eye growth. These two mechanisms are independent of each other. In theory, the combination of these two control methods can play a synergistic role in myopia control.
In conclusion, RLRL therapy and DIMS have gained great interest in the retardation of myopia progression worldwide for its significant efficacy. The present study aims to investigate the efficacy and safety of the combination of RLRL therapy and DIMS spectacle lenses in slowing myopia progression in children. These results would broaden the understanding of myopia progression control efficacy.
Trial status
At the time of submission, recruitment has not begun. The protocol version is 1.0, dated 30 August 2023. The first patient will be recruited on January 1, 2024; the recruitment will be completed by December 31, 2024.
Availability of data and materials
There is no plan for public access to the dataset of this trial now. The principal investigator will supervise the management of the final trial dataset with the statistician. Personal identification information will be anonymized in the final trial dataset.
Abbreviations
- AL:
-
Axial length
- SE:
-
Spherical equivalence
- RLRL:
-
Repeated low-level red light
- DIMS:
-
Defocus-incorporated multiple segments
- OCT:
-
Optical coherence tomography
- mfERG:
-
Multifocal electroretinogram
- OK:
-
Orthokeratology
References
Resnikoff S, Jonas JB, Friedman D, He M, Jong M, Nichols JJ, et al. Myopia - a 21st century public health issue. Invest Ophthalmol Vis Sci. 2019;60:Mi–Mii.
Jan C, Li L, Keay L, Stafford RS, Congdon N, Morgan I. Prevention of myopia, China. Bull World Health Organ. 2020;98:435–7.
Holden BA, Fricke TR, Wilson DA, Jong M, Naidoo KS, Sankaridurg P, et al. Global prevalence of myopia and high myopia and temporal trends from 2000 through 2050. Ophthalmology. 2016;123:1036–42.
Sankaridurg P, Tahhan N, Kandel H, Naduvilath T, Zou H, Frick KD, et al. IMI impact of myopia. Invest Ophthalmol Vis Sci. 2021;62:2.
Bao J, Huang Y, Li X, Yang A, Zhou F, Wu J, et al. Spectacle lenses with aspherical lenslets for myopia control vs single-vision spectacle lenses: a randomized clinical trial. JAMA Ophthalmol. 2022;140:472–8.
Ha A, Kim SJ, Shim SR, Kim YK, Jung JH. Efficacy and safety of 8 atropine concentrations for myopia control in children: a network meta-analysis. Ophthalmology. 2022;129:322–33.
Lawrenson JG, Shah R, Huntjens B, Downie LE, Virgili G, Dhakal R, et al. Interventions for myopia control in children: a living systematic review and network meta-analysis. Cochrane Database Syst Rev. 2023;2:CD014758.
Cho P, Tan Q. Myopia and orthokeratology for myopia control. Clin Exp Optom. 2019;102:364–77.
Gong Q, Janowski M, Luo M, Wei H, Chen B, Yang G, et al. Efficacy and adverse effects of atropine in childhood myopia: a meta-analysis. JAMA Ophthalmol. 2017;135:624–30.
Yuan Y, Zhu C, Liu M, Zhou Y, Yang X, Zheng B, et al. Efficacy of combined orthokeratology and 0.01% atropine for myopia control: the study protocol for a randomized, controlled, double-blind, and multicenter trial. Trials. 2021;22:863.
Wang S, Wang J, Wang N. Combined orthokeratology with atropine for children with myopia: a meta-analysis. Ophthalmic Res. 2021;64:723–31.
Zhu M-J, Feng H-Y, He X-G, Zou H-D, Zhu J-F. The control effect of orthokeratology on axial length elongation in Chinese children with myopia. BMC Ophthalmol. 2014;14:141.
Kinoshita N, Konno Y, Hamada N, Kanda Y, Shimmura-Tomita M, Kaburaki T, et al. Efficacy of combined orthokeratology and 0.01% atropine solution for slowing axial elongation in children with myopia: a 2-year randomised trial. Sci Rep. 2020;10:12750.
Jiang Y, Zhu Z, Tan X, Kong X, Zhong H, Zhang J, et al. Effect of repeated low-level red-light therapy for myopia control in children: a multicenter randomized controlled trial. Ophthalmology. 2022;129:509–19.
Chen Y, Xiong R, Chen X, Zhang J, Bulloch G, Lin X, et al. Efficacy comparison of repeated low-level red light and low-dose atropine for myopia control: a randomized controlled trial. Transl Vis Sci Technol. 2022;11:33.
Tang J, Liao Y, Yan N, Dereje SB, Wang J, Luo Y, et al. Efficacy of repeated low-level red-light therapy for slowing the progression of childhood myopia: a systematic review and meta-analysis. Am J Ophthalmol. 2023;252:153–63.
Dong J, Zhu Z, Xu H, He M. Myopia control effect of repeated low-level red-light therapy in Chinese children: a randomized, double-blind, controlled clinical trial. Ophthalmology. 2023;130:198–204.
Lam CS, Tang WC, Lee PH, Zhang HY, Qi H, Hasegawa K, et al. Myopia control effect of defocus incorporated multiple segments (DIMS) spectacle lens in Chinese children: results of a 3-year follow-up study. Br J Ophthalmol. 2022;106:1110–4.
Lam CSY, Tang WC, Tse DY-Y, Lee RPK, Chun RKM, Hasegawa K, et al. Defocus incorporated multiple segments (DIMS) spectacle lenses slow myopia progression: a 2-year randomised clinical trial. Br J Ophthalmol. 2020;104:363–8.
Wu H, Chen W, Zhao F, Zhou Q, Reinach PS, Deng L, et al. Scleral hypoxia is a target for myopia control. Proc Natl Acad Sci U S A. 2018;115:E7091–100.
Zhao F, Zhang D, Zhou Q, Zhao F, He M, Yang Z, et al. Scleral HIF-1α is a prominent regulatory candidate for genetic and environmental interactions in human myopia pathogenesis. EBioMedicine. 2020;57:102878.
Zhou X, Zhang S, Yang F, Yang Y, Huang Q, Huang C, et al. Decreased choroidal blood perfusion induces myopia in guinea pigs. Invest Ophthalmol Vis Sci. 2021;62:30.
Shinhmar H, Grewal M, Sivaprasad S, Hogg C, Chong V, Neveu M, et al. Optically improved mitochondrial function redeems aged human visual decline. J Gerontol A Biol Sci Med Sci. 2020;75:e49-52.
Acknowledgements
The repeated low-level red light instrument was sponsored by Eyerising, China.
Dissemination plans
The study’s findings will be shared regardless of the effect’s direction. Publications in high-impact, open-access medical journals and talks at national and international medical conferences will serve this purpose.
Funding
This work was supported by funds from Chunhui Project Foundation of the Education Department of China (HZKY20220587), Tianjin Health Technology Research Project (TJWJ2022MS014), and Tianjin Municipal Education Commission Scientific Research Program (Mental Health Education) (2022ZDGX20). These funders had no role in the design of the study, collection, analysis, and interpretation of the data and the writing and decision for publication of the manuscript.
Author information
Authors and Affiliations
Contributions
Hongmei Zhang and Ruihua Wei are the chief investigators; they conceived the study and led the proposal and protocol development. Desheng Song is the lead trial methodologist who is responsible for the analysis and interpretation of the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Before the trial was initiated, this trial received ethical approval from the Institutional Review Board of Tianjin Medical University Eye Hospital (approval number: 2023KY-32).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, H., Song, D. & Wei, R. The efficacy and safety of combination therapy of repeated low-level red light and defocus-incorporated multiple segments spectacle lenses for myopia control in children: the study protocol for a 12-month, randomized, parallel-controlled, and single-center clinical trial. Trials 25, 514 (2024). https://doi.org/10.1186/s13063-024-08210-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-024-08210-w