
Abstract
Background
Colorectal cancer is the fourth most commonly diagnosed cancer in the United States. Approximately 3–10% of the population has an increased risk for colorectal cancer due to family history and warrants more frequent or intensive screening. Yet, < 50% of that high-risk population receives guideline-concordant care. Systematic collection of family health history and decision support may improve guideline-concordant screening for patients at increased risk of colorectal cancer. We seek to test the effectiveness of a web-based, systematic family health history collection tool and decision support platform (MeTree) to improve risk assessment and appropriate management of colorectal cancer risk among patients in the Department of Veterans Affairs primary care practices.
Methods
In this ongoing randomized controlled trial, primary care providers at the Durham Veterans Affairs Health Care System and the Madison VA Medical Center are randomized to immediate intervention or wait-list control. Veterans are eligible if assigned to enrolled providers, have an upcoming primary care appointment, and have no conditions that would place them at increased risk for colorectal cancer (such as personal history, adenomatous polyps, or inflammatory bowel disease). Those with a recent lower endoscopy (e.g. colonoscopy, sigmoidoscopy) are excluded. Immediate intervention patients put their family health history information into a web-based platform, MeTree, which provides both patient- and provider-facing decision support reports. Wait-list control patients access MeTree 12 months post-consent. The primary outcome is the risk-concordant colorectal cancer screening referral rate obtained via chart review. Secondary outcomes include patient completion of risk management recommendations (e.g. colonoscopy) and referral for genetic consultation. We will also conduct an economic analysis and an assessment of providers’ experience with MeTree clinical decision support recommendations to inform future implementation efforts if the intervention is found to be effective.
Discussion
This trial will assess the feasibility and effectiveness of patient-collected family health history linked to decision support to promote risk-appropriate screening in a large healthcare system such as the Department of Veterans Affairs.
Trial registration
ClinicalTrials.gov, NCT02247336. Registered on 25 September 2014.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Background
Colorectal cancer (CRC) is the fourth most commonly diagnosed cancer in the United States, with 140,788 new cases and 52,396 deaths in 2015 [1]. Screening by several modalities has been shown to reduce death from CRC in average-risk individuals [2,3,4,5,6,7] and is recommended by several national organizations [8]. CRC screening via colonoscopy or stool testing is recommended for average-risk individuals beginning at the age of 45 years by the American Cancer Society [9] and beginning at 50 years by the United States Preventive Services Task Force [10] and a Multi-society Task Force [8]. However, different screening modalities and/or frequencies are recommended for patients above population-level risk (hereafter referred to as increased risk), such as those with a personal or family history of colorectal neoplasia or adenomatous polyps [8].
Risk assessment using family health history (FHH) permits tailoring of CRC screening to an individual’s risk level. While collection of FHH is incorporated into multiple clinical guidelines [11, 12], it is rarely used in clinical practice as part of a standardized risk assessment [13]. Challenges to FHH use in clinical practice include physician time constraints during office visits, lack of provider awareness of FHH collection standards, failure of the medical record to support adequate collection and documentation of FHH collection, and provider lack of confidence in identifying patients at increased risk for CRC [14,15,16]. Subsequently, although 3–10% of the United States population [17] is thought to be at increased risk for CRC and, thus, meets criteria for more intensive screening and management, < 50% receive this recommended care [18,19,20].
Systematic FHH collection and provision of clinical decision support (CDS) can improve accuracy of identification and appropriate prevention management of patients at increased risk for disease. For example, Qureshi et al. found that, compared to usual care, systematic FHH collection identified previously undetected primary care patients who were at increased risk for cardiovascular disease (4.8% vs 0.3%) [21]. Systematic FHH collection can also improve appropriate use of strategies for increased-risk management. Orlando et al. found that the use of a web-based FHH risk assessment tool increased the proportion of increased-risk management strategies being delivered appropriately to increased risk patients from 10.5% to 71.8% [22]. Similarly, Scheuner et al. found that a cancer family history toolkit administered in primary care practices increased family history cancer documentation and increased guideline-concordant referral for genetic consultation more than twofold [23].
Integrated health systems, such as the Veterans Administration (VA), can benefit from systematic approaches to FHH collection as they often internally refer patients for procedures and counseling indicated by patient risk. Currently, the VA does not have a system-wide approach to the routine collection and documentation of FHH in the medical record. This lack of systematic documentation can result in poor FHH documentation. For example, at one VA healthcare system, the documentation of FHH for any cancer was 26.6% of patients seen [23]. Another study of 500 consecutive patients at a VA hospital revealed that 7% of fecal occult blood test orders by primary care providers (PCPs) were inappropriate and should have been referred for colonoscopy instead due to a personal or FHH that increased risk of CRC [24]. A standardized FHH collection tool supported by decision support for patients and providers could provide needed structure to improve risk-appropriate CRC management [23].
To address the pressing clinical need for improved risk-identification and risk-appropriate CRC screening, we are conducting a cluster randomized controlled trial (RCT) to evaluate the feasibility and effectiveness of implementing a web-based, systematic FHH collection and decision support platform (MeTree) [25] in VA primary care practices. We will evaluate the effect of the intervention on PCPs’ workflow and health economic outcomes to inform eventual implementation more broadly. To that end, our primary aims are: (1) to determine whether FHH collection via MeTree improves identification of patients at increased risk for CRC; and (2) to evaluate whether patient and provider decision support improves risk-appropriate PCP referrals for, and patient uptake of, CRC screening/surveillance and referral for genetic counseling. Our secondary aims are: (1) to assess experience with clinical decision support reports and effects on primary care provider workflow via qualitative interviews; and (2) to conduct budget-impact analyses of implementing FHH collection and decision support in the VA.
Methods
Study setting and design
We are conducting this RCT at two sites: the Durham VA Health Care System in Durham, NC, including three associated primary care clinics, and the William S. Middleton Memorial Veterans Hospital in Madison, WI. All enrolled patients are veterans receiving care at one of these medical facilities. Institutional review boards at the Durham VA Health Care System and the University of Wisconsin-Madison (the institutional review board of record for the Madison VA hospital) approved this study.
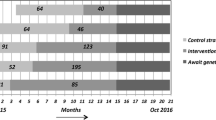
This study is a two-arm, cluster-randomized, type I hybrid implementation-effectiveness trial that aims to test the effectiveness of a clinical intervention (i.e. family history risk assessment with decision support documents) while obtaining data to inform potential future implementation [26]. Enrolled PCPs are randomized to have their patients complete MeTree either upon enrollment (immediate arm) or 12 months later (wait-list control arm). PCP randomization is stratified by site (Durham vs Madison) and provider type (physician versus advanced practice provider [physician assistant or nurse practitioner]) in case there are differences in extent of knowledge about FHH in guideline-concordant care or use of the information in clinical practice. We aim to sub-stratify by age within provider sub-groups (40–49 years vs 50–64 years) to address the potential for intervention effects that differ across age groups given that population-based screening begins at the age of 50 years. We chose a 12-month wait-list control arm as the comparator group to ensure that all patients receive the benefit of risk-appropriate screening. The primary outcome for this study is risk-appropriate CRC screening referral at 12 months. Secondary outcomes include patient completion of CRC screening referral and risk-appropriate genetic consultation, economic outcomes, and barriers and facilitators to intervention use (see Table 1 for additional details of study activities by time point). This trial is registered at clinicaltrials.gov (NCT02247336) (see Additional file 1 for additional trial registration items). Checklist for standard protocol items are listed in Additional file 2.
Study population and recruitment
Primary care provider recruitment and randomization
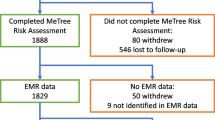
Provider inclusion/exclusion criteria are listed in Table 2. To recruit providers, we attend staff meetings to introduce the study; we do not provide education regarding FHH or CRC during these meetings. PCPs can consent to participate at the meeting or later via email. We use a rolling recruitment strategy to capture any newly hired providers who wish to participate. Providers enroll via email to the site PI in Durham (KG) and a study team member in Madison. The study coordinator (ES) enters consented providers into a study database for randomization assignment. Randomization is at the provider level to avoid contamination by a provider having patients in both study arms (see Fig. 1). Using SAS version 9.4 software, the study statistician developed a randomization scheme for allocating providers to the immediate or wait-list control arm and within stratification groups of provider type (advanced practice provider [nurse practitioner or physician assistant] vs physician). The resulting randomization assignment list was imported into the study’s tracking database. As providers consent to participate, they are assigned to the arm that was randomly associated with the next vacant slot on the randomization list for their provider type. Within the stratification groups, providers are allocated in block sizes (block size is unknown to everyone but study statisticians).

Study flow
Patient recruitment
Patient eligibility is determined using a combination of electronic medical record (EMR) data and telephone screening (see Table 2). We identify patients of enrolled PCPs with an upcoming primary care appointment (at least six weeks out) using EMR appointment schedules. We mail patients a recruitment letter and informational brochure about MeTree. Patients may opt out of the study by calling a toll-free number; otherwise, study personnel may contact potential patient participants by telephone to explain the study and assess interest. During the telephone contact, study staff verify eligibility and willing patients provide verbal consent (see Additional file 3 for informed consent language).
During the recruitment call, consented patients complete a baseline telephone survey (see Table 3) about health screening history and a computer proficiency assessment [28]. After completing the survey, we inform patients of their randomization assignment, determined by whether their PCP was randomized to the immediate versus wait-list control arm. Once randomized, patients remain in their original treatment assignment arm even if they change providers. To be enrolled in the study and thus included in analyses, patients must complete MeTree to determine their CRC risk level. We expect a lower MeTree completion rate for patients in the wait-list control arm compared to the immediate arm because of the planned 12-month delay between baseline survey and MeTree assessment in the control arm. Therefore, we plan to enroll approximately 100 more patients in the control arm than in the immediate arm to enable us to meet our targeted sample size of n = 250 completed MeTree assessments in each arm (see the “Statistical power and sample size” section).
Blinding
Enrolled providers are not informed of their study arm at enrollment. However, we do alert them that their patients will start receiving the intervention either shortly after enrollment or 12 months later. Patient receive their treatment arm assignment immediately after completing the baseline survey. Blinding of outcome assessors cannot be completely ensured as described below under “Measures,” but attempts are made to preserve blinding as much as possible.
Study procedures
Immediate arm
A few weeks before beginning patient enrollment, we provide PCPs randomized to the immediate group with details about the study (e.g. where to find the decision support document in the EMR) via a link to a video of a slide-based presentation with accompanying voice-over. We also provide them with a copy of the slide set. We ask providers not discuss the decision support documents, process, or experience with other clinicians. Study team members send PCPs randomized to the wait-list control group an email with this same information 12 months later. PCPs are responsible for ordering recommended tests as they deem clinically appropriate and per patient agreement after discussion of personal preferences and medical co-morbidities.
Patients assigned to PCPs who are randomized to the immediate arm are offered the choice of completing MeTree online on their own or at the hospital with study personnel. We provide the username and password for MeTree to patients by mail, text message, or the Veterans Health Administration’s Secure Messaging system (MyHealtheVet), or multiple modes, depending on patient preference. We encourage patients to complete MeTree before their upcoming primary care visit with reminder phone calls, text messages, or secure email messages sent up to twice per week. For patients who need assistance, they may choose to complete MeTree in-person by meeting with study personnel before their scheduled appointment or at another convenient time. The number of patients requiring assistance is tracked and recorded in a study database. For patients who complete MeTree on their own, we place the provider-facing report into the patient’s EMR and alert the PCP. Patients who complete MeTree in-person receive a paper copy of the patient-facing decision support documents to share with their PCP at their appointment and the provider-facing decision support document is uploaded into the EMR. Patients in the immediate arm complete an exit survey shortly after completing MeTree (e.g. experience with program and topics discussed with their PCP) and a follow-up survey 12 months later (e.g. updated assessment of personal health screenings and genetic testing) (see Table 3). After MeTree is completed and a clinical decision support document rendered, patients may still enter new information into the system that might develop at a later time; however, any changes in their report would not be transmitted.
Wait-list control arm
At 10–11 months after consent of the first several patients, we contact wait-list PCPs to provide the same education (i.e. informational email and slide set) that was provided to PCPs in the immediate arm. When we disclose the randomization assignment to patients in the wait-list control arm, we inform them that we will contact them 12 months post-consent to complete MeTree. We advise these patients that there is little risk associated with waiting, as CRC takes 7–10 years to develop. During the 12-month follow-up period, neither wait-list control patients nor their providers receive further contact from study personnel.
Ten months after each wait-list control patient provides consent, we mail a reminder letter informing them about the process to complete MeTree. We also telephone these patients to assess their preference for mode of completion using the aforementioned procedures. We follow the same procedures for uploading the PCP decision support document requiring electronic signature from the provider. Wait-list control arm patients complete their exit survey and their 12-month survey at the same time (see Table 3).
All patients receive a small financial compensation after completion of MeTree (baseline or 12 months later, depending on study arm).
Description of MeTree
MeTree is a patient-facing, web-based, FHH collection and clinical decision support platform developed in 2004 [25]. It was designed to include a patient-friendly, interactive interface for entering FHH. MeTree’s algorithms are based on current clinical guidelines used by PCPs (e.g. United States Preventive Services Task Force, National Comprehensive Cancer Network). The current version collects information on personal and FHH for 90 conditions and provides evidence-based, guideline-concordant, patient-specific recommendations for screening on 30 conditions [29]. Additional description of MeTree development, validation, and acceptability has been published [25, 29, 30].
We provide patients with a worksheet to assist them in collecting FHH from their family members before logging into MeTree. The worksheet is downloadable from the MeTree website but is also mailed to patients, depending on timing and preference. The worksheet explains the importance of FHH and guides patients to collect pertinent information from family members. Although this study focuses on CRC as the disease context for the primary outcome, we ask patients to enter their personal and FHH for all diseases included in MeTree. The MeTree decision support document includes decision support for any of the 30 conditions for which the patient may be at increased risk.
To complete MeTree, patients enter their personal and family health history directly into the web-based platform following easy-to-read prompts. Then, the MeTree program analyzes the data using existing guidelines to generate a report for the patient. MeTree also generates a provider report with citations for relevant supporting guidelines that is uploaded in the EMR for provider review.
Measures
We are collecting patient demographics and self-reported cancer screening at baseline and 12 months after consent (see Table 3). We also assess patients’ experiences using MeTree during a brief exit survey that is administered within one month of MeTree completion for both arms. We review the EMR and administrative data to assess the remaining outcomes. Study personnel at both sites conduct chart reviews. Blinding of outcome assessors cannot be ensured because the MeTree decision support document for PCPs is part of the patient’s EMR. However, we attempt to maintain blinding to the extent possible by having study personnel complete chart abstractions without accessing the study database so that they are not directly able to see the randomization status of each patient. To decrease the risk of ascertainment bias and to standardize the abstraction process, a detailed record abstraction protocol with data dictionary is created and training was conducted by a gastroenterologist on the study team (DF).
Outcomes
The primary outcome for this study is a binary outcome of whether risk-appropriate CRC screening referral occurred during the 12 months after consent. In order to measure this outcome, we need to know if a patient is at increased risk for CRC and the risk-appropriate recommendations for screening for that individual (i.e. the outcomes of our first aim). Thus, the primary outcome assessment follows completion of our first aim. Patients’ EMRs are reviewed for CRC-related screening referrals in the 15 months subsequent to consent. We are including an extra three months to capture any delayed uptake in recommended referrals. Specifically, clinical notes and consults are reviewed to identify referrals. For patients in both arms, risk-appropriate referral is based on the CRC risk level determined by the algorithm and recommendations from the MeTree program. Note that for patients determined to be at average risk for CRC by the MeTree program, a risk-appropriate referral may be no referral for CRC screening or surveillance.
Secondary outcomes are binary outcomes for patient completion and uptake of the CRC screening or surveillance referral, and risk-appropriate referral to genetic consultation over the 12-month follow-up period. We are examining patient uptake of CRC screening/surveillance referral for the subset of patients who received a risk-appropriate CRC screening or surveillance referral; patients who have no risk-appropriate referrals are not included in this analysis. A referral is identified by having an order in the record. Referral for CRC screening is identified as “recommended/ordered” and “completed.” “Recommended/ordered” is defined as the placement of an order for CRC screening or mention of such in the medical record, but the ordered screening is not completed. “Completed” refers to an order that was both placed and completed. We also look for the referral, actual order, and completion of risk-appropriate genetic consultation. As patient history can change over a 12-month period, when collecting self-reported screening history during the 12-month phone call, we ask patients if they have had any new relevant FHH diagnoses.
A secondary outcome for the study is baseline risk categorization for CRC in the EMR before study enrollment. We audit the EMR of all patients enrolled in the study to determine whether, at baseline, they were identified as being at increased risk for CRC based on an observation of either: (1) a diagnosis of being at increased risk for CRC (diagnosis code: V16.0); or (2) documentation of being at increased risk for CRC in the EMR. Study staff review progress notes or documentation of FHH of CRC, including the relationship and age at diagnosis or explicit statement of no FHH of CRC. This is accomplished with a text search. Initially, we are validating our search terms in a subset of 25 patients. If necessary, the terms will be adjusted to accommodate those found in the initial 25 patients and validation will be repeated on an additional 25 patients. To decrease the risk of ascertainment bias and to standardize the abstraction, a detailed record abstraction protocol with data dictionary was created and training will be conducted.
To complete follow-up and conduct both exit and 12-month surveys, we will attempt to reach patients by phone until the end of the survey window (one month after MeTree for the exit survey and 58 weeks after consent for the 12-month survey). If we cannot reach patients by phone for these surveys, we will still complete their chart review as described above.
Data management
Our primary and secondary outcome measures are collected in an electronic chart abstraction survey using data entry/management software DatStat Illume (version 5.1 before 29 January 2019 and version 6.1 subsequently). Baseline and 12-month follow-up survey data are also captured electronically in Illume. We developed a centralized tool for capturing data across study sites electronically using a custom patient tracking application and Illume. The tracking application and Illume are housed on a secure database server behind the VA firewall with access limited to key personnel. Data entered by patients into the MeTree online tool are stored on a secure Duke website that meets VA security standards. Throughout enrollment and data collection, we monitor data quality, accuracy, and timeliness.
Analyses
Primary aims
Aim 1 is to determine whether FHH collection via MeTree improves identification of patients at increased risk for CRC by comparing rates of increased risk identification in the medical record before study enrollment (baseline risk) to rates of high-risk identification by MeTree. As patients in both arms use MeTree for risk assessment, the design for this aim is a pre-/post-comparison within patient. We will test whether MeTree risk is higher than baseline risk using a McNemar’s test adjusted for clustering of patients within PCP. Note that the outcome for this aim is used in the primary outcome analysis.
Aim 2, the primary effectiveness aim, is to evaluate whether providing decision support to patients and PCPs improves risk-appropriate PCP referrals for CRC screening/surveillance and, secondarily, whether it improves patient uptake of CRC screening/surveillance and risk-appropriate referrals to genetic counseling. For the referral and uptake outcomes we will use generalized estimating equations (GEE) models to account for clustering of patients within PCP [31]. Because the primary and secondary outcomes are binary, we will use a GEE with a logit link function and an exchangeable correlation structure for each outcome. We will report 95% confidence intervals for the parameter estimates and p values from Wald t-tests with bias-corrected empirical sandwich estimators to maintain type I error control due to small numbers of clusters [32, 33]. Empirical standard errors are robust to misspecifications of the correlation structure.
The primary analyses for this cluster RCT will be conducted on an intent-to-treat basis. The main conclusions drawn from this trial will be based on the pre-specified primary and secondary hypotheses outlined previously and will be tested with two-sided p values at the standard level of 0.05 with effect size estimates and associated 95% confidence intervals reported. For both primary and secondary outcomes, stratification variables (provider type and patient age categorization) will be included in GEE models. Given that the outcomes are determined based on EMR review over a 12-month period, there should be very little missing data. The most likely scenarios to explain missing data are that a patient dies during the study or leaves the VA system before completion of the 12-month follow-up period.
We will conduct descriptive analyses of data acquired through the baseline, exit, and 12-month follow up surveys. Statistical analyses are performed using SAS for Windows (Version 9.4: SAS Institute, Cary, NC, USA) and R (http://www.R-project.org).
Statistical power and sample size
The sample size estimate of n = 250 patients with a completed MeTree assessment per arm (total n = 500) with 40 providers (20 in each arm) is based on 80% power, a type-I error rate of 5%, 4% loss to follow-up, and Z-test for the difference of two proportions adjusted for provider clustering. For the difference-in-proportions sample size calculation, we assumed a risk-appropriate screening referral rate for the wait-list control arm in the range of 60–70% [34]. The sample size for difference in proportions was adjusted by an intraclass correlation coefficient (ICC), a measure of “relatedness” of patients within a PCP to account for patient clustering within provider; we used an ICC of 0.02 [35, 36]. PASS 2008 [37] was used for all sample size calculations. Table 4 gives the estimated difference in proportions under the above stated assumptions that we can detect for the range of baseline referral rates in the wait-list control arm adjusted for clustering of providers and assuming a loss to follow-up of 4%.
For the secondary outcome, uptake of risk-appropriate CRC screening, our sample size is smaller as this outcome is only available for veterans that were referred for a CRC screening/surveillance test. Based on a hypothesized CRC screening/surveillance referral rate of 60% in the control arm and 73% in the immediate arm, the effective sample size after applying the 4% loss to follow-up is approximately 140 in the control arm and 180 in the immediate arm. With this effective sample size, a type-I error rate of 5%, and an ICC of 0.02, we can detect a difference of 15.6% in uptake of CRC screening test rates between arms with 80% power.
Secondary aims
Economic/cost analyses
We will also conduct a budget impact analysis (BIA) of the MeTree system to estimate the real-world financial implications of such an intervention. This analysis is conducted from the perspective of the VA healthcare system. Therefore, costs incurred by patients, such as travel or work absence to receive healthcare services, are not included. We will convert all cost figures to current dollars using the Bureau of Labor Statistics Consumer Price Index Calculator (https://data.bls.gov/cgi-bin/cpicalc.pl). Given that a horizon of 1–5 years is common for BIAs, we will use a three-year time horizon [38].
Our analyses include costs associated with the following elements: the FHH collection intervention; screening for CRC; genetic counseling; and genetic testing. These cost elements will be generated prospectively for both the immediate and wait-list control groups.
To derive intervention cost, we take an implementation perspective. The cost of the intervention will include cost of the computer required for MeTree use in clinic (if patient is met in clinic before appointment), providing the decision support documents, and the labor cost of providing assistance to patients to use MeTree and generate notes in the EMR for each patient’s PCP. We will account for the cost of training the coordinator to facilitate patient use of MeTree and entering notes in the EMR and of educating PCPs on the intervention. We will collect a sample of time required to perform each of these tasks and applying appropriate wage and fringe benefit rates to calculate labor cost. Collecting FHH through MeTree may be time-consuming and lead to longer patient visits. While we do not have a provider survey, we are conducting qualitative interviews with PCPs. During these interviews, we ask PCPs to quantify the impact of the MeTree tool on the length of their clinic visits. If PCPs report an increase or decrease in time spent with patients, then we will apply a wage-per-minute cost to the time difference to derive an incremental visit time cost. Study-related costs, such as the study coordinator’s time to consent and randomize patients, are not included. Development costs, such as designing MeTree and decision support materials, are treated as a one-time “sunk cost” that has already been incurred and are not included in our calculations.
The intervention may differentially impact utilization of healthcare services. We will derive screening costs of fecal occult blood test, flexible sigmoidoscopy, or colonoscopy from the VA’s Managerial Cost Accounting outpatient and inpatient extracts datasets. We will collect a sample of times required to provide CRC genetic counseling and wage and fringe benefit rate for genetic counselors to estimate the cost of providing CRC genetic counseling. For patients who receive genetic testing, we will apply the prevailing market rate for a genetic test at study conclusion to the analysis. Although intervention and screening costs are likely to be higher in the immediate arm, at least some of this cost may be mitigated by cost savings from CRC cases being averted. We will use estimates of the rate and cost of CRC from the literature to incorporate this cost component.
The net budget impact of the intervention is the difference in total costs incurred by patients in the immediate and wait-list control arms. We will also build on our BIA to conduct a cost-consequence analysis. The consequences of clinical interest are the additional number of appropriate referrals for genetic counseling, genetic testing, and additional cases of CRC detected due to the intervention. We will use these cost and consequence estimates from our sample to extrapolate what the budget impact and additional cases of CRC detected would be if the intervention were to be available for the entire VA healthcare system and veteran population.
Qualitative interviews with providers
Our study also includes a secondary aim to assess context for implementation. This aim employs qualitative methods to evaluate PCPs’ experiences using MeTree and how it affects their workflow. This information will provide important context to help interpret quantitative findings on uptake and effectiveness and additionally consider issues around operability and feasibility for future wide scale adoption and implementation. We aim to conduct 20-min semi-structured interviews by phone with 4–5 PCPs at each site. Guided by Weiner’s organizational model of implementation readiness [27, 39,40,41], the interviewer asks participating providers about their use of FHH in clinical decision-making (i.e. innovation-values fit), how they became aware of whether their patients completed MeTree, how they used or did not use the MeTree recommendations, how their patients reacted to the recommendations (i.e. implementation effectiveness), how MeTree affected their workflow and the time required to act on the recommendations, and any barriers to implementing MeTree recommendations (i.e. innovation-task fit). Interviews are audio-recorded and transcribed. Data will be analyzed using conventional content analysis, in which codes, or descriptive labels, will be attached to relevant passages of text in transcripts [42]. A team of two qualitative analysts code data, first working independently, and then meeting to evaluate consistency. Differences will be resolved by discussion and consensus. As is standard practice, analysis will be iterative (i.e. we code and discuss an initial transcript before moving to a second one with an updated coding scheme). The final coding scheme will be applied to all transcripts, recoding transcripts that were coded with earlier versions of the codebook as necessary. Analysts will identify themes by reviewing coded data for patterns about facilitators and barriers to inform future implementation [43].
Monitoring and ethics
This study does not have a data monitoring committee as no medical treatment is being provided as part of the intervention and all patients will eventually receive the intervention. There are no significant anticipated harms from this intervention; however, we track adverse events according to IRB regulations. Adverse events are not assessed systematically; they are recorded if we become aware of them (e.g. a patient mentions an illness during a conversation with a study team member). Significant protocol modifications are enacted only after IRB approval and are updated in trial registration and described in subsequent manuscripts.
Discussion
The systematic collection of a comprehensive FHH combined with readily available, guideline-concordant decision support is crucial to promote timely, patient-centered risk assessment and management. We are conducting a trial to test the effectiveness of such an approach in the VA while gathering information that will inform future implementation.
Our study design offers distinct strengths. First, previous studies of MeTree have not included a control group, precluding a power calculation and determination of effect size. Our study adds to this previous literature as we specifically chose the inclusion of a comparator group to allow for this statistical analysis. Second, in further distinction to previous MeTree studies, we focused on a sex-neutral primary outcome allowing us to explore applicability to men and women patients alike. Third, we are conducting this study in collaboration with developers of the software (co-investigators RW, ERH, and LO), which facilitates informed conversations about risk identification and incorporation into planned analyses. Finally, as part of the shaping of the recruitment process, we sought input from patient stakeholders. We met with the Durham Center for Innovation to Accelerate Discovery and Practice Transformation’s Veteran engagement panel, who reviewed our recruitment materials and provided suggestions for tailoring study procedures to the target population.
This study also has some limitations. First, the comparator arm will not receive risk assignment until 12 months after the immediate intervention arm. This choice is justified by the ethical implications of not providing risk assessment results to patients. Fortunately, the natural progression of CRC is slow and there is little concern about this delay in overall individual patient risk. Second, because randomization takes place at the provider level, the potential patient pool is subject to system-level changes and PCP turnover due to retirement and/or new positions; however, this is expected to be minimal. Finally, all patients enrolled in this study are Veterans receiving care through the Department of Veteran Affairs. While the VA patient population tends to differ in important ways from the general United States population (e.g. fewer women, more medical comorbidities), as a national, integrated system it is comparable to other similarly structured healthcare systems [44].
Conclusion
Results from this trial will determine the effectiveness and feasibility of incorporating patient-collected FHH and decision support on risk-identification and appropriate screening of increased-risk individuals in a large healthcare system such as the VA. Obtaining a detailed and accurate FHH is a critical step in the identification of patients at increased risk for certain cancers, including CRC. Unfortunately, FHH is rarely obtained or documented in a consistent manner that facilitates guideline-concordant, risk-appropriate care. This study will contribute to the body of literature pursuing the advancement of patient-centered, risk-appropriate care provision and the ultimate goal of reducing the morbidity and mortality of health conditions associated with genetic predisposition.
Trial status
-
a.
Protocol version number and date: Protocol version 14, approved by IRB 1-3-2019
-
b.
Date recruitment began: 21 August 2017
-
c.
Approximate date when recruitment will complete: 31 March 2019
Availability of data and materials
The datasets used and/or analyzed during the study will be maintained according to VA regulations and are not currently available for other investigators.
Abbreviations
- BIA:
-
Budget impact analysis
- CDS:
-
Clinical decision support
- CRC:
-
Colorectal cancer
- EMR:
-
Electronic medical record
- FHH:
-
Family health history
- GEE:
-
Generalized estimating equation
- ICC:
-
Intraclass correlation coefficient
- IRB:
-
Institutional review board
- PCP:
-
Primary care provider
- VA:
-
Veterans Administration
References
Centers for Disease Control and Prevention. United states cancer statistics: Data visualizations. Leading cancer cases and deaths, male and female. Atlanta: CDC; 2015. https://gis.cdc.gov/Cancer/USCS/DataViz.html
Atkin WS, Edwards R, Kralj-Hans I, et al. Once-only flexible sigmoidoscopy screening in prevention of colorectal cancer: A multicentre randomised controlled trial. Lancet. 2010;375(9726):1624–33.
Hardcastle JD, Chamberlain JO, Robinson MH, et al. Randomised controlled trial of faecal-occult-blood screening for colorectal cancer. Lancet. 1996;348(9040):1472–7.
Kronborg O, Fenger C, Olsen J, et al. Randomised study of screening for colorectal cancer with faecal-occult-blood test. Lancet. 1996;348(9040):1467–71.
Mandel JS, Bond JH, Church TR, et al. Reducing mortality from colorectal cancer by screening for fecal occult blood. Minnesota colon cancer control study. N Engl J Med. 1993;328(19):1365–71.
Mandel JS, Church TR, Bond JH, et al. The effect of fecal occult-blood screening on the incidence of colorectal cancer. N Engl J Med. 2000;343(22):1603–7.
Schoen RE, Pinsky PF, Weissfeld JL, et al. Colorectal-cancer incidence and mortality with screening flexible sigmoidoscopy. N Engl J Med. 2012;366(25):2345–57.
Levin B, Lieberman DA, McFarland B, et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: A joint guideline from the american cancer society, the us multi-society task force on colorectal cancer, and the american college of radiology. Gastroenterology. 2008;134(5):1570–95.
Wolf AMD, Fontham ETH, Church TR, et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the american cancer society. CA Cancer J Clin. 2018;68(4):250–81.
Bibbins-Domingo K, Grossman DC, Curry SJ, et al. Screening for colorectal cancer: Us preventive services task force recommendation statement. Jama. 2016;315(23):2564–75.
Gupta S, Provenzale D, Regenbogen SE, et al. Nccn guidelines insights: Genetic/familial high-risk assessment: Colorectal, version 3.2017. J Natl Compr Cancer Netw. 2017;15(12):1465–75.
Rex DK, Johnson DA, Anderson JC, et al. American college of gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol. 2009;104(3):739–50.
Lowery JT, Ahnen DJ, Schroy PC 3rd, et al. Understanding the contribution of family history to colorectal cancer risk and its clinical implications: A state-of-the-science review. Cancer. 2016;122(17):2633–45.
Eichler K, Zoller M, Tschudi P, et al. Barriers to apply cardiovascular prediction rules in primary care: A postal survey. BMC Fam Pract. 2007;8:1. https://doi.org/10.1186/471-2296-8-1.
Sabatino S, McCarthy E, Phillips R, et al. Breast cancer risk assessment and management in primary care: Provider attitudes, practices, and barriers. Cancer Detect Prev. 2007;31(5):375–83.
Suther S, Goodson P. Barriers to the provision of genetic services by primary care physicians: A systematic review of the literature. Genet Med. 2003;5(2):70–6.
Henrikson NB, Webber EM, Goddard KA, et al. Family history and the natural history of colorectal cancer: Systematic review. Genet Med. 2015;17(9):702–12.
Bronner K, Mesters I, Weiss-Meilnik A, et al. Do individuals with a family history of colorectal cancer adhere to medical recommendations for the prevention of colorectal cancer? Familial Cancer. 2013;12(4):629–37.
Lowery JT, Horick N, Kinney AY, et al. A randomized trial to increase colonoscopy screening in members of high-risk families in the colorectal cancer family registry and cancer genetics network. Cancer Epidemiol Biomark Prev. 2014;23(4):601–10.
Tsai MH, Xirasagar S, Li YJ, et al. Colonoscopy screening among us adults aged 40 or older with a family history of colorectal cancer. Prev Chronic Dis. 2015;12:E80.
Qureshi N, Armstrong S, Dhiman P, et al. Effect of adding systematic family history enquiry to cardiovascular disease risk assessment in primary care: A matched-pair, cluster randomized trial. Ann Intern Med. 2012;156(4):253–62.
Orlando LA, Wu RR, Myers RA, et al. Clinical utility of a web-enabled risk-assessment and clinical decision support program. Genet Med. 2016;18(10):1020–8.
Scheuner MT, Hamilton AB, Peredo J, et al. A cancer genetics toolkit improves access to genetic services through documentation and use of the family history by primary-care clinicians. Genet Med. 2014;16(1):60–9.
Fisher DA, Judd L, Sanford NS. Inappropriate colorectal cancer screening: Findings and implications. Am J Gastroenterol. 2005;100(11):2526–30.
Orlando LA, Buchanan AH, Hahn SE, et al. Development and validation of a primary care-based family health history and decision support program (MeTree). N C Med J. 2013;74(4):287–96.
Curran GM, Bauer M, Mittman B, et al. Effectiveness-implementation hybrid designs: Combining elements of clinical effectiveness and implementation research to enhance public health impact. Med Care. 2012;50(3):217–26.
Weiner BJ, Lewis MA, Linnan LA. Using organization theory to understand the determinants of effective implementation of worksite health promotion programs. Health Educ Res. 2009;24(2):292–305.
Boot WR, Charness N, Czaja SJ, et al. Computer proficiency questionnaire: Assessing low and high computer proficient seniors. Gerontologist. 2015;55(3):404–11.
Orlando LA, Wu RR, Beadles C, et al. Implementing family health history risk stratification in primary care: Impact of guideline criteria on populations and resource demand. Am J Med Genet C Semin Med Genet. 2014;166c(1):24–33.
Wu RR, Orlando LA, Himmel TL, et al. Patient and primary care provider experience using a family health history collection, risk stratification, and clinical decision support tool: A type 2 hybrid controlled implementation-effectiveness trial. BMC Fam Pract. 2013;14:111.
Zeger SL, Liang KY. An overview of methods for the analysis of longitudinal data. Stat Med. 1992;11(14–15):1825–39.
Li P, Redden DT. Small sample performance of bias-corrected sandwich estimators for cluster-randomized trials with binary outcomes. Stat Med. 2015;34(2):281–96.
Huang S, Fiero MH, Bell ML. Generalized estimating equations in cluster randomized trials with a small number of clusters: Review of practice and simulation study. Clin Trials. 2016;13(4):445–9.
Long MD, Lance T, Robertson D, et al. Colorectal cancer testing in the national veterans health administration. Dig Dis Sci. 2012;57(2):288–93.
Adams G, Guillford M, Ukoumunne O, et al. Patterns of intra-cluster correlation from primary care research to inform study design and analysis. J Clin Epidemiol. 2004;57(8):785–94.
Donner A, Klar N. Design and analysis of cluster randomization trials in health research. New York: Oxford University Press; 2000.
Hintze J. Pass 2008. Kaysville: NCSS, LLC; 2008.
Sullivan SD, Mauskopf JA, Augustovski F, et al. Budget impact analysis-principles of good practice: Report of the ISPOR 2012 Budget Impact Analysis Good Practice II Task Force. Value Health. 2014;17(1):5–14.
Helfrich CD, Weiner BJ, McKinney MM, et al. Determinants of implementation effectiveness: Adapting a framework for complex innovations. Med Care Res Rev. 2007;64(3):279–303.
Klein K, Sorra J. The challenge of innovation implementation. Acad Manag Rev. 1996;21(4):1055–80.
Weiner BJ, Haynes-Maslow L, Kahwati LC, et al. Implementing the move! Weight-management program in the veterans health administration, 2007-2010: A qualitative study. Prev Chronic Dis. 2012;9:E16.
Hsieh HSS. Three approaches to qualitative content analysis. Qual Health Res. 2005;15(9):1277–88.
Bradley EH, Curry LA, Devers KJ. Qualitative data analysis for health services research: Developing taxonomy, themes, and theory. Health Serv Res. 2007;42(4):1758–72.
Wong ES, Wang V, Liu CF, et al. Do veterans health administration enrollees generalize to other populations? Med Care Res Rev. 2016;73(4):493–507.
Acknowledgements
We would like to thank the following for their contributions to this project: Rachel R Adler ScD, RD; Avi Alkon; Jamiyla Bolton, MS; Kate Cronin MPH; Elizabeth Jeanes, MPH; Christine Kolehmainen MD; Erinn Mullan, APNP; Kerui Xu, PhD; and Jennifer Zervakis, PhD. We also thank Stacy Lavin for her editorial assistance. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs or the United States Government.
Funding
This study was funded by VA HSR&D IIR # 12–378. The funder had no role in study design; collection, management, analysis, and interpretation of data; writing of the report; or the decision to submit this report for publication. The funder also will not have ultimate authority over any of these activities. Dr. Voils’ time was also supported by VA HSR&D RCS #14–443. Dr. Goldstein’s time was supported by VA HSR&D CDA #13–263. This work was supported by the Durham Center of Innovation to Accelerate Discovery and Practice Transformation (ADAPT), (CIN 13–410) at the Durham VA Health Care System.
Author information
Authors and Affiliations
Contributions
CV was the primary lead for the conceptualization and design of this study, provided leadership for study conduct, and contributed substantially to this manuscript; KG contributed to the conduct of the intervention and contributed substantially to this manuscript; DF, LO, MS, TR-B, NS, ERH, and DP contributed to the conceptualization and design of the study and provided ongoing input during study conduct and the writing of this manuscript; CC and JG contributed to the design and development of the analysis plan as well as study conduct and contributed substantially to writing this manuscript; VW contributed to the conceptualization and design of this study, provided ongoing input during study conduct, and contributed significantly to this manuscript; SD contributed to the concept and design of this study; RN provided ongoing input during this study and contributed significantly to the writing of this manuscript; ES was a major contributor to the conduct of this study, acquiring data and assisting in drafting this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was reviewed and approved by the IRB at both the Durham VA Health Care System and Madison VA Medical Center. Informed Consent will be obtained from all study patients.
Consent for publication
Not applicable.
Competing interests
Drs. Orlando, Hauser, and Wu, and Ms. Rakhra-Burris are co-founders of MeTree&You, which is working towards providing the MeTree platform as a clinical service. Neither they nor the company received any financial gain from participating in this research study other than financial support for effort extended on research activities for this study. The other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
World Health Organization trial registration dataset. (DOCX 15 kb)
Additional file 2:
SPIRIT 2013 Checklist: Recommended items to address in a clinical trial protocol and related documents*. (DOC 122 kb)
Additional file 3:
Informed consent language. (DOCX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Goldstein, K.M., Fisher, D.A., Wu, R.R. et al. An electronic family health history tool to identify and manage patients at increased risk for colorectal cancer: protocol for a randomized controlled trial. Trials 20, 576 (2019). https://doi.org/10.1186/s13063-019-3659-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-019-3659-y