
Abstract
Background
Cancer-related fatigue (CRF) is still undertreated in most patients, as evidence for pharmacological treatments is limited and conflicting. Also, the efficacy of the pharmacological agents relative to each other is still unclear. Therefore, medications that may potentially contribute to improving CRF will be investigated in this head-to-head trial. Our main objective is to compare the efficacy of methylphenidate vs. bupropion vs. ginseng vs. amantadine vs. placebo in patients with advanced cancer.
Methods
The 5-EPIFAT study is a 5-arm, randomized, multi-blind, placebo-controlled, multicenter trial that will use a parallel-group design with an equal allocation ratio comparing the efficacy and safety of four medications (Methylphenidate vs. Bupropion vs. Ginseng vs. Amantadine) versus placebo for management of CRF. We will recruit 255 adult patients with advanced cancer who experience fatigue intensity ≥ 4 based on a 0–10 scale. The study period includes a 4-week intervention and a 4-week follow-up with repeated measurements over time. The primary outcome is the cancer-related fatigue level over time, which will be measured by the functional assessment of chronic illness therapy-fatigue (FACIT-F) scale. To evaluate safety, the secondary outcome is the symptomatic adverse events, which will be assessed using the Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events in cancer clinical trials (PRO-CTCAE). Also, a subgroup analysis based on a decision tree-based machine learning algorithm will be employed for the clinical prediction of different agents in homogeneous subgroups.
Discussion
The findings of the 5-EPIFAT trial could be helpful to guide clinical decision-making, personalization treatment approach, design of future trials, as well as the development of CRF management guidelines.
Trial registration
IRCT.ir IRCT20150302021307N6. Registered on 13 May 2023.
Similar content being viewed by others

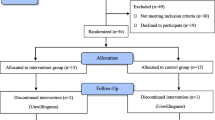

Introduction
According to the clinical practice guideline of the National Comprehensive Cancer Network (NCCN), cancer-related fatigue (CRF) is defined as “a distressing, persistent, subjective sense of physical, emotional, and/or cognitive tiredness or exhaustion related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning” [1]. CRF affects 60 to 90% of cancer patients [2,3,4] and is the most common and debilitating symptom reported in people with advanced cancer [5]. It has been identified as a high-priority research area in the oncology setting (among the top 5) by the National Cancer Institute [6].
Although CRF is a complex, multidimensional and multifactorial problem that significantly affects patients’ quality of life and survival [7,8,9,10,11], it is still undertreated in most patients, mainly due to lack of any effective treatment [12]. Hence, CRF management is an important priority for patients and a serious challenge for clinicians in palliative cancer care [11, 13]. Although there are clinical guidelines for the management of CRF, it is still unclear which treatment method is most effective [14]. In general, treatment options for improving CRF are limited [15]. It has been shown that some non-pharmacological approaches (such as physical activity, massage therapy, etc.) are effective in controlling CRF. However, many patients do not have the desire or the ability to be committed to such treatments on a regular basis, and this is particularly true with patients with advanced cancer [16,17,18]. Therefore, pharmacological interventions can be considered a helpful approach, but evidence in this regard is limited and recommendations are often contradictory [19,20,21]. In fact, pharmacological strategies for the treatment of CRF have not yet been established, nor is there any consensus on pharmacological management [9, 19]. As a result, more research on pharmacological treatments of CRF is needed, especially in patients with advanced cancer [20]. Past trials have focused only on one pharmacological agent, and head-to-head clinical trials to compare the efficacy of different medications in CRF treatment are rare [15, 20, 22]. A Cochrane systematic review on pharmacological therapies for fatigue associated with palliative care strongly recommends that future trials should compare one anti-fatigue drug against another, along with a placebo [15].
Methylphenidate, bupropion, and ginseng have shown positive effects on improving CRF in some past trials [23,24,25,26,27,28,29,30,31]. Although these medications have been tested for years, their efficacy is still unclear, and there is no consensus among clinicians regarding their effectiveness in CRF treatment [15, 19, 20, 31]. Also, the above-cited Cochrane review study reports that amantadine appears to be promising in improving fatigue associated with multiple sclerosis, but whether this drug can also relieve fatigue in cancer patients has not been shown and should be investigated [15]. Therefore, the medications that will be tested in this trial are those that may potentially be effective in improving CRF.
To the best of our knowledge, no prior trial of pharmacological therapies for CRF has been conducted to compare the efficacy of different medications. Hence, we designed a head-to-head trial using subgroup analysis based on a decision tree-based machine learning algorithm for clinical prediction. The findings of the 5-EPIFAT trial could be helpful to guide clinical decision-making, personalize treatment approach, design future trials, and develop CRF management guidelines. The objectives of this study include [1] comparison of the efficacy of different pharmacological agents with placebo in managing CRF; [2] comparison of the efficacy of pharmacological agents with each other in managing CRF; [3] comparison of the efficacy of pharmacological agents in homogeneous subgroups of patients with CRF; and [4] comparison of the safety of treatment with pharmaceutical agents versus placebo and each other.
Method
This 5-EPIFAT trial protocol is according to the Standard Protocol Items: Recommendations for Interventional Trial (SPIRIT) 2013 statement. All items from the WHO trial registry data set are found within the protocol accessible at https://irct.behdasht.gov.ir/trial/69613.
Study design
This 5-EPIFAT study is a 5-arm, longitudinal, randomized, multi-blind, placebo-controlled, multicenter, superiority trial that will use a parallel-group design with an equal allocation ratio comparing the efficacy of four palliative medications (methylphenidate vs. bupropion vs. ginseng vs. amantadine) versus placebo in management of CRF among adult advanced cancer patients on active treatment. The study period will be 8 weeks (a 4-week intervention and a 4-week post-intervention follow-up) with repeated measurements over time.
Settings
This trial will be conducted in 5 academic sites (3 hospitals and 2 outpatient oncology clinics) in 3 provinces of Iran (Khuzestan located in the southwest of Iran, Tehran located in the north-central Iran, and Yazd located in the center of Iran). More details of the study sites are available at https://irct.behdasht.gov.ir/trial/69613.
Participants
A convenience sampling method will be used in this trial. Adult patients aged 18 years or older with advanced cancer (which is unlikely to be cured with treatment) who report moderate to severe CRF levels and are of different ethnicities and socio-economic status suffering from different cancer types will be included in the study. Eligibility criteria were set to maximize the generalizability of findings and prioritize participant safety. The eligibility criteria and withdrawal criteria are listed in Table 1.
Intervention
In this study, four groups will receive medicine, and one group will be given placebo for 4 weeks. All medications will be prepared in opaque and identical (in terms of size, shape, and color) capsules by the study pharmacist, and then they will be packed in sufficient quantities in opaque and identical cans. In all groups, the participants will start taking one capsule in the morning within the first week. After that, in case the participants tolerate the medication well and do not report significant adverse events (AEs) attributed to the medication, they will take two capsules daily (morning and evening) in the second and third weeks. Afterwards, in the fourth week, one capsule will be given to the participants in the morning in order to reduce the possibility of withdrawal symptoms. Figure 1 shows the guiding algorithm of nurses who will contact participants regarding medication dosage adjustment. All AEs will be handled appropriately. In case of severe or serious AEs, the medication will be discontinued, and the participant will be withdrawn from further treatment. The criteria for discontinuing allocated intervention are listed in Table 1.

Algorithm guiding nurses who contact patients regarding dosage adjustment
The study arms include the following: Arm 1, study participants will receive oral methylphenidate for 4 weeks. Each capsule contains 10 mg of methylphenidate. Methylphenidate will be started in the first week with a dose of 10 mg daily (in the morning). If the patient tolerates it well, the dose of 10 mg twice a day (morning and evening) will be continued in the second and third weeks. Then, in the fourth week, the dose is again reduced to 10 mg daily (in the morning) to reduce the possibility of withdrawal symptoms; Arm 2, study participants will receive oral bupropion sustained-release for 4 weeks. Each capsule contains 150 mg of bupropion. Bupropion will be started in the first week with a dose of 150 mg daily (in the morning). If the patient tolerates it well, the dose of 150 mg twice a day (morning and evening) will be continued in the second and third weeks. Then, in the fourth week, the dose is again reduced to 150 mg daily (in the morning) to reduce the possibility of withdrawal symptoms; Arm 3, study participants will receive oral amantadine for 4 weeks. Each capsule contains 100 mg of amantadine. Amantadine will be started in the first week with a dose of 100 mg daily (in the morning). If the patient tolerates it well, the dose of 100 mg twice a day (morning and evening) will be continued in the second and third weeks. Then, in the fourth week, the dose is again reduced to 100 mg daily (in the morning) to reduce the possibility of withdrawal symptoms; Arm 4, study participants will receive oral Panax ginseng for 4 weeks. Each capsule contains 500 mg of ginseng. Ginseng will be started in the first week with a dose of 500 mg daily (in the morning). If the patient tolerates it well, the dose of 500 mg twice a day (morning and evening) will be continued in the second and third weeks. Then, in the fourth week, the dose returns to 500 mg daily (in the morning); Arm 5, study participants will receive an oral placebo. The placebo will be started in the first week with one capsule daily (in the morning). After that, two capsules daily (morning and evening) will be given to patients in the second and third weeks. Then, in the fourth week, the patients will again receive one capsule (in the morning). All patients will receive usual care and treatment regimens during the study.
Pharmacological agents
Although methylphenidate, bupropion, and ginseng have been investigated in various studies for the management of CRF, the findings are contradictory, and their clinical effectiveness is still in doubt and requires further research [12, 15, 19, 20]. Some studies have also shown that amantadine has promising effectiveness in improving fatigue associated with chronic diseases, but this needs to be investigated in cancer patients as well [15]. Therefore, the medications that will be evaluated in this 5-EPIFAT trial are those that we assume will be effective in improving CRF. Also, due to the uncertain effectiveness of the medications in this trial, we also included a placebo group. Comparing the effectiveness of these agents against each other requires showing their superiority over the placebo in the first place, and then over each other in the second place. In Table 2, a brief review of the medications selected for the 5-EPIFAT study is given.
Outcomes and data collection
The tools will be given to patients in the form of a diary. Demographic and clinical data will be collected using a researcher-developed checklist at baseline (before random allocation). To evaluate the efficacy, the main outcome is the cancer-related fatigue level over time, which will be measured using the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) self-reported scale version 4 [38]. CRF level will be measured at baseline, weekly during the 4-week intervention period, and at weeks 6 and 8 as a follow-up in order to evaluate the potential durability of the effect of each medication on improving fatigue [The intervention termination is the 4th week, and follow-up termination is the 8th week]. We will compare the trend of changes in mean fatigue level over time (from baseline to the eighth week) between the trial arms. FACIT-F is a multidimensional and validated patient-reported outcome that includes 13 questions with a recall period of 7 days. Questions are scored based on a Likert scale from 0 to 4 (from “not at all” to “very much”). To score the FACIT-F, all questions are summed up to create a single fatigue score ranging from 0 to 52, and negative items are reversely scored. Higher scores represent better results (i.e., less fatigue). The FACIT-F has been widely applied in various cancer populations, and results demonstrate satisfactorily psychometric properties [38, 39]. We obtained the license for using FACIT in this study. Considering that the intensity of fatigue varies at different times of the day, we will ask all participants to complete the questionnaire at a certain time of the day (i.e., between 2 and 6 pm).
To evaluate safety, the secondary outcome is the symptomatic adverse events, which will be assessed using the Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events in cancer clinical trials (PRO-CTCAE) [40]. PRO-CTCAE is a validated tool that measures the frequency, severity, or interference of symptoms experienced by patients participating in cancer clinical trials. This tool is designed to assess symptomatic toxicities experienced by patients. The frequency, intensity, interference, degree, and presence of 78 symptomatic side effects are evaluated in this tool. The recall period of this tool is 1 week, which also matches well with its daily report, so it will be completed weekly by the patients [40, 41]. PRO-CTCAE will be completed at baseline, and weekly during the 4-week intervention period. We will compare the frequency of adverse events between the trial arms from week 1 to week 4. Safety analysis will be performed for all patients who receive at least one dose of the medication. The pharmacological agents studied in this trial have known safety profiles and are already approved and used for other conditions. Therefore, we do not need laboratory indicators and clinical examinations to diagnose side effects, and we will simply collect patient-reported adverse events.
Trial handling
In this study, blinded nurses will make phone calls to all patients to remind them to complete the diary and adjust the drug dose in weeks 1 to 4 (with a weekly schedule) and weeks 6 and 8 (or more if necessary). Also, during the second and fourth weeks, the patients will be visited in-person only for the purpose of checking how they complete the diary and take the medications and ensuring their safety (no data collection will be done during the in-person visits). Nurses are required to determine the reasons for participants who discontinue or deviate from intervention protocols and record them in the checklist. At the end of the eighth week, the data diaries will be collected and imported into the statistical software. Also, the data entry process will be periodically double-checked by the statistician to prevent registration errors. The trial implementation process is shown step by step in Fig. 2.

The step by step process of 5-EPIFAT trial
Recruitment
Eligible patients will be continuously recruited until the target sample size is reached. Oncologists and oncology nurses will screen patients at 5 sites, and refer the patients who meet the initial criteria. Then, the patients will be subjected to detailed evaluation (including demographic and clinical characteristics, history, and medical record) by trained study staff for final confirmation of eligibility, obtain written informed consent, and collect baseline data.
Assignment and blinding
A permuted block randomization allocation algorithm with an equal allocation ratio (1:1:1:1:1) will be generated by the statistician before enrollment using the package blockrand [42]. The statistician will then send the randomization list to the study pharmacist and randomization provider. The pharmacist will not have any role whatsoever in data collection or analysis. Neither will the randomization provider play a role in the rest of the trial. Each participant giving written consent to participate in the study will be identified with a unique two-part hash code. This code is a combination of the unique number of each center and the patient’s number. In each center, the first recruited patient will be assigned number 1, and consecutive numbers will be assigned to the next participants. At each site, a blinded research assistant will send a randomization request to the randomization provider, who will immediately receive the order via email, assign the participants to one of the 5 study groups (identified by a code), and provide them with the medication package (these assistants will not be involved in the rest of the trial). Other personnel of the research team will be blind until the end of data analysis. Also, for the purpose of blinding, the medications will be placed by the study pharmacist in completely identical opaque capsules in similar opaque boxes differently coded with preprinted medication code labels (a specific 3-letter code will be considered for each of the five medications used in the study). The medication codes and randomization list will be kept by the study pharmacist until the trial is closed (only in case of a serious adverse event, the patient’s medication code will be broken). The exact details of blinding are listed in Table 3.
Participant timeline
Table 4 shows the details of the participant timeline (SPIRIT diagram).
Sample size
Using the package WebPower in R software, a sample size of 205 subjects was calculated based on comparing study groups assuming a repeated measures ANOVA with a 5% level of significance, 80% power, an effect size equal to 0.57 [31], the number of groups (5-arm), and the number of repeated measures (7 time points) [43]. Finally, assuming a drop-out rate of 20%, the sample size was set to be 255 subjects who were supposed to be randomly assigned in each arm (n = 51 each).
Statistical methods
Analysis plan
Descriptive statistics of qualitative and quantitative variables will be shown as frequency (percentage) and mean ± standard deviation (SD), respectively. Analysis of variance (ANOVA) and Kruskal–Wallis test will be used to assess the difference between quantitative variables across study groups (including age and body mass index). Chi-square test and trend chi-square test will be used to test the difference of frequency distribution of qualitative variables across study groups (including sex, ethnicity, type of cancer, and type of treatment). The normality of data will be evaluated using the Shapiro–Wilk test. Two-tailed p-values less than 0·05 will be regarded as statistically significant, and data analysis will be done using free statistical software R. The interpretation of the results will be based on intention-to-treat (ITT) analysis.
Primary outcome (fatigue level)
If there is a significant interaction effect between the study groups and the time factor to assess the changes in the primary outcome variable over time (from the baseline to the eighth week), then the study groups will be compared within each time point using ANOVA based on the Bonferroni correction to adjust the type I error. Since the primary outcome variable does not have a normal distribution, the changes over time points will be assessed using nonparametric repeated measure analysis in the nparLD package [44]. In addition, if there were not the same number of measurements for each participant over time, we would use the linear mixed-effect model to compare the treatment groups using the lme4 package because this model can deal with any degree of imbalance in the longitudinal data [45]. The waterfall and spider plots will be used to represent each individual patient’s response and the individual changes in response over time relative to baseline using the waterfall and ggplot2 packages, respectively [46,47,48]. All time points will be used for analysis.
Secondary outcome (adverse events)
The assessment of safety will be based on the frequency of adverse events from week 1 to week 4 (number [percentage] of participants reporting each type of event in each subgroup of adverse events), and comparison between groups will be made using chi-square test.
Subgroup analysis
The linear mixed-effect model tree (LMEMT) using the glmertree package will be employed to predict the efficacy of medications in different subgroups. This method is a modern flexible tree algorithm for subgroups in longitudinal data [49]. Tree-based algorithms are supervised non-parametric algorithms and are one of the most popular machine learning tools for modeling and clinical prediction. These methods have some notable advantages over parametric models such as easy interpretability due to the graphical display of the results, which is one of the attractive properties of tree models, without the need to determine assumptions about the functional form of the data, deal with nonlinear relationships and high-order interactions, and extract homogeneous subgroups of observations. As a powerful method of machine learning, tree algorithms can identify homogeneous subgroups of patients who need different treatment strategies, and they can also guide the clinician in decision-making [50].
Missing data
Longitudinal data collected from the same subjects over time is frequently used in clinical trials. In these studies, the researchers often encounter major challenges including missing values. To alleviate this problem, we will use the Copy Mean method to impute monotone/dropout and non-monotone/intermittent missing data of longitudinal quantitative response variables [51, 52]. To understand the computational strategy related to this method, a time-dependent variable is recorded at t time points for each subject. In this case, a trajectory for cluster i is defined as the sequence yi. = (yi1, yi2, …, yit). Let yik show a missing value for cluster i at a specific time point k. yik is non-monotone missing if time points as a < k < b exist and yia and yib are not missing. yik is monotone missing if for all time points h > k, yih is missing. The copy-mean method is based on two steps for the imputation of missing observations. First, the missing values are imputed using the LOCF method to provide an initial approximation of these values (In the LOCF method, yik is estimated by the last observed value of the trajectory of interest). Then the mean trajectory of the population is used to refine the initial approximation in the previous step. Let,
(\({\overline{y} }_{.1}\), …, \({\overline{y} }_{.t}\)): the mean trajectory of a population.
yik: the first missing value of ith trajectory.
yikLOCF: the imputed value for yik using LOCF method for all time points k ≥ d.
(\({\overline{y} }_{.1}\) LOCF, …, \({\overline{y} }_{.t}\) LOCF): the mean trajectory of a population with missing values using the LOCF method.
AVk: the average variation at kth time point and is equal to \({\overline{y} }_{.k}\) ‒ \({\overline{y} }_{.k}\) LOCF
The missing value yik is obtained from the Copy Mean LOCF by adding AVk to the imputed value for yik using the LOCF method (yikLOCF + AVk).
Interim analyses
No interim analyses of efficacy towards the primary endpoint will be performed because we do not expect any of the trial medications to have dramatic effects compared with other medications.
Ethics, monitoring, and responsibilities
This study was registered in IRCT.ir on 13/05/2023, https://irct.behdasht.gov.ir/trial/69613. The 5-EPIFAT protocol is approved by the Institutional Review Board (IRB) of Ahvaz Jundishapur University of Medical Sciences (Ref. ID: IR.AJUMS.REC.1401.587). All sites received local approval from their IRBs. The trial will be evaluated by an independent Data and Safety Monitoring Committee (DSMC). DSMC members will be selected by the ethics committee and will independently monitor the entire trial process and data. The DSMC will conduct both on-site and remote monitoring. The DSMB and the IRB must approve any modifications to the protocol. Amendments are first notified to the funder, the IRB, and the DSMC. When approved, the PI will notify the centers, and a copy of the revised protocol will be sent to the PI to add to the Investigator Site File. Also, amendments will be updated in the IRCT clinical trial registry. To double-check the details, go over the timeline table, and finally approve the protocol, the principal investigators (PI), DSMC, and IRB representative attended the first open meeting. Regular DSMC meetings will be held at quarterly intervals during the trial period. The Site Principal Investigator (PI) is responsible for leading the clinical trial team, overseeing the conduct of clinical trial at a site in accordance with the approved protocol, and is responsible for adequately supervising the team members. Also, the PIs are responsible for monitoring and coordinating the process of gathering, entering, safeguarding data during the trial. The PIs must report any potential protocol deviations and the incidence of serious adverse events to the main site within 24 h. Trained study staff are responsible for running the trial according to the protocol, which are monitored regularly and weekly by the PI or their representative at each study site. In case of any severe adverse event, the DSMC will be notified within 48 h by a short statement and then a fully detailed report will be prepared. In addition, the patient is referred for side effects control free of charge. The electronic version of the data will be encrypted and stored with a double password, and only the main authors (MM, HK, MJ) will have access to the dataset. Finally, the 5-EPIFAT trial data will be presented in peer-reviewed journals and at national and international conferences. The protocol is accessible in IRCT.ir. Data and statistical codes will be available upon reasonable request from the corresponding author.
Discussion
Fatigue is one of the most distressing and common symptoms experienced by cancer patients, and its management poses serious challenges for medical oncologists, nurses, and palliative care specialists. Treatment options for cancer-related fatigue are still emerging [53]. Overall, the evidence for the use of pharmacological agents for CRF is still not very solid [20]. Because the exact pathophysiological mechanism of CRF is not known, it is difficult to find effective pharmacological treatments for managing CRF [53]. Therefore, more clinical studies are needed to find palliative pharmacotherapy options for the treatment of CRF, especially studies that compare the effectiveness of different drugs. More particularly, further clinical evidence is still needed concerning the efficacy of palliative pharmacotherapy for treating CRF, particularly studies comparing the efficacy of different medications [27]. The 5-EPIFAT trial is the first multi-arm, head-to-head randomized trial for CRF treatment, comparing the efficacy of methylphenidate vs. bupropion vs. ginseng vs. amantadine vs. placebo in patients with advanced cancer on active treatment phase.
The 5-EPIFAT study is a multi-arm trial. Evaluating more than one intervention simultaneously increases the chance of finding an effective intervention. The use of a multi-arm design, as opposed to 2-arm trials, provides the possibility of comparing the effectiveness of different interventions with each other [33, 54]. The 5-EPIFAT study has a repeated measures design by virtue of which the efficacy of medications could be observed, which increases statistical power [55].
Conducting a trial with a cross-over design will be time-consuming due to the multi-arm nature of the study, and due to the possibility of some medications having no effectiveness compared to the placebo, the attrition rate is expected to be high in some groups, and there will be bias. On the other hand, although the agents used to have a short half-life and a cross-over design requires a short wash-out period, it is possible that the medications effective in improving fatigue have a long-lasting effect and the patient experiences less fatigue long after stopping the drug. Therefore, there is concern about carryover effects even if the wash-out period is taken into consideration. However, a parallel-group design does not have the mentioned problems and is more suitable.
All the medications used in this trial have a rapid onset of possible effects, so they are comparable with each other, and a 4-week period of use is sufficient to see their effects. In order to have a more realistic world, we will set relatively broad inclusion criteria, and the exclusion criteria will be chosen with the aim of patient safety. Different types of cancer will be taken into account because CRF is a syndrome that results from increased inflammatory cytokines and tumor byproducts regardless of tumor type. This pathophysiology of CRF is more related to the interaction between the cancer and the host than to any specific histology. Also, the frequency and severity of CRF in patients with different types of tumors are to a large extent the same [35].
In addition, the 5-EPIFAT trial will detect treatment-subgroup interactions with subgroup analysis using a decision-tree-based machine learning method [49] to guide clinical decision-making for CRF management in order to achieve the best effectiveness.
We expect that this study will lead to future evidence-based treatment of CRF in patients with advanced cancer, and to the design of clinical trials and guidelines. Even if the findings are negative, the results of this study will still be critical for further efforts to elucidate the pathophysiology of new therapeutic targets for CRF.
Trial status
Participant recruitment commenced on September 2023 (protocol version 2, dated August 2023) and is ongoing. Up to now, 45 participants have been recruited. Recruitment is expected to be completed in September 2024.
Statement of patient and public involvement
The public and patients were not consulted about how research protocols are written. At the conclusion of the study, social media will be used to share the findings with the patients.
Declaration of authorship
The standard criteria to determine authorship for publishing our trial results will have significant contributions to the work’s conception or design, or to the gathering, processing, or interpretation of data; composing the work or critically editing it to remove any significant intellectual content; and the final approval of the version to be published, along with a commitment to take responsibility for every aspect of the work and make sure that any concerns about the integrity or accuracy of any part of it are duly looked into and addressed. Our team is dedicated to transparently discussing and reaching consensus on the distinct contributions of every possible author at the beginning of the research phase. We will guarantee that all individuals meeting the specified criteria are acknowledged as authors. Furthermore, individuals who assist in the project but do not qualify for authorship will be appropriately recognized in the acknowledgments section of the publication.
Availability of data and materials
The final dataset and the code for statistical analysis will be available upon reasonable request.
Abbreviations
- CRF:
-
Cancer-related fatigue
- NCCN:
-
National Comprehensive Cancer Network
- AE:
-
Adverse event
- ASCO:
-
American Society of Clinical Oncology
- FACIT-F:
-
Functional assessment of chronic illness therapy-fatigue
- PRO-CTCAE:
-
Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events
- SPIRIT:
-
Standard Protocol Items: Recommendations for Interventional Trials
- DSMC:
-
Data and Safety Monitoring Committee
- PI:
-
Principal investigator
References
Berger AM, Mooney K, Alvarez-Perez A, Breitbart WS, Carpenter KM, Cella D, et al. Cancer-related fatigue, Version 2.2015. J Natl Compr Canc Netw. 2015;13(8):1012–39.
Ebede CC, Jang Y, Escalante CP. Cancer-related fatigue in cancer survivorship. Med Clin North Am. 2017;101(6):1085–97.
Mohandas H, Jaganathan SK, Mani MP, Ayyar M, Rohini Thevi GV. Cancer-related fatigue treatment: an overview. J Cancer Res Ther. 2017;13(6):916–29.
Spathis A, Fife K, Blackhall F, Dutton S, Bahadori R, Wharton R, et al. Modafinil for the treatment of fatigue in lung cancer: results of a placebo-controlled, double-blind, randomized trial. J Clin Oncol. 2014;32(18):1882–8.
Mo J, Darke AK, Guthrie KA, Sloan JA, Unger JM, Hershman DL, et al. Association of fatigue and outcomes in advanced cancer: an analysis of four SWOG treatment trials. JCO Oncol Pract. 2021;17(8):e1246–57.
Symptom Management and Quality of Life Steering Committee. National Cancer Institute. Strategic Priorities: Symptom Management and Quality of Life Steering Committee. (SxQoL SC Strategic Priorities) 2015. [Available from: https://www.cancer.gov/about-nci/organization/ccct/steering-committees/2015-SxQoLSC-StrategicPriorities.
Cedenilla Ramón N, Calvo Arenillas JI, Aranda Valero S, Sánchez Guzmán A, Moruno MP. Psychosocial interventions for the treatment of cancer-related fatigue: an umbrella review. Curr Oncol. 2023;30(3):2954–77.
Miladinia M, Baraz S, Ramezani M, Malehi AS. The relationship between pain, fatigue, sleep disorders and quality of life in adult patients with acute leukaemia: during the first year after diagnosis. Eur J Cancer Care (Engl). 2018;27(1):e12762.
Animaw L, Woldegiorgis Abate T, Endeshaw D, Tsegaye D. Fatigue and associated factors among adult cancer patients receiving cancer treatment at oncology unit in Amhara region, Ethiopia. PLoS ONE. 2023;18(1):e0279628.
Dolgoy ND, O’Krafka P, McNeely ML. Cancer-related fatigue in head and neck cancer survivors: Energy and functional impacts. Cancer Treatment Res Commun. 2020;25:100244.
Bower JE, Bak K, Berger A, Breitbart W, Escalante CP, Ganz PA, et al. Screening, assessment, and management of fatigue in adult survivors of cancer: an American Society of Clinical Oncology Clinical Practice Guideline Adaptation. J Clin Oncol. 2014;32(17):1840–50.
Thong MSY, van Noorden CJF, Steindorf K, Arndt V. Cancer-related fatigue: causes and current treatment options. Curr Treat Options Oncol. 2020;21(2):17.
Jim HSL, Hoogland AI, Han HS, Culakova E, Heckler C, Janelsins M, et al. A randomized placebo-controlled trial of bupropion for cancer-related fatigue: study design and procedures. Contemp Clin Trials. 2020;91: 105976.
Mustian KM, Alfano CM, Heckler C, Kleckner AS, Kleckner IR, Leach CR, et al. Comparison of pharmaceutical, psychological, and exercise treatments for cancer-related fatigue: a meta-analysis. JAMA Oncol. 2017;3(7):961–8.
Mücke M, Cuhls H, Peuckmann-Post V, Minton O, Stone P, Radbruch L. Pharmacological treatments for fatigue associated with palliative care. Cochrane Database Syst Rev. 2015;2015(5):Cd006788.
van Waart H, van Harten WH, Buffart LM, Sonke GS, Stuiver MM, Aaronson NK. Why do patients choose (not) to participate in an exercise trial during adjuvant chemotherapy for breast cancer? Psychooncology. 2016;25(8):964–70.
Miladinia M, Jahangiri M, Kennedy AB, Fagerström C, Tuvesson H, Safavi SS, et al. Determining massage dose-response to improve cancer-related symptom cluster of pain, fatigue, and sleep disturbance: a 7-arm randomized trial in palliative cancer care. Palliat Med. 2023;37(1):108–19.
Miladinia M, Voss JG, Molavynejad S, Malehi AS, Zarea K, Nouri EM, et al. Slow-stroke back massage compared with music therapy for leukemia-related pain and fatigue: a randomized controlled trial. JCO oncology practice. 2021;17(11):e1614–21.
Fabi A, Bhargava R, Fatigoni S, Guglielmo M, Horneber M, Roila F, et al. Cancer-related fatigue: ESMO Clinical Practice Guidelines for diagnosis and treatment. Ann Oncol. 2020;31(6):713–23.
Klasson C, Helde Frankling M, Lundh Hagelin C, Björkhem-Bergman L. Fatigue in Cancer Patients in Palliative Care-A Review on Pharmacological Interventions. Cancers. 2021;13(5):985. https://doi.org/10.3390/cancers13050985.
Sun X, Chen Y, Cheung WK, Wu IX, Xiao F, Chung VC. Pharmacological interventions for the management of cancer-related fatigue among cancer survivors: systematic review and meta-analysis. Integr Cancer Ther. 2021;20:15347354211038008.
Tomlinson D, Robinson PD, Oberoi S, Cataudella D, Culos-Reed N, Davis H, et al. Pharmacologic interventions for fatigue in cancer and transplantation: a meta-analysis. Current oncology (Toronto, Ont). 2018;25(2):e152–67.
Ashrafi F, Mousavi S, Karimi M. Potential Role of Bupropion Sustained Release for Cancer-Related Fatigue: a Double-Blind, Placebo-Controlled Study. Asian Pac J Cancer Prev. 2018;19(6):1547–51.
Barton DL, Liu H, Dakhil SR, Linquist B, Sloan JA, Nichols CR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105(16):1230–8.
Cullum JL, Wojciechowski AE, Pelletier G, Simpson JS. Bupropion sustained release treatment reduces fatigue in cancer patients. Can J Psychiatry. 2004;49(2):139–44.
Kim JW, Han SW, Cho JY, Chung IJ, Kim JG, Lee KH, et al. Korean red ginseng for cancer-related fatigue in colorectal cancer patients with chemotherapy: a randomised phase III trial. Eur J Cancer. 2020;130:51–62.
Pedersen L, Lund L, Petersen MA, Sjogren P, Groenvold M. Methylphenidate as needed for fatigue in patients with advanced cancer. a prospective, double-blind, and placebo-controlled study. J Pain Symptom manage. 2020;60(5):992–1002.
Pourmohamadi K, Ahmadzadeh A, Latifi M. Investigating the effects of oral ginseng on the cancer-related fatigue and quality of life in patients with non-metastatic cancer. Int J Hemat Oncol Stem Cell Res. 2018;12(4):313–7.
Richard PO, Fleshner NE, Bhatt JR, Hersey KM, Chahin R, Alibhai SM. Phase II, randomised, double-blind, placebo-controlled trial of methylphenidate for reduction of fatigue levels in patients with prostate cancer receiving LHRH-agonist therapy. BJU Int. 2015;116(5):744–52.
Roth AJ, Nelson C, Rosenfeld B, Scher H, Slovin S, Morris M, et al. Methylphenidate for fatigue in ambulatory men with prostate cancer. Cancer. 2010;116(21):5102–10.
Salehifar E, Azimi S, Janbabai G, Zaboli E, Hendouei N, Saghafi F, et al. Efficacy and safety of bupropion in cancer-related fatigue, a randomized double blind placebo controlled clinical trial. BMC Cancer. 2020;20(1):158.
Centeno C, Rojí R, Portela MA, Santiago AD, Cuervo MA, Ramos D, et al. Improved cancer-related fatigue in a randomised clinical trial: methylphenidate no better than placebo. BMJ Support Palliat Care. 2022;12(2):226–34.
Grayling MJ, Wason JMS. A web application for the design of multi-arm clinical trials. BMC Cancer. 2020;20(1):80.
Shi Z-Y, Zeng J-Z, Wong AST. Chemical Structures and Pharmacological Profiles of Ginseng Saponins. Molecules. 2019;24(13):2443.
Yennurajalingam S, Tannir NM, Williams JL, Lu Z, Hess KR, Frisbee-Hume S, et al. A Double-Blind, Randomized, Placebo-Controlled Trial of Panax Ginseng for Cancer-Related Fatigue in Patients With Advanced Cancer. J Natl Compr Canc Netw. 2017;15(9):1111–20.
Nourbakhsh B, Revirajan N, Morris B, Cordano C, Creasman J, Manguinao M, et al. Safety and efficacy of amantadine, modafinil, and methylphenidate for fatigue in multiple sclerosis: a randomised, placebo-controlled, crossover, double-blind trial. The Lancet Neurology. 2021;20(1):38–48.
Nourbakhsh B, Revirajan N, Waubant E. Treatment of fatigue with methylphenidate, modafinil and amantadine in multiple sclerosis (TRIUMPHANT-MS): Study design for a pragmatic, randomized, double-blind, crossover clinical trial. Contemp Clin Trials. 2018;64:67–76.
Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63–74.
Cai T, Chen J, Ni F, Zhu R, Wu F, Huang Q, et al. Psychometric properties of the Chinese version of the functional assessment of chronic illness therapy–fatigue (FACIT–F) among patients with breast cancer. Health Qual Life Outcomes. 2023;21(1):91.
Basch E, Reeve BB, Mitchell SA, Clauser SB, Minasian LM, Dueck AC, et al. Development of the National Cancer Institute’s patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE). J Natl Cancer Inst. 2014;106(9):dju244.
Dueck AC, Mendoza TR, Mitchell SA, Reeve BB, Castro KM, Rogak LJ, et al. Validity and Reliability of the US National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). JAMA Oncol. 2015;1(8):1051–9.
Snow G, Snow MG. Package ‘blockrand’. The Comprehensive R Archive Network. 2013.
Zhang Z, Mai Y, Yang M, Zhang MZ. Package ‘WebPower’. Basic and Advanced Statistical Power Analysis Version. 2018;72.
Noguchi K, Latif M, Konietschke F, Gel YR, Noguchi MK. Package ‘nparLD’. 2022.
Bates D, Maechler M, Bolker B, Walker S, Christensen RHB, Singmann H, et al. Package ‘lme4’. http://lme4.r-forge.r-project.org. 2009.
Howard JP, Howard MJP. Package ‘waterfall’. 2016.
Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag, 2016. https://ggplot2.tidyverse.org.
Mercier F, Consalvo N, Frey N, Phipps A, Ribba B. From waterfall plots to spaghetti plots in early oncology clinical development. Pharm Stat. 2019;18(5):526–32.
Fokkema M, Smits N, Zeileis A, Hothorn T, Kelderman H. Detecting treatment-subgroup interactions in clustered data with generalized linear mixed-effects model trees. Behav Res Methods. 2018;50(5):2016–34.
Loh W-Y, He X, Man M. A regression tree approach to identifying subgroups with differential treatment effects. Stat Med. 2015;34(11):1818–33.
Genolini C, Jacqmin-Gadda H. Copy mean: a new method to impute intermittent missing values in longitudinal studies. Open J Stat. 2013;3(04):26.
Genolini C, Lacombe A, Écochard R, Subtil F. CopyMean: a new method to predict monotone missing values in longitudinal studies. Comput Methods Programs Biomed. 2016;132:29–44.
Sleight AG, Crowder SL, Skarbinski J, Coen P, Parker NH, Hoogland AI, et al. A New Approach to Understanding Cancer-Related Fatigue: Leveraging the 3P Model to Facilitate Risk Prediction and Clinical Care. Cancers. 2022;14(8):1982.
Juszczak E, Altman DG, Hopewell S, Schulz K. Reporting of Multi-Arm Parallel-Group Randomized Trials: Extension of the CONSORT 2010 Statement. JAMA. 2019;321(16):1610–20.
Goulet M-A, Cousineau D. The Power of Replicated Measures to Increase Statistical Power. Adv MethodsPract Psychol Sci. 2019;2(3):199-213.
Acknowledgements
The authors would like to thank the Nursing Care Research Center in Chronic Diseases in affiliation with Ahvaz Jundishapur University of Medical Sciences for supporting this trial.
Funding
This study is fully funded by a research grant from the Ahvaz Jundishapur University of Medical Sciences (Grant number: NCRCCD-0116). The funder will monitor all steps of the trial. The funder will not be involved in the implementation of the trial procedures.
Author information
Authors and Affiliations
Contributions
MoM, HK, KZ, MGH, and RG participated in the conception of the trial. MoM, HK, RG, AI, SJW, SC, and CF had a role in designing the trial. MoM, MJ, HK, AF, and MGH participated in plans for data analysis. MoM, HK, MJ, MaM, and RG drafted the initial version of the manuscript. All authors contributed to revising the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The trial protocol was approved by the Institutional Review Board (IRB) of Ahvaz Jundishapur University of Medical Sciences (Ref. ID: IR.AJUMS.REC.1401.587). All sites received local approval from their IRBs. Written informed consent will be obtained from all participants. All methods will be carried out in accordance with relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Miladinia, M., Jahangiri, M., White, S.J. et al. 5-EPIFAT trial protocol: a multi-center, randomized, placebo-controlled trial of the efficacy of pharmacotherapy for fatigue using methylphenidate, bupropion, ginseng, and amantadine in advanced cancer patients on active treatment. Trials 25, 230 (2024). https://doi.org/10.1186/s13063-024-08078-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-024-08078-w