
Abstract
Background
Electroencephalography (EEG)-based brain-computer interfaces (BCIs) allow to modulate the sensorimotor rhythms and are emerging technologies for promoting post-stroke motor function recovery. The Promotoer study aims to assess the short and long-term efficacy of the Promotoer system, an EEG-based BCI assisting motor imagery (MI) practice, in enhancing post-stroke functional hand motor recovery. This paper details the statistical analysis plan of the Promotoer study.
Methods
The Promotoer study is a randomized, controlled, assessor-blinded, single-centre, superiority trial, with two parallel groups and a 1:1 allocation ratio. Subacute stroke patients are randomized to EEG-based BCI-assisted MI training or to MI training alone (i.e. no BCI). An internal pilot study for sample size re-assessment is planned. The primary outcome is the effectiveness of the Upper Extremity Fugl-Meyer Assessment (UE-FMA) score. Secondary outcomes include clinical, functional, and user experience scores assessed at the end of intervention and at follow-up. Neurophysiological assessments are also planned. Effectiveness formulas have been specified, and intention-to-treat and per-protocol populations have been defined. Statistical methods for comparisons of groups and for development of a predictive score of significant improvement are described. Explorative subgroup analyses and methodology to handle missing data are considered.
Discussion
The Promotoer study will provide robust evidence for the short/long-term efficacy of the Promotoer system in subacute stroke patients undergoing a rehabilitation program. Moreover, the development of a predictive score of response will allow transferring of the Promotoer system to optimal clinical practice. By carefully describing the statistical principles and procedures, the statistical analysis plan provides transparency in the analysis of data.
Trial registration
ClinicalTrials.gov NCT04353297. Registered on April 15, 2020.
Similar content being viewed by others

Introduction
Stroke is a leading cause of long-term disability with substantial social costs [1,2,3]. After experiencing a stroke, individuals often suffer from upper limb motor dysfunction, which leads to long-term disability [4]. The extent of upper extremity function is a crucial predictor of the patient’s ability to return to work [5]. Despite extensive therapy, variations in upper limb recovery patterns continue to significantly impact rehabilitation outcomes [6], and cost-effective post-stroke rehabilitation programs for the upper limbs are critically needed.
Electroencephalography (EEG)-based brain-computer interface (BCI) is an emerging technology that enables the direct translation of brain activity into physical motion [7] and is considered as a promising tool to promote functional motor recovery of the upper limbs after a stroke [8].
In a pilot study, an EEG-based BCI-assisted MI training (BCI-MI) administered using a BCI system fully compliant with rehabilitation requirements—the Promotoer system—proved to be useful in enhancing post-stroke functional hand motor recovery [9]. However, robust evidence for short- and long-term efficacy and availability of quantifiable indices (predictors) of patient response is needed for BCI-MI application to clinical practice. To this end, a randomized controlled study, named the Promotoer study, was conceived in subacute stroke participants.
The primary objective of the “Promotoer” trial is to determine whether the BCI-MI administered using a BCI system fully compatible with a clinical setting (the Promotoer) is superior to a non-BCI assisted MI training (Control-MI) in improving hand motor function outcomes in sub-acute stroke patients admitted to the hospital for their standard rehabilitation care. The secondary objectives of this study are as follows: (i) to assess the long-term efficacy of the BCI-based intervention on hand motor function outcome (6 months follow-up) and (ii) to determine if the clinical improvement is accompanied by a long-lasting neuroplasticity change. Moreover, the study aims to identify biomarkers and potential predictors of patient responses to the BCI-Promotoer training.
The protocol of the study, providing detailed information on background, study design, experimental intervention, and clinical procedures, was published previously [10]. The study was approved by the Independent Ethical Committee of the Fondazione Santa Lucia (FSL) IRCCS, Rome, Italy (CE/PROG.755), and is being conducted in accordance with the Helsinki Declaration and current national regulation on clinical trials, with high priority given to patient safety. The study has been registered at www.clinicaltrials.gov (NCT04353297), and its status is recruiting.
In this article, we report the details of the statistical analysis plan (SAP) of the Promotoer study (SAP Version 1.0 July 13, 2023), based on information contained in the PROMOTOER Clinical Protocol Version 1.0 [10], by carefully describing statistical principles and procedures for the analysis of data and presentation of the study results. The document follows the guidelines for the content of statistical analysis plans in clinical trials [11] and is in full compliance with the International Conference on Harmonisation ICH E9 guidance [12].
Study methods
Trial design
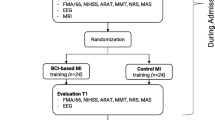
The Promotoer trial is designed as a randomized, controlled, assessor-blinded, single-centre, superiority trial with two parallel groups with a 1:1 allocation ratio. The two intervention groups are the BCI-assisted MI training (experimental group, BCI-MI) and the MI training not supported by BCI (control group, Control-MI). Both interventions will comprise a total of twelve 45-min sessions with a delivery of three times per week. Training will be completed in 4–6 weeks. The study interventions are conceived as add-on regimen to standard rehabilitation care (details are given in the published study protocol [10]).
Participant recruitment, intervention delivery, and data collection will take place at Fondazione Santa Lucia IRCCS, Rome, Italy.
Randomization
Participants are assigned to either the experimental group (BCI-MI) or the control group (Control-MI) in a 1:1 ratio through a randomized allocation process. This randomization process has been stratified by the side of the stroke lesion (left or right hemisphere) and the baseline score of UE-FMA/60, with 60 as the maximum score (< 19, severe; from 20 to 47, moderate) [13].
To carry out the randomization, a list has been generated using permuted blocks of varying sizes through the Ralloc procedure of the Statistical software STATA (https://www.stata.com/). The randomization list was centrally generated by the trial statistician at Istituto Superiore di Sanità (ISS, Rome Italy) and then securely managed. To maintain the integrity of the randomization process, numbered, opaque, sealed envelopes were prepared to replicate the randomization list for each specific stratum. These envelopes were delivered to the clinician responsible for participant randomization at Fondazione Santa Lucia IRCCS.
When a patient becomes eligible for randomization, the clinician in charge submits a randomization form containing the relevant stratification data and the date of the request. Then, the clinician responsible for participant randomization at Fondazione Santa Lucia IRCCS assigns to the patient the envelope with the number corresponding to the progressive order of randomization of the patient in his/her stratum.
Sample size
The results of the pilot study [8] demonstrated that the BCI-MI group had significantly higher effectiveness in terms of FMA scores compared to the control group (mean ± SD: 44 ± 34.7 vs. 19.8 ± 19.8). This effect size was considered clinically relevant and used for sample size calculation, which was carried out with a one-sided alpha level of 5% for the test of superiority, a statistical power of 80%, and a t-test for independent groups. The calculation revealed that each group should consist of 18 participants. To account for potential loss to follow-up, a dropout rate of 25% was considered, leading to a total of 48 patients required for the study. The sample size calculation was conducted using the power twomeans module in the Stata/MP 17.0 statistical software.
Framework
The primary objective of the trial is to evaluate the superiority of BCI-MI training (BCI-MI) against non-BCI assisted MI training (Control-MI) in improving hand motor function outcomes in sub-acute stroke patients admitted to the hospital for their standard rehabilitation care. Thus, the research hypothesis is settled in a superiority framework.
Statistical interim analysis and stopping guidance
To check the congruity of nuisance parameters used in sample size calculation, an internal pilot study (IPS) for sample-size recalculation was planned on data of primary outcome in the first 20 participants. Specifically, estimates of standard deviations of the effectiveness of the FMA score at the end of intervention training (T1) in the intervention group will be obtained from masked IPS data and used in the sample size procedure specified in the “Sample size” section, considering other assumptions and data as unchanged. Only upward adjustments of the initially planned sample size will be allowed, according to restricted IPS [14]. R code used for masked calculation of standard deviations is reported in the Appendix.
No interim efficacy analysis is planned. Pockock method for control of family-wise type I might be implemented by using the software RPACK (Confirmatory Adaptive Clinical Trial Design and Analysis, R package), if an interim analysis is required due to changes in the recruitment conditions.
Timing of final analysis
The final analyses will be performed after completion of the last visit of the last enrolled patient. The time schedule of trial assessments is reported in Table 1. Data for analyses will be extracted from the electronic case report form (eCRF) used for data entry and monitoring during the clinical trial.
Blinding will be removed in two steps: analysis will be performed upon the study’s completion and the database’s freezing, using group assignments as group A and group B. When all analyses have been performed and the final report drafted, BCI-MI and Control-MI will be un-blinded.
Statistical principles
Confidence intervals and P values
The primary hypothesis testing will be performed at a one-sided alpha of 0.05. The significance level used for the primary outcome is consistent with that used in the sample size calculation. Hypothesis testing on any of the secondary outcomes will be performed at a two-sided alpha of 0.05. Treatment effects will be reported through mean differences for continuous variables and odds ratio for binary variables, with two-sided 95% confidence intervals.
Adjustment for multiplicity will be considered in post-hoc comparisons of secondary analyses, specifically in comparisons among groups in each evaluation time (T1, T2, T3, and T4). Bonferroni’s correction criteria will be applied, and exact P-values will be reported.
Adherence and protocol deviations
Adherence to study interventions (BCI-MI or Control-MI) is fixed as a minimum of 9 training sessions (out of 12) delivered within 6 weeks. Moreover, protocol deviations include loss at follow-up (withdrawal/drop-out/death for any cause) and/or missed assessment for primary outcome at T1. The number of patients with adherence to the study intervention and the number of patients showing deviations from the planned study intervention will be reported for each intervention group. Deviations will be described for each patient by intervention group.
Major protocol deviations are missing adherence to study interventions.
Analysis populations
The as-randomized (AR) population includes all randomized patients, regardless of subsequent withdrawal from training sessions or deviation from the study protocol.
The intention-to-treat (ITT) population is defined as all randomized participants who will perform (minimum) one training session (both for BCI-MI training and Control-MI training intervention groups).
The per-protocol (PP) population includes those randomized participants without major protocol deviations, namely patients with adherence to the study intervention.
The user experience (UE) population includes all patients randomized and assessed for user experience.
Trial population
Screening and eligibility
Screening for eligibility and enrolment in the study are detailed in the study protocol [10]. Briefly, eligible candidates are patients of any gender, aged 18 to 80 years, with a first-ever unilateral stroke confirmed by magnetic resonance imaging (MRI), that occurred from 1 to 6 months before admission to the Fondazione Santa Lucia IRCCS site for rehabilitation care. The eligible patients must show hemiplegia/hemiparesis of the upper extremity and a UE-FMA/60 score (out of a maximum 60 score) lower or equal to 47 [participants with severe symptoms (scores from 0 to 19) and moderate symptoms (score from 20 to 47) will be included]. The main exclusion criteria include dementia, severe neglect, or severe aphasia, severe spasticity (Modified Ashworth Scale > 4 at shoulder/elbow/wrist), a UE-FMA/60 score greater than > 47, a Token Test (TT) score lower or equal to 29, and concomitant neurological disorders. All eligibility criteria must be met by the time of the screening visit.
Recruitment, follow-up, and withdrawal
Patients who meet the eligibility criteria and have provided informed consent will be randomized and will undergo the planned evaluations at T0 (see Table 1). Any participant who enrolled in the study can withdraw from the study at any time, for any reason, without prejudice or consequence. The status of participants will be monitored during each scheduled training session and evaluation visit. In cases where a patient misses a planned evaluation visit, a phone call will be made to check their status.
A CONSORT flow diagram specifically designed for the Promotoer study (Fig. 1) will be used to summarize the numbers of patients who were randomized, received their allocated intervention, withdrew, or were lost to follow-up and were included in population analysis at T1 and T4 evaluation visits.

Promotoer CONSORT flow diagram (CONSORT 2010)
The timing of withdrawals and loss to follow-up, whenever possible, will be presented as time-to-event data and analysed using Kaplan–Meier curves, stratified by intervention group. The Log-rank test will be employed to assess any potential differences between the two curves. Furthermore, the reasons for the loss of participants during the study will be described for each intervention group.
The data collected from participants who have withdrawn from the study will be included in the analysis sets, unless the participants expressly withdraw their consent for the use of data collected until that time point.
Baseline patient characteristics
Following the time schedule shown in Table 1, patient characteristics in the two randomized groups will include:
-
Demographical data (sex, age at enrolment)
-
Clinical data and functional assessments collected at the screening visit for eligibility (type and side of lesion, TT score, FMA/60 upper limb section score)
-
Functional assessments completed at evaluation time T0 after the signature of the informed consent, (MEP, TMT, TAP, Edinburgh handedness inventory score, UE-FMA 66, NIHSS, ARAT, MMT for shoulder/elbow/wrist, NRS for pain for affected upper limb, and the MAS for spasticity score for shoulder/elbow/wrist)
All continuous variables will be summarized by descriptive statistics of n, mean, standard deviation, minimum, maximum, median, and interquartile range. Shapiro Wilk’s test will be used to assess the normal distribution of variables. Categorical data will be summarized by frequencies and percentages. Test of statistical significance will not be undertaken for baseline characteristics; rather, the clinical importance of any imbalance will be noted.
A table detailing baseline characteristics will be compiled for the ITT population (as outlined in the “Analysis populations” section). Additionally, separate tables of baseline characteristics will be generated for the AR and PP populations. These tables will be useful to assess the success of randomization by examining the balance of prognostic factors among the different intervention groups and to verify whether any attrition during the study has introduced a selection bias or has disrupted the balance that was initially achieved through randomization.
Analysis
Outcome definitions
The primary outcome is the “effectiveness” of the UE-FMA score at T1. The UE-FMA is widely recommended for the evaluation of sensorimotor impairments of the upper extremity in stroke rehabilitation research [9, 15, 16]. It ranges from 0 to 66 (best score). The effectiveness of the UE-FMA score is defined as the proportion of potential improvement that could be achieved after the intervention and is calculated as score at T1 minus score at T0, divided by the maximum score (66) minus score at T0, multiplied by 100 (as specified in [9]) [formula: 100 × (UE-FMA at T1 − UE-FMA at T0)/(66 − UE-FMA at T0)]. Thus, if a patient achieves the highest possible score after the intervention, the effectiveness is 100%. This approach allows to normalize the data, taking into account baseline data. The secondary outcomes are presented in Table 2. Electroencephalogram (EEG) and MRI data will be considered in a separate document.
Analysis methods for primary and secondary outcomes
The primary analysis will be carried out on the primary outcome in the ITT population—namely effectiveness of the UE-FMA at T1. In the secondary analysis, the analysis of the primary outcome will be carried out in the PP population. The secondary outcomes will be compared between groups both in the ITT and PP populations.
For continuous variables (effectiveness of UE-FMA score, of ARAT score, of NIHSS score, of MMT score, NRS, QCM, Workload NASA-TLX, SUS, QUEST, VAS Mood, VAS Satisfaction) descriptive statistics (mean and standard deviation, median and interquartile range) will be reported by intervention group for each evaluation time/training session. t-test for independent groups will be used for comparisons of the BCI-MI group vs Control-MI group in each evaluation time/training session. Shapiro–Wilk’s test will be used to assess the normal distribution, and the Mann–Whitney test will be used for comparison if the normality assumption is violated.
To determine whether and to what extent the long-term efficacy of the BCI intervention on the UE-FMA score can be maintained after the end of the intervention, we will use a repeated measures analysis of variance (ANOVA). ANOVA will treat the group as a between-subject factor and time as a within-subject factor, assessing the effectiveness of the UE-FMA scores at T1, T2, T3, and T4.
All categorical variables will be summarized (by number and frequency) in each intervention group and compared between groups by Fisher’s test, at each evaluation time (T2, T3, T4).
To identify clinical and/or neurophysiological determinants of MCID (Table 2) in the participant response to an intervention at T1, a forward-stepwise binary logistic regression will be performed including the intervention group, demographical, clinical, and neurophysiological parameters. Odds ratios and 95% confidence intervals will be calculated. To reduce the dimensional space of available variables, variable selection algorithms (e.g. least absolute shrinkage and selection operator—LASSO regression) will be used to select the most informative features to be included in the predictive model. A probability score will be defined to assess the likelihood of good recovery based on obtained determinants of response. A further logistic regression analysis will be performed including only the BCI-MI group. The same strategies of analysis will be used for the evaluation of MCID at all subsequent evaluation times (T2, T3, T4).
Statistical analysis of the MAS score will be detailed for shoulder, elbow, and wrist. Descriptive statistics on frequency distributions will be reported for each time point (T1, T2, T3 and T4) and compared between intervention groups by chi-square test. Moreover, a change in the score between T0 and T1, and changes along the follow-up (e.g. T2 vs T0; T3 vs T0, T3 vs T0, and T4 vs T0) will be computed and defined as improved/stable/deteriorated. Such changes will be compared in the intervention groups using a chi-square test or Fisher’s exact test.
Scores obtained in user experience assessments for QCM (separately for Mastery confidence, Incompetence/Fear-to Fall, Challenge and Interest), Workload NASA-TLX, SUS, QUEST, VAS Mood, and VAS Satisfaction, will be described for each experimental group and comparisons will be carried out using the methodologies adopted for other continuous variables. Analyses will be carried out by training sessions.
Subgroup analyses
To investigate the possible effect of the number/amount of training sessions received, comparisons among sub-groups of BCI-MI participants defined according to the number of completed training sessions (< 9 vs > = 9) will be carried out on primary and secondary outcomes using the same statistical methodologies adopted in the primary and secondary analyses.
Moreover, to assess the robustness of the effects of BCI-MI intervention, explorative subgroup analyses [27] will be carried out on the primary outcome in subgroups of participants identified by the following: baseline score of UE-FMA/60 with 60 as maximum score (< 19, severe; from 20 to 47, moderate); side of stroke lesion (left/right hemisphere); age at enrolment (< = 50 years vs > 50 years); time from stroke to admission to Fondazione Santa Lucia IRCCS site for rehabilitation care (< = 3 months vs > 3 months). Estimates and confidence intervals will be obtained for each subgroup and will be presented through forest plots. Moreover, for each subgroup variable, statistical tests for interaction [12, 28] will be carried out using ANOVA including the intervention group (BCI-MI vs Control-MI) and its interaction with the subgroup covariate. Exact P-values of the test of interaction will be reported to allow detection of signals for further inspection.
Missing data
The primary analysis will be performed on all available cases. During the conduction of the trial, data collection will be monitored to minimize missing data for the primary outcome. Moreover, we have planned sensitivity analyses for addressing missing data [29] pertaining to the primary outcome. These analyses will allow to evaluate the robustness of study findings.
The following data will be obtained separately for each intervention group:
-
Proportion of participants with missing data for the primary outcome
-
Comparison of baseline characteristics between participants with available data and participants with missing data for the primary outcome
If no important difference in baseline characteristics is observed between participants with available data and participants with missing data for the primary outcome, comparisons of intervention groups for the primary outcome will be carried out using multiple imputation techniques for missing data [30]. The estimation model will include variables used in the stratification of patients at randomization (baseline score of UE-FMA/60 and side of stroke lesion), age at enrolment and gender; 20 imputations will be obtained.
In case of a relevant amount of missing data (more than 25% of dropouts), a revision of the SAP will be necessary.
Safety analysis
BCI-MI and Control-MI interventions are not expected to yield adverse effects, as they are non-invasive procedures without the administration of drugs. Moreover, both interventions will be administered by trained professionals, including physiotherapists and neurophysiology technicians with expertise in EEG recordings, ensuring the safety and well-being of the participants. Previous experience with BCI training delivery in subacute stroke participants is encouraging since training was well-tolerated by the participants and no dropouts were reported.
Statistical software
Analyses will be carried out by the STATA 17 and R software (version 4.3.0).
Data collection and management
The study data are collected and managed using REDCap electronic data capture tools [31, 32] hosted at Istituto Superiore di Sanità (Rome, Italy). An ad hoc REDCap template was developed in line with the case report form (CRF) of the Promotoer study. This includes two sections: (1) the “Baseline and Randomization Section” including demographical and clinical baseline data, randomization data, and training session data, and (2) the “Outcomes Section” including baseline neurophysiological assessments and clinical and functional outcomes data at the planned evaluation times. Section 1 data are filled in by unblinded personnel, and Sect. 2 data are filled in by outcome assessors blinded to assigned intervention.
Automated export procedures for seamless data downloads to STATA and R statistical packages are available.
Discussion
The Promotoer study is a randomized controlled study conceived to produce robust evidence for short and long-term efficacy of the EEG-based BCI-assisted MI training (the Promotoer system) and identify accurate indices (predictors) of response to this intervention in subacute stroke patients undergoing rehabilitation. Rigorous methodologies have been adopted in the study design, such as clear definitions of the experimental and control interventions, randomization of patients to the intervention groups, and blinded outcome assessment. The effect size considered in the sample size calculation was derived from the findings of the pilot study [8] and was considered clinically relevant and useful to support the translation of the Promotoer system to the clinical practice in neurorehabilitation. The trial has been registered on the ClinicalTrials.gov website (NCT04353297), and a detailed study protocol [10] has been developed according to SPIRIT guidance [33]. This paper describes the SAP of the Promotoer study, which was developed following the Guidelines for the Content of Statistical Analysis Plans in Clinical Trials [11]. Study methods, statistical principles, trial populations, and analysis issues have been detailed in line with the objectives of the study and the study design, to support transparency and reproducibility of data analysis.
Conclusion
There is a strong need to improve the quality of conduct and reporting of research studies in rehabilitation [34], and a Randomized Controlled Trial Rehabilitation Checklists (RCTRACK) project is ongoing aiming at producing a specific reporting guideline for randomized controlled trials (RCTs) in rehabilitation [35].
Registration of the study, production of the study protocol and statistical analysis plan, and adherence to reporting guidelines are well-known, good clinical research practices [36, 37]. However, it is the responsibility of researchers to actively integrate them into their everyday research practices. The availability of SAP ensures the integrity and credibility of study results, guarding against selective reporting of outcomes and analyses, facilitating reproducible research, and increasing trust in science. Producing SAP has a pivotal role in upholding the quality of rehabilitation research, enhancing the reliability of findings, and ultimately contributing to evidence-based decision-making.
Trial status
Recruitment status: ongoing recruitment.
Recruitment start date: 24 November 2020 (first randomization on 27 November 2020).
Availability of data and materials
Not applicable.
Abbreviations
- ANOVA:
-
Analysis of variance
- AR:
-
As randomized
- ARAT:
-
Action research arm test
- BCI:
-
Brain-computer interface
- CRF:
-
Case report form
- CTC:
-
Clinical trial centre
- EEG:
-
Electroencephalography
- EMA:
-
European medicines agency
- FDA :
-
Food and drug administration
- FSL:
-
Fondazione Santa Lucia
- FMA:
-
Fugl-meyer assessment
- ICH:
-
International council for harmonisation
- IPS :
-
Internal pilot study
- IRCCS:
-
Istituti di Ricovero e Cura a Carattere Scientifico
- ISS:
-
Istituto Superiore di Sanità
- ITT:
-
Intention-to-treat
- LASSO :
-
Least absolute shrinkage and selection operator
- MAS:
-
Modified ashworth scale
- MCID:
-
Minimal clinically important difference
- MEP:
-
Motor evoked potential
- MI:
-
Motor imagery
- MMT :
-
Manual muscle test
- MRI:
-
Magnetic resonance imaging
- NASA-TLX:
-
NASA task load index
- NIHSS:
-
National institute of health stroke scale
- NRS:
-
Numeric rating scale
- PP:
-
Per-protocol
- QCM:
-
Questionnaire on current motivation
- QUEST:
-
Quebec user evaluation of satisfaction with assistive technology
- RCT:
-
Randomized controlled trial
- SAP:
-
Statistical analysis plan
- SD:
-
Standard deviation
- SPIRIT:
-
Standard protocol items: recommendations for interventional trials
- SUS:
-
System usability score
- TAP:
-
Test of attentional performance
- TMS:
-
Transcranial magnetic stimulation
- TMT:
-
Trail making test
- TT:
-
Token test
- UE:
-
User experience
- UE-FMA:
-
Fugl-meyer assessment for upper extremity
- VAS:
-
Visual analogue scale
References
Kim AS, Cahill E, Cheng NT. Global Stroke Belt. Stroke. 2015;46(12):3564–70.
Johnson CO, Nguyen M, Roth GA, Nichols E, Alam T, Abate D, et al. Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):439–58.
Meijeren-Pont W, Tamminga S, Goossens P, Groeneveld I, Arwert H, Meesters J, et al. Societal burden of stroke rehabilitation: costs and health outcomes after admission to stroke rehabilitation. J Rehabil Med. 2021;53(6):jrm00201.
Lang CE, Bland MD, Bailey RR, Schaefer SY, Birkenmeier RL. Assessment of upper extremity impairment, function, and activity after stroke: foundations for clinical decision making. J Hand Ther. 2013;26(2):104–15.
Coupar F, Pollock A, Rowe P, Weir C, Langhorne P. Predictors of upper limb recovery after stroke: a systematic review and meta-analysis. Clin Rehabil. 2012;26(4):291–313.
Raffin E, Hummel FC. Restoring motor functions after stroke: multiple approaches and opportunities. Neuroscientist. 2018;24(4):400–16.
Wolpaw JR, Birbaumer N, McFarland DJ, Pfurtscheller G, Vaughan TM. Brain–computer interfaces for communication and control. Clin Neurophysiol. 2002;113(6):767–91.
Pichiorri F, Mattia D. Brain-computer interfaces in neurologic rehabilitation practice. In Handb Clin Neurol. 2020;168:101–16.
Pichiorri F, Morone G, Petti M, Toppi J, Pisotta I, Molinari M, et al. Brain-computer interface boosts motor imagery practice during stroke recovery. Ann Neurol. 2015;77(5):851–65.
Mattia D, Pichiorri F, Colamarino E, Masciullo M, Morone G, Toppi J, et al. The Promotoer, a brain-computer interface-assisted intervention to promote upper limb functional motor recovery after stroke: a study protocol for a randomized controlled trial to test early and long-term efficacy and to identify determinants of response. BMC Neurol. 2020;20(1):254.
Gamble C, Krishan A, Stocken D, Lewis S, Juszczak E, Doré C, et al. Guidelines for the Content of Statistical Analysis Plans in Clinical Trials. JAMA. 2017;318(23):2337.
ICH Topic E 9 Statistical Principles for Clinical Trials Step 5 NOTE FOR GUIDANCE ON STATISTICAL PRINCIPLES FOR CLINICAL TRIALS [Internet]. 1998. Available from https://www.ema.europa.eu/en/ICH-E9-statistical-principles-for-clinical-trials
Woodbury ML, Velozo CA, Richards LG, Duncan PW. Rasch analysis staging methodology to classify upper extremity movement impairment after stroke. Arch Phys Med Rehabil. 2013;94(8):1527–33.
Wittes J, Brittain E. The role of internal pilot studies in increasing the efficiency of clinical trials. Stat Med. 1990;9(1–2):65–72.
Fugl-Meyer AR, Jääskö L, Leyman I, Olsson S, Steglind S. The post-stroke hemiplegic patient. 1. a method for evaluation of physical performance. Scand J Rehabil Med. 1975;7(1):13–31.
Hernández ED, Forero SM, Galeano CP, Barbosa NE, Sunnerhagen KS, Alt MM. Intra- and inter-rater reliability of Fugl-Meyer Assessment of Lower Extremity early after stroke. Braz J Phys Ther. 2021;25(6):709–18.
Shah S, Vanclay F, Cooper B. Efficiency, effectiveness, and duration of stroke rehabilitation. Stroke. 1990;21(2):241–6.
Sivan M, O’Connor R, Makower S, Levesley M, Bhakta B. Systematic review of outcome measures used in the evaluation of robot-assisted upper limb exercise in stroke. J Rehabil Med. 2011;43(3):181–9.
Bohannon RW, Smith MB. Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther. 1987;67(2):206–7.
Lyle RC. A performance test for assessment of upper limb function in physical rehabilitation treatment and research. Int J Rehabil Res. 1981;4(4):483–92.
Brott T, Adams HP, Olinger CP, Marler JR, Barsan WG, Biller J, et al. Measurements of acute cerebral infarction: a clinical examination scale. Stroke. 1989;20(7):864–70.
Morone G, Pisotta I, Pichiorri F, Kleih S, Paolucci S, Molinari M, et al. Proof of principle of a brain-computer interface approach to support poststroke arm rehabilitation in hospitalized patients: design, acceptability, and usability. Arch Phys Med Rehabil. 2015;96(3):S71–8.
Kleih SC, Nijboer F, Halder S, Kübler A. Motivation modulates the P300 amplitude during brain–computer interface use. Clin Neurophysiol. 2010;121(7):1023–31.
Riccio A, Leotta F, Bianchi L, Aloise F, Zickler C, Hoogerwerf EJ, et al. Workload measurement in a communication application operated through a P300-based brain–computer interface. J Neural Eng. 2011;8(2): 025028.
Bangor A, Kortum PT, Miller JT. An empirical evaluation of the system usability scale. Int J Hum Comput Interact. 2008;24(6):574–94.
Demers L, Weiss-Lambrou R, Ska B. The Quebec User Evaluation of Satisfaction with Assistive Technology (QUEST 2.0): an overview and recent progress. Technol Disabil. 2002;14(3):101–5.
EMA/CHMP Guideline on the investigation of subgroups in confirmatory clinical trials [Internet]. 2019. Available from: Guideline on the investigation of subgroups in confirmatory clinical trials (europa.eu)
Lagakos SW. The challenge of subgroup analyses — reporting without distorting. N Engl J Med. 2006;354(16):1667–9.
EMA/CHMP. Guideline on missing data in confirmatory clinical trials [Internet]. 2010. Available from: guideline-missing-data-confirmatory-clinical-trials_en.pdf (europa.eu)
Sterne JAC, White IR, Carlin JB, Spratt M, Royston P, Kenward MG, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ. 2009;338(jun29 1):b2393–b2393.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–81.
Harris PA, Taylor R, Minor BL, Elliott V, Fernandez M, O’Neal L, et al. The REDCap consortium: building an international community of software platform partners. J Biomed Inform. 2019;95: 103208.
Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, et al. SPIRIT 2013 Statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200.
Arienti C, Armijo-Olivo S, Minozzi S, Tjosvold L, Lazzarini SG, Patrini M, et al. Methodological issues in rehabilitation research: a scoping review. Arch Phys Med Rehabil. 2021;102(8):1614-1622.e14.
Negrini S, Chan L, Ferriero G, Frontera WR, Heinemann AW. Current evidence from the Randomized Controlled Trials Rehabilitation Checklist (RCTRACK) reporting guideline project. Arch Phys Med Rehabil. 2021;102(8):1665–7.
Schwab S, Janiaud P, Dayan M, Amrhein V, Panczak R, Palagi PM, et al. Ten simple rules for good research practice. PLoS Comput Biol. 2022;18(6): e1010139.
Sauerbrei W, Bland M, Evans SJW, Riley RD, Royston P, Schumacher M, et al. Doug Altman: Driving critical appraisal and improvements in the quality of methodological and medical research. Biom J. 2021;63(2):226–46.
Acknowledgements
We would like to acknowledge the contribution of the following members of the project team:
• Neurophysiology technicians
• Marco Secci, Fondazione Santa Lucia, IRCCS, Rome Italy
• Giovanni Nicolai, Fondazione Santa Lucia, IRCCS, Rome Italy
• Giorgio Tartaglia, Fondazione Santa Lucia, IRCCS, Rome Italy
We would like to acknowledge the contribution of Mimma Tripaldi for administrative and technical support.
Funding
This project is funded by the Italian National Ministry of Health (MoH), Ricerca Finalizzata 2018 (grant # RF-2018–12365210 to Marco Molinari).
Author information
Authors and Affiliations
Contributions
MP and MC conceived, planned, finalised, and wrote the statistical analysis plan, with review by JT, EC, and FP. MP and MC wrote the manuscript with a review by DM. DM, in collaboration with FP, GM, and MM, led the study conceptualization, design, and application for funding; MM is the grant holder. DM manages the trial. FP, GM, and DM are responsible for all medical and neurological and neurophysiological issues that arise during participant screening, intervention delivery, and follow-up. EC and FC provide leadership in the design and implementation of the BCI technology; JT provides leadership in the design and implementation of the advanced biosignal (EEG-EMG) processing; AB provides advice on neuropsychology and behavioural evaluation and participates in study participant evaluation; FTamburella and ML provide advice on the functional motor evaluation and participate in study evaluation of participants. FTomaiuolo is responsible for neuroimaging data analysis. DM, FP, GM, MM, and MP contributed to the statistical plan of the study. FS, MP, FP, and EM developed the REDCAP template for data collection and trial monitoring. FS is REDCap Administrator for Istituto Superiore di Sanità, Rome, Italy. DC is an IT system administrator at Istituto Superiore di Sanità, Rome, Italy. MP generated the randomization list. DM was the clinician responsible for the randomization assignment at FSL. MM is the chief investigator of the Promotoer project. DM is the clinical lead of the Promotoer study. MP is the senior statistician responsible. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval for the study was obtained from the institutional review board, the Independent Ethical Committee of the Fondazione Santa Lucia, IRCCS, Rome, Italy (https://www.hsantalucia.it/en/ricerca/ethics-committee); study protocol # CE/PROG.755 11–06–19. All participants provided a written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendix
Appendix
Script for blinded estimation of the standard deviation of the effectiveness of UE-FMA/66 at T1 by the intervention group in R language.
# Load the dataset (3 columns: record id, endpoint value, treatment).
load(“db.Rdata”).
data = db[,c(“record_id”,“endpoint”,“prot”)].
# Create a group variable (empty vector).
data$group = 0.
# Randomly sample one unit from the population.
rand = sample(data$record_id,1).
# Assign group 1 to this unit.
data$group[data$record_id = = rand] = 1.
# Group 1 is assigned to all units that come from the same treatment as the selected patient.
data$group = ifelse(data$prot = = data$prot[data$record_id = = rand],1,0).
# Remove the treatment column from the dataset.
data = data[,-3].
# Compute the standard deviation of the endpoint (stratified by intervention group).
sd = aggregate(data$endpoint, by = list(data$group), FUN = sd).
# Print the results.
sd.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cipriani, M., Pichiorri, F., Colamarino, E. et al. The Promotoer, a brain-computer interface-assisted intervention to promote upper limb functional motor recovery after stroke: a statistical analysis plan for a randomized controlled trial. Trials 24, 736 (2023). https://doi.org/10.1186/s13063-023-07773-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-023-07773-4