
Abstract
Background
Cough variant asthma (CVA) is one of the leading causes of chronic coughing. The main treatment is currently anti-inflammatory medication. However, the coughing may return or be aggravated and lung function may deteriorate once the anti-inflammatory treatment is stopped. The effect of Chinese herbal medicine (CHM) on chronic coughing is remarkable, but high-quality evidence supporting its effectiveness is still lacking. This trial aims to evaluate the safety and efficacy, especially the long-term efficacy, of CHM plus anti-inflammatory medications for the treatment of CVA.
Methods/design
A randomized placebo-controlled double-blind trial will be conducted. It will consist of a 3-month intervention followed by a 6-month follow-up period.
The target sample size is 60 patients with CVA who are between 18 and 70 years old. The eligible subjects will be allocated randomly into the experimental or control group in a ratio of 1:1. Patients in the experimental group will take CHM granules (4.9 g twice daily), while patients in the control group will be given a matched placebo. An administration of salmeterol/fluticasone propionate combination for 12 weeks will be the basic therapy for the two groups. The primary outcome is the cough visual analog scales (CVAS). The secondary outcomes include quality of life, rate of symptom relapse, lung function, and blood tests. A safety assessment will also be performed during the trial.
Discussion
The evidence gathered by the trial will be a valuable addition to informing treatment options for patients with CVA.
Trial registration
http://www.chictr.org.cn, ID: ChiCTR-IOR-16009148. Registered on 3 September 2016.
Similar content being viewed by others

Background
Cough variant asthma (CVA) is the most common cause of chronic cough [1,2,3]. According to a Chinese epidemiological survey, CVA accounts for 1/3 of cases of chronic cough [4]. In Japan, this proportion reaches 42.2% [5]. Long-term and constant coughing seriously affects the quality of life of patients with CVA. Such patients tend to be more depressed and anxious than classic asthma outpatients, on average [6]. Currently, maintenance therapy with a combination of inhaled corticosteroid and long-acting β2-agonist (ICS/LABA) is recommended for the treatment of CVA [7,8,9]. After discontinuing the anti-inflammatory treatment, cough symptom scores may increase, and lung function and eosinophilic airway inflammation may become aggravated and return to their baseline levels. The cumulative relapse rate of symptoms is as high as 67% after discontinuing treatment for 6 months [7].
Chinese herbal medicine (CHM) is widely used for CVA in China. It is reported that CHM plus ICS/LABA is better at reducing cough symptom scores, improving lung function, and improving quality of life compared with ICS/LABA alone [10,11,12]. Studies have also suggested that CHM plus ICS/LABA could further reduce levels of serum inflammatory factors, such as IL-10, IL-17, and IL-4/IFN-γ, compared to treatment with ICS/LABA alone [13, 14]. Furthermore, CHM plus ICS/LABA is better at reducing levels of airway neurotransmitters in the sputum than ICS/LABA alone [11]. Another study showed that compared to ICS/LABA alone, CHM plus ICS/LABA was superior at inhibiting airway remodeling in patients with CVA [15]. Although many clinical studies have proven that CHM plus ICS/LABA is superior to ICS/LABA alone for the treatment of CVA, the methodological quality of these studies was low because of the absence of sample size calculations, lack of definite random methods, unclear use of blinding methods, and so on. Furthermore, in only one study did participants undergo a follow-up period after stopping treatment [12]. How do cough symptoms, lung function, and airway inflammation change after stopping treatment with CHM plus ICS/LABA? Do these outcomes show a greater improvement after treatment with CHM plus ICS/LABA compared to after treatment with ICS/LABA alone? These questions remain to be determined.
A CHM prescription that was developed by Professor Zhang consists of ephedra (mahuang), asarum (xixin), fructus schisandrae (wuweizi), folium eriobotryae (pipaye), radix peucedani (qianhu), pummelo peel (huajuhong), folium perillae (zisuye), radix sileris (fangfeng) and radix asteris (ziwan). From the theory of traditional Chinese medicine, this prescription can warm the lungs, dispel wind, relieve coughing, and reduce the production of sputum. Most Chinese herbs in the prescription have been proven to be useful in the treatment of asthma. For example, it has been proven that the effective components of ephedra (mahuang) can decrease the secretion of IL-5, IL-13, and IL-8, alleviate the infiltration of eosinophils in the bronchoalveolar lavage fluid, inhibit airway remodeling, and inhibit the proliferation of airway smooth muscle cells [16,17,18,19]. Asarum (xixin) and fructus schisandrae (wuweizi) are often used in combination in Chinese medicine. Animal experiments have shown that the effective components of these two herbs may prolong the coughing latent period and decrease coughing frequency [20]. An extract of fructus schisandrae can also increase antioxidant levels and reduce oxidative damage. This also suggests that it may be helpful in the treatment of asthma [21]. It has been reported that folium eriobotryae (pipaye) has anti-inflammatory and immunoregulatory effects [22, 23]. An injection of folium eriobotryae can reduce the number of CD4+ cells, enhance the function of CD8+ cells, and regulate the CD4+/CD8+ imbalance [24]. The Th17/Treg cell balance has recently been discovered to be important in the pathogenesis of asthma. It has been reported that radix peucedani (qianhu) can alleviate airway inflammation and reduce airway hyperresponsiveness by regulating the imbalance of Th17/Treg cells [25]. In China, pummelo peel (huajuhong) is one of the most widely used Chinese herbs for respiratory diseases. Animal experiments have shown that pummelo peel can eliminate phlegm and relieve coughing, while also having anti-inflammatory, antioxidant, and immunomodulatory effects [26,27,28,29]. In addition, the other herbs in the prescription, folium perillae (zisuye), radix sileris (fangfeng), and radix asteris (ziwan), have anti-inflammatory effects as well. In general, the prescription may alleviate the symptoms or causes of asthma in multiple ways.
Based on a review of the literature and clinical observation, we predicted that our prescription plus ICS/LABA may be better at relieving coughing, improving lung function, and alleviating airway inflammation compared to ICS/LABA alone, even after the discontinuation of treatment. An observational study with a small sample size of 20 was carried out at a respiratory outpatient department. The control group was treated with ICS/LABA, while the experimental group was treated with ICS/LABA plus CHM. During the 12-week treatment period, the cough visual analog scales (CVAS) decreased from 5.13 ± 1.42 (mean ± standard deviation) to 1.21 ± 0.93 in the control group and from 5.11 ± 1.10 to 0.8 ± 0.69 in the experimental group. However, at the end of the 24-week follow-up phase, the CVAS increased to 4.50 ± 2.72 in the control group and 2.36 ± 2.54 in the experimental group. In addition, the cumulative percentages of patients whose symptoms worsened were 60% in the control group and 20% in the experimental group at the end of the follow-up period. We observed that ICS/LABA plus CHM was better for the treatment of CVA than ICS/LABA alone, not only during the 12-week treatment phase but also during the 24-week follow-up phase. Therefore, we have designed a randomized controlled trial to evaluate the long-term efficacy and safety of CHM plus ICS/LABA for the treatment of CVA.
Methods/design
Study design
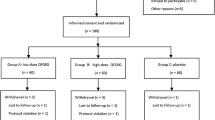
This is a single-center double-blind randomized controlled trial. The trial has been approved by the Institutional Ethics Committee of Guangdong Provincial Hospital of Chinese Medicine (GPHCM; B2016–097-01) and registered in the Chinese Clinical Trial Registry (ChiCTR-IOR-16009148). The trial aims to investigate the additional benefit and safety of oral CHM compared to salmeterol/fluticasone propionate combination (SFC) alone for the treatment of CVA, with special consideration of long-term efficacy. This study protocol conforms to the Standard Protocol Items: Recommendations for Interventional Trials guidelines (Fig. 1 and Additional file 1).

Spirit figure
Trial procedure
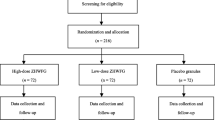
The entire trial consists of a screening assessment, a 2-week run-in period (weeks −2 and −1), a 12-week treatment period (weeks 1 to 12), and a 24-week follow-up period (weeks 13 to 36) (Fig. 2).

Trial flow chart. CHM Chinese herbal medicine, SFC salmeterol/fluticasone propionate combination
Potential participants will be invited to an initial assessment (first visit) consisting of screening for inclusion and exclusion criteria. If eligible, participants will be asked for informed consent and will undergo assessments of CVAS and quality of life, as well as a full blood count, blood tests on kidney and liver function, an electrocardiogram, and a chest radiograph examination. Then they will commence a 2-week run-in period. During this period, the participants’ use of asthma-related medication and CHM will be restricted. Participants will be provided with a salbutamol inhaler (Ventolin 5 mg/ml, GlaxoSmithKline, UK), to be used as required during this period to ease coughing until the treatment period begins. In the 2-week run-in period, the participants will be required to record the severity of their cough each day using the CVAS. After the run-in period, participants will undergo another set of baseline assessments (second visit), including assessments of quality of life, lung function, and blood tests.
After the baseline assessments, participants will be randomly allocated to receive 12 weeks of CHM plus SFC or placebo plus SFC. CVAS will be self-reported by the participants and recorded daily in a diary during the treatment period. Face-to-face assessments with blinded assessors will be scheduled at weeks 4 (third visit), 8 (fourth visit), and 12 (fifth visit). These will collect additional data, including lung function assessments and blood tests, as well as evaluations of quality of life, safety, and drug usage. Participants will be interviewed by telephone at weeks 2, 6, and 10 to determine adherence to the medication regulations and to report adverse events (AEs). The frequency of visits and phone calls can be increased if necessary.
During the 24-week follow-up phase, participants will continue to record CVAS daily in their diaries and document their utilization of health resources (including drug therapy use). They will attend follow-up assessments at weeks 24 and 36 (sixth and seventh visits). At each visit, quality of life, lung function, AEs, and drug usage will be assessed and participants will undergo blood tests.
Participants
The participants will be recruited from the respiratory outpatient department of GPHCM. Poster, newspaper, and Internet advertisements will also be used for recruitment. Participants will be included only if they satisfy all the following inclusion criteria: (1) the diagnostic criteria for CVA [9], (2) between 18 and 70 years of age, and (3) CVAS ≥ 2.
Patients with one or more of the following conditions will be excluded: (1) systematic corticosteroid treatment, bronchodilator treatment, or CHM treatment in the last 4 months; (2) serious acute or chronic organic disease or mental disorder; (3) pregnant, breastfeeding, or planning a pregnancy; (4) known sensitivity to the intervention drugs of the trial; and (5) participating in other trials or participation in a trial within the last month.
Interventions
Experimental group
The oral CHM granule will be produced by Tian Jiang Pharmaceutical Co., Ltd. (Jiangyin, Jiangsu, China). The herbs in the prescription will be mixed, cooked, filtered, and pressure spray-dried to form granules. The granules will be packaged into small single-dose sachets, each weighing 4.9 g. Participants in the experimental group will take two sachets twice a day for 12 weeks, dissolving each dose in warm boiled water. At the same time, patients will take SFC (50/250 μg, GlaxoSmithKline, England) twice a day for 12 weeks.
Control group
Placebo granules will be produced and packaged by the same manufacturer. They will consist of starch with no active ingredients. The placebo will be matched as closely as possible in appearance and taste to the real granules by adding various edible pigments. The dosage and administration of the placebo and SFC will be the same as in the experimental group.
Randomization, allocation concealment, and blinding
Eligible patients will be randomly allocated into the experimental group or the control group in a ratio of 1:1 through the central web-based interactive randomization service system run by GPHCM. A block randomization procedure will be performed by the Key Unit of Methodology in Clinical Research at GPHCM using SAS 9.2 software (SAS Institute Inc., Cary, USA).
The allocation will be concealed until the trial has closed and the analysis has completed. The patients, investigators, outcome assessors, and statisticians will not know the intervention allocation. The randomization schedule and blinding codes will be kept strictly confidential until the statistical analysis has completed. Blinding will be ensured by using a matched placebo granule identical in color, shape, and taste to the intervention granule. The quality of the CHM granule and the matched placebo will be rigorously controlled according to Chinese good manufacturing practices, and will be tested and verified by the researchers.
Outcome measures
Primary outcome
The primary outcome is CVAS change from baseline to the end of the follow-up phase (week 36). CVAS is used to assess the participants’ perceptions of the severity of their cough. Participants will be asked to draw a vertical line intersecting a 10-cm horizontal line to indicate the severity of their cough: zero is no cough while 10 cm is the worst cough imaginable [30].
Secondary outcomes
Quality of life
The total score in the Chinese version of the Leicester Cough Questionnaire [31] will be used to assess the quality of life of the participants (at weeks 1, 4, 8, 12, 24, and 36).
Lung function
Lung function will be determined mainly by measuring the forced expiratory volume in the first second (FEV1), the ratio of forced vital capacity occupied by forced expiratory volume in the first second (FEV1/FVC), and cumulative dose of bronchial activator to reduce FEV1 by 20% (PD20-FEV1) (at weeks 1, 12, 24, and 36).
Blood tests
The blood tests will include peripheral blood eosinophil count, eosinophil cationic protein, IgE, IFN-γ, and IL-4 (at weeks 1, 12, 24, and 36).
Other secondary outcomes
The rate of symptom relapse will be assessed at week 36. Overall health resource utilization will be recorded, including doctor visits, hospitalizations, and medication usage.
Safety assessment
Blood and urine samples, liver function, renal function, electrocardiograms, and chest X-rays will be examined at the end of the run-in period and at weeks 4 and 12. Researchers will pay attention to abnormal changes in the results.
An AE is any adverse medical event that occurs during the trial, regardless of whether it is related to the drugs of the trial. Patients will be required to report all AEs at each visit. The effects, degree, time of occurrence, duration, treatment process, and follow-up of AEs will be recorded on case report forms. All AEs will be reviewed to assess the causal effects of the interventions, according to the standards set by the Uppsala Monitoring Centre of the World Health Organization [32].
Termination and withdrawal
The trial will be terminated for any participant who develops one or more of the following conditions during the trial: (1) CVA develops into typical asthma, (2) intolerable side effects, or (3) serious acute or chronic organic disease. Any participant can withdraw from the trial for any reason at any time without prejudicing current or future treatments. The investigators will try to contact any withdrawn or terminated participants to complete the final assessment. Reasons for withdrawal or termination and the last medication time will be recorded. All withdrawn and terminated cases will be reported and analyzed.
Compliance
The compliance evaluation will be conducted using the pill counting method. Each participant will be asked to keep a daily medication diary. Investigators will telephone the participants every 2 weeks to check their compliance. At the subsequent visit, the participants will show the investigator their diaries and report their compliance with the doctor’s advice. Ongoing support, such as free registration and treatment advice, will be provided to the participants in the follow-up phase.
Sample size estimation
The sample size calculation for the primary outcome needs to be statistically and clinically significant. Based on previous literature [7] and our observational study, a sample size of 24 participants in each group can achieve 90% power to dismiss from a two-sided type I error of 0.05 to detect a superiority margin difference of 2.5 in this trial with two-tailed P values < 0.05 indicating significance. The total sample size was adjusted to 30 participants in each group to allow for 20% loss to follow-up and non-compliance.
Data management and quality control
The investigators in the research team were required to attend a training workshop before recruitment. Each one received a copy of the study protocol and they were asked to adhere to the protocol during the study period. The investigators responsible for collecting data received training on how to perform CVAS and quality of life assessments. The lung function and blood tests will be done by medical professionals.
Data will be collected and recorded on case report forms. All data will be entered into a predesigned password-protected database by personnel blinded to group allocation. Data entry will be performed continually throughout the trial using the double-entry method, with any corrections or changes of data written in the case report forms documented and dated. Data quality will be checked regularly by a qualified specially assigned person. The Guangdong International Clinical Research Center of Chinese Medicine (Guangzhou, China), which is independent of the sponsor and does not have any competing interests, will be responsible for monitoring the data. The Department of Science Research of GPHCM, which is independent of the sponsor and investigators, will perform data audits in the middle of the trial.
Statistical analysis
The data analysis will be carried out by an independent statistician using PASW Statistic 18.0 (IBM SPSS Inc., Armonk, New York, USA). Continuous data will be presented as means and standard deviations, or 95% confidence intervals. Dichotomous data, such as relapse of symptoms, will be presented as risk ratios and 95% confidence intervals. Two-tailed P values < 0.05 are considered to be statistically significant.
Baseline characteristics will be described using standard statistical analysis methods. The categorical variables will be analyzed via chi-squared tests or Fisher’s exact analyses, while continuous variables will be analyzed using t-tests. The main analyses include an intention-to-treat analysis and a per-protocol subject analysis for the primary outcomes. The last observation carried forward method will be used in the intention-to-treat analysis for missing data imputation. CVAS will be calculated as monthly averages, which will be compared with the average for the 2-week run-in period. Quality of life, lung function, and blood tests will be compared with the measurements from the end of the run-in period. Differences in changes from baseline to each time point between the two groups will be compared using analyses of variance. Paired t-tests will be used to compare data from before and after treatment in the same group. In the analysis of factors affecting the outcome changes, mixed-effects linear or logistic regression models will be performed, adjusting for baseline characteristics and other variables. Descriptive analyses and chi-squared tests or Fisher’s exact tests will be used for the safety data analysis. Any causality between AEs and the interventions will be taken into account.
Discussion
To our knowledge, this is the first clinical study to investigate the long-term efficacy of CHM plus ICS/LABA for CVA. Compared to previous studies, we are focusing on CVAS, lung function, and airway inflammation, not only for the treatment phase but also during the 6-month follow-up phase.
In clinical practice, compliance with asthma medications is poor, especially for the inhaled glucocorticoids used for maintenance therapy. It is reported that only 52.4% of patients with CVA regularly inhaled ICS/LABA within the past 6 months [33]. This can lead to undertreatment of any underlying inflammation and increased risk of exacerbation. If the CHM granule plus ICS/LABA treatment is found to be safe and effective in the long term for CVA patients, it will be a great help in resolving this problem. The results of this trial will provide high-quality evidence of the efficacy of CHM in the treatment of CVA.
Trial status
Recruitment into this study started on 1 January 2017, following protocol version 001/20160603. The final participants are expected to complete the 6-month follow-up assessment in mid-2018.
Abbreviations
- AE:
-
Adverse event
- CHM:
-
Chinese herbal medicine
- CVA:
-
Cough variant asthma
- CVAS:
-
Cough visual analog scales
- FEV1:
-
Forced expiratory volume in 1 s
- GPHCM:
-
Guangdong Provincial Hospital of Chinese Medicine
- ICS/LABA:
-
Inhaled corticosteroid (glucocorticoid) and long-acting β2-agonist
- PEF:
-
Morning and evening peak expiratory flow
- SFC:
-
Salmeterol/fluticasone propionate combination
References
Irwin RS, Corrao WM, Pratter MR. Chronic persistent cough in the adult: the spectrum and frequency of causes and successful outcome of specific therapy. Am Rev Respir Dis. 1981;123:413–7.
Liu GL, Lin JT. The spectrum and clinical features of causes for chronic cough. Chin J Tuberc Respir Dis. 2009;32:422–5.
Si SY, Peng QF, Shi X, Kong LF. The spectrum and clinical features of causes for chronic cough in Shengyang and surrounding areas. Chin J Tuberc Respir Dis. 2010;33:862–3.
Lai KF, Chen RC, Lin JT, Huang KW, Shen HH, Kong LF, et al. A prospective, multi-center survey on causes of chronic cough in China. Chest. 2013;143:613–20.
Niimi A, Ohbayashi H, Sagara H, Yamauchi K, Akiyama K, Takahashi K, et al. Cough variant and cough-predominant asthma are major causes of persistent cough: a multicenter study in Japan. J Asthma. 2013;50:932–7.
Saito N, Itoga M, Tamaki M, Yamamoto A, Kayaba H. Cough variant asthma patients are more depressed and anxious than classic asthma patients. J Psychosom Res. 2015;79:18–26.
Tagaya E, Kondo M, Kirishi S, Kubota N, Tamaoki J. Effects of regular treatment with combination of salmeterol/fluticasone propionate and salmeterol alone in cough variant asthma. J Asthma. 2015;52:512–8.
Bao W, Chen Q, Lin Y, Liu H, Zhao G, Chen Z, et al. Efficacy of procaterol combined with inhaled budesonide for treatment of cough-variant asthma [J]. Respirology. 2013;18(Suppl 3):53–61.
Asthma association of Chinese medical association respiratory branch. Diagnosis and treatment of chronic cough. Chin J Tuberc Respir Dis. 2016;39:1–32.
Liu Y. Effects of Qingjin Runfei Decoction assisted salmeterol/fluticasone combination in the treatment of cough variant asthma. Mod J Integr Tradit Chin West Med. 2016;25:2106–7.
Lu F, Wang SC, Chen ZB. Majing Zhike Granule in treating 37 patients with cough variant asthma of wind exuberance syndrome. Fujian J TCM. 2017;48:1–3.
Gan DK. Clinical evaluation of Fuzheng Qufeng Granule combined with Salmeterol Cardon pulp inhalation in treating Qixu Fengsheng cough variant asthma. Henan: Henan University of Traditional Chinese Medicine; 2017. p. 1–36.
Zhou Q, Li HX, Wang L, Liu RB, Zhao GY. Effect of Shufeng Xuanfei Decoction on airway inflammatory factors in patients with cough variant asthma. J Chin Med Mater. 2017;40:2974–6.
Ma JR, Zhang CX, Han Y, Ba YY, Liu SY. Budesonide/Formoterol combination combined with Chinese herb medicine in the treatment of elderly cough variant asthma. Chongqing Med J. 2017;46:2962–5.
Han B, Huang ZY. Effect of inhalant combined with Dingchuan Zhike decoction therapy on the airway remodeling, inflammation and PARC/CCL-18 pathways in patients with cough variant asthma. J Hainan Med Univ. 2017;23:1052–5.
Li ZY, Deng J, Xiong B, Xiong Y, Wang SP. Effect of ephedrine on expression of interleukin-8 in human bronchial epithelial cells. Shandong Med J. 2016;56:19–21.
Wang J, Xiong Y, Xiong B, Wang SP. Effects of aerosolized aqueous extract of ephedra on airway inflammation in asthmatic mice. Chongqing Med J. 2013;42:304–7.
Xiong Y, Xiong B, Wang SP, Yang LT, Li YK, Sun XW. The effect of aqueous extract of ephedra on the airway inflammation and epithelial STAT1 expression of guinea pig with asthma. Med J West China. 2008;20:913–6.
Jiong HJ, Wang CD, Song S, Gan L, He GY. Effects of ephedrine on proliferation of bronchial smooth muscle cells. J Biol. 2008;25:27–9.
Wu JB, Zhe XP, Zhang YY, Qiu MF, Jia W. Screening of the Anti-Asthma Active Sites for the Drug Partnership Containing Fructus Schisandrae and Asarum. Chin Arch Tradit Chin Med. 2013;31:121–3.
Chen X, Feng J. Effects of Fructus Schisandrae extract on blood antioxidant activity in asthmatic rats. China Pharmaceuticals. 2013;22:25–7.
Guo L, Zhang XB. Effect and mechanism of Folium Eriobotryae triterpenic acid on chronic bronchitis in rats. J Chin Med Mater. 2015;38:2166–8.
Ge JF, Li J, Hu CM, Jin Y, Lv XW, Peng L, Zhang L, Yu SC. Immunomodulatory effect of triterpenic acid in Folium Eriobotryae. Chin Pharmacol Bull. 2006;22:1194–8.
Wei Y, Tang HF, Li XH, Zhu XY. Effect of Folium Eriobotryae Injecion on T lymphocyte subsets in asthmatic mice. JETCM. 2013;22:1151–2.
Xiong YY, Shi WJ, Yu H, Zhang XL, Miu CG, Ma ST. Regulation of Praeruptorin C on the Imbalance of Th17/Treg in a Mouse Asthmatic Model. Chin J Mod Appl Pharm. 2015;32:671–6.
Yan C, Wu H, Nie Y C, et al. Mucoactive effects of naringin in lipopolysaccharide-induced acute lung injury mice and beagle dogs. Environ Toxicol and Pharmacol, 2014;38:279-87.
Nie Y C, Wu H, Li P B, et al. Anti-inflammatory effects of naringin in chronic pulmonary peutrophilic inflammation in cigarette smoke-exposed rats. J of Med Food, 2012;15:894-900.
Xu X F. The study of processing technology and quality standards of exocarpium decocting pieces. Guangzhou: Guangzhou University of Traditional Chinese Medicine; 2012. P. 1-103.
Dong H P, Jiang M S, Zhu W J. Immunomodulatory effect of Pummelo Peel on mice. Chin Traditional Patent Med, 2010;32:491-3.
Lacourciere Y, Brunner H, Irwin R, Karlberg B, Ramsay L, Snavely D, et al. Effects of modulators of the renin-angiotensin-aldosterone system on cough. J Hypertens. 1994;12:1387–93.
Ma W, Yu L, Li X, Lu H, Qiu Z. Changes in health-related quality of life and clinical implications in Chinese patients with chronic cough. Cough. 2009;5:7.
WHO, “The use of the WHO-UMC system for standardized case causality assessment.” http://www.who.int/medicines/areas/quality_safety/safety_efficacy/WHO%20causality%20assessment.pdf
Ge M, Zhu X, Wang J, Guo YD, Cheng SH, Li YK. Compliance analysis of inhaled medication in patients with cough variant asthma. Front Med. 2016;6:142–3.
Acknowledgements
We thank the staff working in the lung function room for their help with subject recruitment.
Funding
This trial is supported by the special project for scientific research and construction of the national Chinese medicine clinical research base, funded by the State Administration of Traditional Chinese Medicine, China (TDZX2015200). The funding body did not participate in the study design or the collection, management, analysis, and interpretation of data. The funding body has provided only financial support for the study.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Dissemination policy
Our results will be published in peer-reviewed journals, regardless of whether they are positive or negative.
Author information
Authors and Affiliations
Contributions
RF and YL designed the trial and drafted the manuscript. DW, ZW, LJ, and RY contributed to the design and made critical revisions. AO was responsible for the statistical analysis. ZZ developed the CHM formulation and the matched placebo and assisted in designing and managing the trial. All authors read and approved the final version of this manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The trial has been approved by the Institutional Ethics Committee of GPHCM (B2016–097-01). Any protocol amendments must be reported to the Department of Science Research of GPHCM and agreed by the ethics committee. Each participant will receive information on this research, including the potential benefits and risks. The investigators must obtain informed consent from a patient before they are randomized. The personal information of the enrolled participants will be kept confidential. Only relevant investigators can access this information.
Consent for publication
Only anonymized data obtained in the observational study will be reported. Informed consent has been obtained from the patients for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
SPIRIT Checklist. (DOC 118 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fan, Rr., Wen, Zh., Wang, Dw. et al. Chinese herbal medicine for the treatment of cough variant asthma: a study protocol for a double-blind randomized controlled trial. Trials 20, 3 (2019). https://doi.org/10.1186/s13063-018-3073-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-3073-x