
Abstract
Background
There is a widely recognized need for more pragmatic trials that evaluate interventions in real-world settings to inform decision-making by patients, providers, and health system leaders. Increasing availability of electronic health records, centralized research ethics review, and novel trial designs, combined with support and resources from governments worldwide for patient-centered research, have created an unprecedented opportunity to advance the conduct of pragmatic trials, which can ultimately improve patient health and health system outcomes. Such trials raise ethical issues that have not yet been fully addressed, with existing literature concentrating on regulations in specific jurisdictions rather than arguments grounded in ethical principles. Proposed solutions (e.g. using different regulations in “learning healthcare systems”) are speculative with no guarantee of improvement over existing oversight procedures. Most importantly, the literature does not reflect a broad vision of protecting the core liberty and welfare interests of research participants. Novel ethical guidance is required. We have assembled a team of ethicists, trialists, methodologists, social scientists, knowledge users, and community members with the goal of developing guidance for the ethical design and conduct of pragmatic trials.
Methods
Our project will combine empirical and conceptual work and a consensus development process. Empirical work will: (1) identify a comprehensive list of ethical issues through interviews with a small group of key informants (e.g. trialists, ethicists, chairs of research ethics committees); (2) document current practices by reviewing a random sample of pragmatic trials and surveying authors; (3) elicit views of chairs of research ethics committees through surveys in Canada, UK, USA, France, and Australia; and (4) elicit views and experiences of community members and health system leaders through focus groups and surveys. Conceptual work will consist of an ethical analysis of identified issues and the development of new ethical solutions, outlining principles, policy options, and rationales. The consensus development process will involve an independent expert panel to develop a final guidance document.
Discussion
Planned output includes manuscripts, educational materials, and tailored guidance documents to inform and support researchers, research ethics committees, journal editors, regulators, and funders in the ethical design and conduct of pragmatic trials.
Similar content being viewed by others

Background
Pragmatic versus explanatory trials
Pragmatic trials aim to determine if an intervention works in real-world settings, so that results can be generalized to everyday practice and support decision-making by patients, providers, and health system leaders; contrastingly, explanatory trials aim to determine if and how an intervention works under well-defined and highly controlled conditions [1]. While differences between explanatory and pragmatic attitudes in trials were first highlighted in a seminal article by Schwartz and Lellouch five decades ago [2], interest in pragmatic trials has increased dramatically in recent years [3]. Accordingly, reporting guidelines for pragmatic trials have been recently published [4].
In practice, trials are seldom purely pragmatic or purely explanatory, but various design choices can make a trial more or less pragmatic. The PRagmatic Explanatory Continuum Indicator Summary (PRECIS-2) was developed to help trialists identify explicit design choices that can shift a trial towards being more pragmatic [5]. PRECIS-2 has nine dimensions along which a trial can be scored from very explanatory to very pragmatic. In brief, trials that are more pragmatic have broader eligibility criteria, recruit participants at the time of presentation, include a diverse range of settings that mirror real-world circumstances, do not require highly specialized training or research personnel, give healthcare providers flexibility in how the intervention is delivered, require no special strategy for monitoring protocol compliance, follow and monitor patients as in routine clinical practice, have clinically meaningful and patient-centered outcomes, and include all randomized patients in analysis.
Explanatory trials, more often conducted on innovative medical products and devices for regulatory purposes, usually do not fully explicate the benefits and harms of interventions relative to existing alternatives and hence do not address the central question of what is likely the best (among the available) options for particular patients. In contrast, pragmatic trials test a much wider range of interventions, including diagnostic, preventive, therapeutic, and delivery system interventions. They may test new interventions against current routine interventions or the comparative effectiveness of different routine interventions head-to-head. When designed appropriately, they may address not only whether an intervention works, but more importantly, for whom and under what conditions. They may test different quality and service improvement interventions as well as knowledge translation interventions. Pragmatic trials therefore offer an important opportunity to improve patient health and health system outcomes by reducing variations in care, improving uptake of evidence-based practice, and reducing costs. For these reasons, the need for more pragmatic trials has been identified as a priority by governments worldwide [6,7,8,9,10].
This manuscript presents the study protocol for a four-year, interdisciplinary, mixed methods research project with the ultimate goal to develop internationally accepted guidance for the ethical design and conduct of pragmatic trials. Although there are many innovative observational study designs which can be used to evaluate interventions and produce new knowledge that informs decision-making [11], here we focus exclusively on intervention studies that use randomization. We allow for a broad range of intervention types including diagnostic, preventive, therapeutic, knowledge translation, and delivery system interventions. See Table 1 for a brief glossary of terms used in this manuscript.
Ethical issues in pragmatic trials
Trials undertaken in real-world settings raise substantial ethical issues that have not yet been fully addressed [12,13,14,15]. These ethical issues arise not only from the push towards a greater degree of pragmatism (e.g. along the nine PRECIS-2 dimensions), but are closely tied to the types of interventions, as well as the choice of study design. In addition to established study designs which use patient randomization (see Example 1 below), pragmatic trials include some emerging designs and approaches which capitalize on methodological and statistical innovations as well as the availability of registries and routinely collected health data (e.g. cohort multiple randomized controlled designs [16], randomized registry trials [17], cluster cross-over trials [18], and stepped wedge cluster randomized trials [18, 19]). The cohort multiple randomized design and randomized registry trials are examples of pragmatic trial designs that facilitate the evaluation of usual care interventions embedded within routine settings, but their ethical implications remain unclear. Cluster randomization [20] may be used to randomize entire medical practices or hospitals to differing interventions and is a natural choice for evaluating service delivery or other health system level interventions; however, with the push towards more pragmatic trials, this design is increasingly being used to evaluate individual-level interventions (i.e. interventions that, in theory, could have been evaluated using traditional patient randomized designs). While the Ottawa Statement on the Ethical Design and Conduct of Cluster Randomized Trials [21] provides explicit guidance for cluster randomized trials, the use of this design in the case of usual care individual-level interventions raises additional ethical issues (see Example 2 below); moreover, the stepped wedge cluster randomized design raises its own unique ethical issues (Example 3).
Table 2 describes a preliminary framework of nine ethical issues; the pragmatic, intervention, and design characteristics that give rise to these issues; and their potential implications for researchers and research ethics committees. This framework was developed based on an initial scoping review [12] and will be further developed and refined during our project.
Examples of pragmatic randomized controlled trials
For illustrative purposes, we present three examples of pragmatic trials that raise important ethical issues needing guidance.
Example 1: patient-randomized trial comparing usual care interventions (SUPPORT)
The SUPPORT trial [22] sought to determine the optimal level of supplemental oxygen on incidence of retinopathy of prematurity and mortality in preterm infants. Infants were randomized to high and low oxygen saturations, both of which fell within the range used routinely in practice. Lower oxygen levels were found to reduce retinopathy but increase mortality, leading the authors to urge caution in their use. In response to an anonymous complaint by parents, the US Office for Human Research Protections launched an investigation into the trial [23]. It determined that investigators failed to adequately inform parents of “reasonably foreseeable risks” as consent materials did not list visual impairment and death as research risks. This incited considerable debate in the literature [24,25,26,27] centered almost exclusively on considerations of risk and risk communication and reflecting a preoccupation with US regulations. A central ethical issue raised by the SUPPORT trial is: Should usual care interventions be considered part of research or clinical practice? Other issues raised are: What constitutes usual care? Can a study be considered “minimal risk” if its endpoints include serious impairments such as blindness or death? Do parents need to be informed that their child is participating in a trial? At what level of detail do the study interventions need to be disclosed to participants? What potential benefits and harms ought to be disclosed? What type of research ethics review is appropriate?
Example 2: cluster randomized registry trial comparing usual care individual-level interventions implemented as policy interventions at the cluster-level (FLUID)
The FLUID trial is a pragmatic cluster randomized cross-over trial comparing resuscitation with Ringer’s Lactate versus normal saline on death and hospital readmissions in hospitalized patients, with outcomes assessed using health administrative data [28]. Both fluids are considered usual care interventions that have been available for decades and are administered to many hospitalized patients. While fluid administration is an individual-level intervention, the study becomes feasible only when interventions are implemented as hospital policies (i.e. at the cluster level) with a waiver of patient informed consent. In particular, it is essential to have only one type of study fluid available throughout the hospital to minimize the risk of contamination due to a patient receiving both types of study fluids in different areas of the hospital. It would also be logistically challenging and very costly to recruit and randomize individual patients within all areas of participating hospitals. In contrast, the availability of routinely collected data available for all patients permits the conduct of a very cost-efficient trial involving hospitals across the province, without the need to recruit patients for data collection. However, the FLUID trial raises several ethical issues: What is an appropriate justification for adopting cluster randomization? When individual-level interventions are implemented institution-wide as a policy in a cluster randomized trial, may one proceed without patient consent? Do patients need to be notified about the trial; if so, how? Is consent required for the use of routinely collected participant data?
Example 3: stepped wedge trial of a quality improvement intervention (Surgical Checklist Trial)
The Surgical Checklist Trial was a stepped wedge trial randomizing surgical units in two hospitals in Norway to evaluate the impact of the World Health Organization Surgical Safety Checklist on morbidity, mortality, and length of hospital stay [29]. The stepped wedge design is characterized by the fact that clusters (here, surgical units) cross gradually and in random sequence from the control to the intervention condition, with all clusters exposed to the intervention by the end of the trial. As stated by the investigators, the 19-item checklist consists of an oral confirmation by surgical teams of the completion of the basic steps for ensuring safe delivery of anesthesia, prophylaxis against infection, effective teamwork, and other essential practices in surgery. Compliance with the checklist was assessed prospectively by nurses. All patient outcomes were collected from hospital administrative databases. The regional research ethics committee advised the investigators that the study was considered clinical service improvement and that research ethics approval and patient informed consent were not required. The Surgical Checklist Trial raises several ethical issues: Should trials evaluating quality and service improvement be considered research? What are the ethical implications if health system leaders conduct (potentially less robust) quality improvement studies rather than rigorous pragmatic randomized controlled trials or refrain from conducting important studies due to perceived ethical barriers? Should health professionals targeted by study interventions be considered research participants? If study participation poses no more than minimal risk, is patient or provider consent required? What is the harm–benefit balance of exposing all surgical units to the intervention by the end of the study?
Challenges to the conventional framework for research ethics
As illustrated by our three examples, pragmatic trials raise important ethical issues that have not yet been satisfactorily addressed. Existing ethical and regulatory frameworks were developed primarily for trials with explanatory aims, i.e. focusing on efficacy and safety of experimental interventions for marketing approval. With the move towards the conduct of more pragmatic trials, existing ethics guidance is becoming more difficult to interpret and apply and may not be sufficient to address these new ethical issues. On the one hand, the absence of clear guidance may put trials at risk of being accused of exposing participants to inadequate protections (real or perceived) (e.g. the SUPPORT trial [23]). It is essential to avoid undermining the trust that patient communities have in the research enterprise. On the other hand, strict enforcement of conventional rules is likely to pose unnecessary obstacles, undermine scientific quality or impede improvements in patient health and health system outcomes. Unless these ethical issues are addressed, important research with large potential healthcare benefits may be impeded or may go ahead without adequate safeguards.
Objectives
The overarching goal of our project is to develop guidance for the ethical design and conduct of pragmatic trials. Specific objectives are to:
-
1.
Systematically identify ethical issues arising from pragmatic trials;
-
2.
Document ethical practices in completed and ongoing pragmatic trials;
-
3.
Elicit views and experiences of trialists, methodologists, chairs of research ethics committees, trial participants, and health system leaders;
-
4.
Develop novel ethical solutions informed by ethical analyses;
-
5.
Generate ethics guidance through a consensus process with an independent expert panel;
-
6.
Disseminate tailored guidance to stakeholders.
Methods
Overview
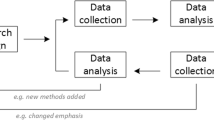
Our project, summarized in Fig. 1, consists of both empirical and conceptual work and concludes with a consensus development process and knowledge translation. The empirical work, to be completed over the first three years, consists of five studies:
-
1.
Key informant interviews with a small group of 24–40 experts;
-
2.
Identification and review of a random sample of 300 completed and ongoing pragmatic trials;
-
3.
Survey of trialists (investigators of the 300 pragmatic trials);
-
4.
Survey of a random sample of chairs of research ethics committees in Canada, USA, UK, France, and Australia; and
-
5.
Embedded focus group discussions and a community survey with trial participants (e.g. patients) and gatekeepers (i.e. those who have ability to allow or deny access to trial participants).

Overview of project phases
All outputs will inform the ethical analysis in the conceptual work.
The conceptual work will be an ethical analysis of the identified ethical issues from the empirical work, resulting in a series of publications outlining proposed solutions with principles, policy options, and rationales. The empirical and conceptual work will proceed concurrently to allow the empirical and ethical analyses to be mutually informative.
The consensus process, occurring upon completion of the empirical and conceptual work, will involve identifying an international expert panel of trialists, ethicists, chairs of research ethics committees, regulators, funders, and community representatives and organizing a consensus conference attended by the panel, as well as invited researchers, representatives of funding agencies, regulators, journal editors, and patient and community groups. In addition, we will engage in e-consultation with the broader research community, funders, regulators, journal editors, and patient groups. The panel will produce the final ethics guidance document which will be disseminated to stakeholders by the research team.
Empirical studies
Study 1: key informant interviews
The objective of this study is to conduct interviews with a small group of pragmatic trial experts and stakeholders (trialists, ethicists, methodologists, chairs of research ethics committees, health system leaders, quality improvement experts, and patient representatives on research study teams) to generate a thorough understanding of types of ethical issues arising in the practice of pragmatic trials from a variety of perspectives. Informants will be selected using a purposive sampling strategy, augmented through snowball sampling. Potential interviewees will be selected across a broad range of jurisdictions and clinical areas to reflect a range of experiences, including lower- and middle-income countries. The sample size will be determined to ensure representation from all targeted stakeholders and by when saturation is reached (i.e. when new interviews cease to provide fresh information) [30,31,32,33,34]. Based on our prior experience with similar studies [35], we anticipate that 24–40 interviews will be required. Interviews are expected to last 1 h and will be audio recorded. A semi-structured interview guide will allow participants to respond freely, to illustrate concepts, and to present perspectives that the interviewer can probe further. To monitor the progress of the interviews and permit follow-up of issues that may emerge from the data, interviewing, transcription, and analysis will proceed concurrently. The interview guide may evolve as a typology of ethical issues begins to emerge. Recordings will be transcribed and verified before analysis. Data will be imported into a qualitative software package (NVivo 11) to facilitate thematic coding, evaluation, and analysis. The results will be used to formulate a typology of ethical issues arising from pragmatic trials, to be addressed in the conceptual work. It will also inform data extraction and questionnaire items for studies 2–5.
Study 2: review of published trials
The objectives of this study are to select and review a random sample of recently completed and ongoing trials that have more pragmatic (than explanatory) aims, to describe ethical characteristics, identify ethical challenges reported, the circumstances under which they arise, and how they are being addressed. We anticipate challenges in identifying a sample of “pragmatic” trials given wide variation in definitions and inconsistent and unreliable reporting of trial design. We will develop objective and reproducible criteria to characterize a trial as having pragmatic aims, as well as the conditions under which those criteria apply. We will work with an information scientist to develop a sensitive and specific electronic search strategy to identify a sample of trials. To develop and validate an electronic search strategy, we will use a multi-pronged approach to identify a gold standard set of trials meeting our criteria for testing the search strategy, including: (1) pragmatic trials conducted by the investigator team and our extensive networks; (2) pragmatic trials identified in the key informant interviews; (3) databases of funded pragmatic trials in Canada, the USA, UK, France, and Australia; (4) a database of pragmatic trials maintained by the PRECIS group [36]; (5) demonstration projects by the National Institutes of Health Collaboratory [37] and PCORnet [38]; and (6) trials included as exemplars in recent publications about pragmatic trials. The proportion of these pragmatic trials retrieved by the search strategy will be calculated and used to refine the search strategy if necessary as done in our previous work [39]. Once validated, the search strategy will be used to select a random sample of 300 pragmatic trials. As the ethical landscape may have been changing in recent years, we will include completed trials as well as study protocols for ongoing trials. Eligibility criteria, to be refined, will include trials or study protocols published in the past five years by investigators in Canada, UK, USA, France, and Australia, including trials conducted in lower- and middle-income countries. Items for extraction will be generated based on team discussion and key informant interviews. After pilot testing, two reviewers will independently extract data from each trial report. Discrepancies between reviewers will be identified and resolved by discussion with a third reviewer if required. Prevalence of ethical issues and practices arising from pragmatic design features, study design, and type of study interventions will be described overall and within subgroups of interest where feasible (e.g. over time, between countries, study sponsors). Methodological and reporting quality of the included trials will be collected and evaluated against major methodological and reporting criteria as done in our previous work [40, 41]. The preliminary ethics framework will be updated as extractions proceed.
Study 3: survey of trialists
The objective of this study is to gather more detailed information about practices and experiences identified in our review of pragmatic trials. After pilot testing, we will administer a survey consisting of open- and closed-ended items to corresponding authors of the sample of 300 trials. The primary mode of survey administration will be web-based, but alternatives (paper, telephone) will be considered to increase the response rate. A series of contacts (pre-notifications, notifications, and reminders) based on Dillman’s recommendations for the implementation of mail and Internet surveys will be used [42]. The survey will be used to characterize the ethical conduct, review, and reporting of pragmatic trials from the perspectives of trialists. We will offer respondents a $30 gift certificate or donation on behalf of the respondent in appreciation for their time. We will use questionnaire personalization—a previously published methodology developed by our team—to gather more detailed information about aspects of the published trial [43]. The anticipated response rate, based on previous experience with this population and methodology, is 65% [44]. Results from the survey will be compared to results from the published trial to assess adequacy of reporting of ethical issues and to describe implications of design choices (e.g. impact of alternative consent models on study recruitment and risks of selection bias). Potential non-response bias will be assessed by comparing characteristics of respondents and non-respondents using information in trial reports. We will describe the use of gatekeepers (i.e. individuals or bodies that represent the interests of community members, communities, or organizations [45]), use of consent waivers or alternative consent models (e.g. “streamlined consent” [12]), and details about information conveyed to participants in each study arm. We will describe the type of ethics review required, perceived impact on the timing of implementation, ethical and scientific quality of the trial, and uniformity of process and decisions in multicenter trials. We will explore the possibility of requesting informed consent documents and research ethics application forms and protocols for a subset of trials. Results will inform the ethics framework for analysis in the conceptual work.
Study 4: survey of research ethics committee chairs
The objective of this study is to gather information on the views, practices, and experiences of research ethics committee chairs in Canada, the USA, UK, France, and Australia. We will aim to select all research ethics committees that review clinical trials in Canada (approximately 200), the UK (approximately 100), Australia [46] (approximately 200), France (approximately 39) [47], and a random sample of 200 from over 9000 Institutional Review Board Organizations in the USA [48]. We chose these five countries primarily based on logistical considerations: our team members have connections with research ethics organizations in these countries which will help facilitate participation. Canada does not maintain a list of research ethics committees. We will use a strategy previously developed by our team to identify eligible committees [49]. It involves integrating internet searches with a list of Institutional Review Board Organizations maintained by the US Office of Human Research Protections. Given that biomedical and non-biomedical Institutional Review Boards are not differentiated in this list, we will use stratification by National Institutes of Health funding levels to increase the efficiency of identifying Boards with relevant experience reviewing clinical research.
Questionnaire items will be informed by the preliminary ethics framework, key informant interviews, and results from the trialist survey. Questionnaires will consist of open- and closed-ended items and include a series of scenarios. After pilot testing, a series of contacts (notifications and reminders) based on Dillman’s recommendations for Internet surveys will be used [42]. We will likely encounter challenges in ensuring an adequate response rate; based on previous experience with surveying this population, we expect a response rate of approximately 35% [49]. We will deliberately keep questionnaires short and adhere to recent recommendations for improving response rates [42]. Results will be summarized using descriptive statistics and compared across subgroups (e.g. country, size and type of committee, years of experience). Where feasible, potential non-response bias will be assessed by comparing characteristics of respondents and non-respondents using information on research ethics committee websites. Questionnaires will be prepared in both English and French. Open-ended responses will be analyzed thematically. Results will inform the ethics framework for analysis in the conceptual work and knowledge translation activities to research ethics committees.
Study 5: focus group discussions and community survey
The objective of this study is to gather information on the views and experiences of trial participants or prospective trial participants (e.g. patients), gatekeepers (organizational leadership, medical directors), and communities. Our research team members are involved in 15–20 pragmatic trials at any one time. We will identify one ongoing or recently completed pragmatic trial in each country, ensuring a range of types of pragmatic trials. We will conduct focus groups with eligible trial participants (with permission from the responsible research ethics committees and the chief investigator). We anticipate five focus groups with patients and five with gatekeepers. Focus groups will be 1–2 h in length and involve six participants per group. A semi-structured discussion guide will be used to gather information on participants’ experiences with the trial including recruitment, informed consent, perceived benefits and harms, any privacy concerns, and satisfaction with the trial. Among eligible prospective participants, we will explore potential reasons for non-participation. Discussions will be recorded, transcribed verbatim, and verified by the facilitator before analysis in NVivo 11 [50]. To monitor progress and permit follow-up of issues that may emerge from the data, discussions, transcription, and analysis will proceed concurrently [51]. For focus groups not conducted in French, interview guides will be developed in English by the study team and then translated into French by bilingual members of the research team. To verify the accuracy of the translation, all guides will be independently back-translated. All focus group discussions will be transcribed in the language in which the group was conducted. Non-English transcripts will be translated into English and then independently back-translated to the original language. A Canadian pragmatic trial will be used to design a survey of community members targeted by the trial (e.g. diabetes patients, hospital patients). Quantitative and qualitative analyses will be used to summarize results from the community survey. Results will be used to inform the ethical analysis in the conceptual work.
Ethical analysis
The ethical analysis will be an intensive process run concurrently with the empirical work and will extend over a period of three years. Conceptual work in bioethics is not amenable to the degree of a priori methodological specification that is expected of empirical research. Reproducibility is an indispensable feature of rigorous science, necessitating the clear statement of hypotheses and experimental methods upfront. Rigorous conceptual work in ethics begins with the articulation of clear and important questions and is realized in the construction of careful and clear analysis of the relevant concepts and of ethical arguments in peer-reviewed publications and policy reports [52]. The ethical analysis in this project will be based on an evolving framework of ethical issues developed using results from an extensive literature search conducted in preparation for this proposal (see Table 2) and revised using results from the five empirical studies. For each identified set of core ethical issues, an in-depth and written ethical analysis will be prepared. An extensive review of the scholarly literature will document and critically analyze arguments proffered for and against ethical positions. The ethical analysis will seek to synthesize foundational documents, regulations, and arguments in the literature into a coherent solution. Where disagreement among the various sources cannot be resolved by critical analysis, the contours of the ethical dispute will be documented. The ethical analysis will result in a series of background documents laying out proposed solutions with principles, policy options, and rationale and will also be submitted for peer-reviewed publication. These documents will be used as background materials in the consensus process.
Expert panel and consensus process
An international expert panel will be convened to develop ethics guidance. The composition of the panel is expected to be 4–6 pragmatic trialists and methodologists, 4–6 ethicists and chairs of research ethics committees, 2–4 quality improvement experts and health system leaders, 1–2 regulators, 1–2 journal editors, and 2–4 community members. No more than one-third of the members of the panel will be drawn from the research team and adequate representation from a broad range of countries including lower- and middle-income countries will be sought. The panel will be provided with output from the empirical studies as well as the documents prepared during the ethical analysis one month in advance of a three-day consensus conference which will also be attended by invited researchers and representatives from major funding bodies, regulators, journal editors, and community groups. The conference will consist of both open and closed sessions. At the open sessions, proposed ethics guidance with supporting ethical analysis will be presented by the research team and comments and discussion invited from attendees. The panel will then meet in closed sessions to discuss and formulate draft guidance. Based on previous experience with the consensus process [21], we anticipate that at the beginning of the meeting, the panel will set rules for debate, handling of disagreements, and how to achieve consensus. We do not expect to use a majority voting system but anticipate that agreement will be reached through discussion, with documentation of disagreements where they exist. After the meeting, a draft guidance document will be produced by a writing committee and refined after further discussion with the panel. An e-consultation process will be launched to invite comments from the broader research community, funders, regulators, journal editors, and community groups. Based on results from this process, the writing committee will make revisions and produce the final consensus guidance.
Discussion
The goal of our international, interdisciplinary collaboration is to develop, publish, and promote the uptake of guidance for the ethical design and conduct of pragmatic randomized controlled trials. We seek to create a novel approach to the ethics of pragmatic trials that improves upon the existing literature, which has been criticized for lacking convincing arguments grounded in ethical principles [53], including claims based on erroneous assumptions [54], appealing mainly to regulations in particular jurisdictions [55,56,57], offering speculative solutions with no guarantee of improvement over existing oversight procedures [58], and failing to reflect a broad vision of protecting the liberty and welfare interests of research participants [59]. The proposed process is informed and enriched by our previous experience with developing ethics guidance for cluster randomized trials [21, 60]. The planned output includes manuscripts, educational materials, and tailored guidance documents. Our proposed project is novel in that it: (1) involves close collaboration between clinical trialists, ethicists, and methodologists; (2) combines concurrent empirical and ethical analysis in a mutually informative approach; (3) integrates views and experiences of stakeholders (e.g. trialists, chairs of research ethics committees, health system leaders, community members); and (4) aims to generate guidance rooted in internationally accepted ethical principles rather than regulation specific to one jurisdiction. We expect that the study outputs will be of interest to a wide range of knowledge users including trialists, healthcare professionals, ethicists, research ethics committees, journal editors, regulators, health system leaders, research funders, and patient groups. Guidance will facilitate the conduct of research important to patients, clinicians, and the healthcare system, while upholding the highest ethical standards in research. While the scope of the planned guidance is intended to be international, some of the empirical studies (e.g. focus groups) will be geographically restricted based on logistical and feasibility considerations.
Our knowledge translation strategy will be guided by the Canadian Institutes of Health Research Guide to Knowledge Translation Planning [61]. We will use our considerable informal networks nationally and internationally to disseminate our findings. Work stemming from the research will be submitted for presentation at national and international conferences and meetings targeting specific stakeholder groups (e.g. journal editors, funders, regulators). The final ethics guidance document will be published in a major journal with summaries published simultaneously in other major journals. Educational material for researchers and research ethics committees will be developed. Social media will be used to communicate results to the public.
Abbreviations
- CRT:
-
Cluster randomized trial
- PRECIS – 2:
-
PRagmatic Explanatory Continuum Indicator Summary
- QI:
-
Quality improvement
- RCT:
-
Randomized controlled trial
- REC:
-
Research Ethics Committee
References
Zwarenstein M. ‘Pragmatic’ and ‘Explanatory’ attitudes to randomized trials. J R Soc Med. 2016;110:208–18.
Schwartz D, Lellouch J. Explanatory and pragmatic attitudes in therapeutical trials. J Clin Epidemiol. 2009;62:499–505. https://doi.org/10.1016/j.jclinepi.2009.01.012.
Patsopoulos NA. A pragmatic view on pragmatic trials. Dialogues Clin Neurosci. 2011;13:217–24.
Zwarenstein M, Treweek S, Gagnier JJ, Altman DG, Tunis S, Haynes B, et al. Improving the reporting of pragmatic trials: an extension of the CONSORT statement. BMJ. 2008;337:a2390. https://doi.org/10.1136/bmj.a2390.
Loudon K, Zwarenstein M, Sullivan F, Donnan P, Treweek S. PRECIS-2: a tool to improve the applicability of randomised controlled trials. Trials. 2013;14:O28. https://doi.org/10.1186/1745-6215-14-s1-o28.
Innovative Clinical Trials Initiative. http://www.cihr-irsc.gc.ca/e/49773.html. Accessed 01 May 2018.
Woodcock A. Commentary: view from the frontline of pragmatic trials. BMJ. 2017;357:j2837. https://doi.org/10.1136/bmj.j2837.
Pragmatic clinical trials: Testing treatments in the real world. https://www.nia.nih.gov/research/blog/2017/06/pragmatic-clinical-trials-testing-treatments-real-world. Accessed 01 May 2018.
NIH Health Care Systems Research Collaboratory. Demonstration Projects for Pragmatic Clinical Trials (UG3/UH3). https://grants.nih.gov/grants/guide/rfa-files/RFA-RM-16-019.html. Accessed 01 May 2018.
Australian Clinical Trials Alliance (ACTA). Report on the 2014 national summit of investigator-initiated clinical trials networks. Melbourne; 2014. http://www.clinicaltrialsalliance.org.au/wp-content/uploads/2014/10/ACTA_Summit_RPT14_Print_LR.pdf. Accessed 24 Sept 2018.
Frieden TR. Evidence for health decision making - beyond randomized, controlled trials. N Engl J Med. 2017;377:465–75. https://doi.org/10.1056/NEJMra1614394.
Goldstein CE, Weijer C, Brehaut JC, Fergusson DA, Grimshaw JM, Horn AR, et al. Ethics issues in pragmatic randomized controlled trials: a review of the recent literature identifies gaps in argumentation. BMC Med Ethics. 2018;19(14). https://doi.org/10.1186/s12910-018-0253-x.
Califf RM, Sugarman J. Exploring the ethical and regulatory issues in pragmatic clinical trials. Clin Trials. 2015;12:436–41. https://doi.org/10.1177/1740774515598334.
Sugarman J, Califf RM. Ethics and regulatory complexities for pragmatic clinical trials. JAMA. 2014;311:2381–2. https://doi.org/10.1001/jama.2014.4164.
Kalkman S, van Thiel GJ, Grobbee DE, van Delden JJ. Pragmatic randomized trials in drug development pose new ethical questions: a systematic review. Drug Discov Today. 2015;20:856–62. https://doi.org/10.1016/j.drudis.2015.03.005.
Relton C, Torgerson D, O’Cathain A, Nicholl J. Rethinking pragmatic randomised controlled trials: introducing the “cohort multiple randomised controlled trial” design. BMJ. 2010;340:c1066. https://doi.org/10.1136/bmj.c1066.
Lauer MS, D’Agostino RB Sr. The randomized registry trial - the next disruptive technology in clinical research? N Engl J Med. 2013;369:1579–81. https://doi.org/10.1056/NEJMp1310771.
Bellomo R, Forbes A, Akram M, Bailey M, Pilcher DV, Cooper DK. Why we must cluster and cross over. Crit Care Resusc. 2013;15:155–7.
Hemming K, Haines TP, Chilton PJ, Girling AJ, Lilford RJ. The stepped wedge cluster randomised trial: rationale, design, analysis, and reporting. BMJ. 2015;350:h391. https://doi.org/10.1136/bmj.h391.
Donner A, Klar N. Design and analysis of cluster randomization trials in health research. London: Arnold; 2000.
Weijer C, Grimshaw JM, Eccles MP, McRae AD, White A, Brehaut JC, et al. The Ottawa statement on the ethical design and conduct of cluster randomized trials. PLoS Med. 2012;9:e1001346. https://doi.org/10.1371/journal.pmed.1001346.
Support Study Group of the Eunice Kennedy Shriver NICHD Neonatal Research Network. Target ranges of oxygen saturation in extremely preterm infants. N Engl J Med. 2010;362:1959–69. https://doi.org/10.1056/NEJMoa0911781.
Office for Human Research Protections. Letter to the University of Alabama at Birmingham. 2013. https://www.hhs.gov/ohrp/sites/default/files/ohrp/detrm_letrs/YR13/mar13a.pdf. Accessed 24 Sept 2018.
Annas GJ, Annas CL. Legally blind: the therapeutic illusion in the SUPPORT study of extremely premature infants. J Contemp Health L Pol’y. 2013;30:1–36.
Lantos JD, Feudtner C. SUPPORT and the ethics of study implementation: lessons for comparative effectiveness research from the trial of oxygen therapy for premature babies. Hast Cent Rep. 2015;45:30–40. https://doi.org/10.1002/hast.407.
Macklin R, Shepherd L. Informed consent and standard of care: what must be disclosed. Am J Bioeth. 2013;13:9–13. https://doi.org/10.1080/15265161.2013.849303.
Macklin R, Shepherd L, Dreger A, Asch A, Baylis F, Brody H, et al. The OHRP and SUPPORT — another view. N Engl J Med. 2013;369:e3.
McIntyre L, Taljaard M, McArdle T, Fox-Robichaud A, English S, Martin C, et al. FLUID trial: a protocol for a hospital-wide open-label cluster crossover pragmatic comparative effectiveness randomized pilot trial. BMJ Open. 2018;8:e022780.
Haugen AS, Softeland E, Almeland SK, Sevdalis N, Vonen B, Eide GE, et al. Effect of the World Health Organization checklist on patient outcomes: a stepped wedge cluster randomized controlled trial. Ann Surg. 2015;261:821–8. https://doi.org/10.1097/SLA.0000000000000716.
Patton MQ. Qualitative evaluation and research methods. Newbury Park, CA: Sage; 2002.
Patton M. Qualitative research and evaluation methods. 3rd ed. Thousand Oaks, CA: Sage Publications; 2002.
Lincoln Y, Guba E. Naturalistic Inquiry. New York, NY: Sage; 1985.
Krueger RA, Casey MA. Focus groups: a practical guide for applied research. 3rd ed. Thousand Oaks, CA: Sage; 2000.
Bowling A. Research methods in health. 2nd ed. Maidenhead: Open University Press; 2004.
McRae A, Bennett C, Belle Brown J, Weijer C, Boruch R, Brehaut J, et al. Researchers’ perceptions of ethical challenges in cluster randomized trials: a qualitative analysis. Trials. 2013;14:1.
Trials. https://www.precis-2.org/Trials. Accessed 01 May 2018.
Rethinking Clinical Trials: A Living Textbook of Pragmatic Clinical Trials. http://www.rethinkingclinicaltrials.org/. Accessed 01 May 2018.
PCORnet, the National Patient-Centered Clinical Research Network. https://www.pcornet.org/. Accessed 01 May 2018.
Taljaard M, McGowan J, Grimshaw JM, Brehaut JC, McRae A, Eccles MP, et al. Electronic search strategies to identify reports of cluster randomized trials in MEDLINE: low precision will improve with adherence to reporting standards. BMC Med Res Methodol. 2010;10:15.
Ivers NM, Taljaard M, Dixon S, Bennett C, McRae A, Taleban J, et al. Impact of CONSORT extension for cluster randomised trials on quality of reporting and study methodology: review of random sample of 300 trials, 2000-8. BMJ. 2011;343:d5886. https://doi.org/10.1136/bmj.d5886.
Taljaard M, McRae AD, Weijer C, Bennett C, Dixon S, Taleban J, et al. Inadequate reporting of research ethics review and informed consent in cluster randomised trials: review of random sample of published trials. BMJ. 2011;342:d2496. https://doi.org/10.1136/bmj.d2496.
Dillman DA, Smyth JD, Christian LM. Internet, phone, mail, and mixed-mode surveys. The tailored design method. 4th ed. Hoboken, NJ: Wiley; 2014.
Taljaard M, Chaudhry SH, Brehaut JC, Weijer C, Grimshaw JM. Mail merge can be used to create personalized questionnaires in complex surveys. BMC Res Notes. 2015;8:574. https://doi.org/10.1186/s13104-015-1570-5.
Taljaard M, Chaudhry SH, Brehaut JC, Weijer C, Boruch R, Donner A, et al. Survey of consent practices in cluster randomized trials: improvements are needed in ethical conduct and reporting. Clin Trials. 2014;11:60–9.
Gallo A, Weijer C, White A, Grimshaw JM, Boruch R, Brehaut JC, et al. What is the role and authority of gatekeepers in cluster randomized trials in health research? Trials. 2012;13:116.
Human Research Ethics Committees (HRECs). https://www.nhmrc.gov.au/health-ethics/human-research-ethics-committees-hrecs. Accessed 01 May 2018.
National Information: France. http://www.eurecnet.org/information/france.html. Accessed 01 May 2018.
Office for Human Research Protections (OHRP) Database for Registered IORGs & IRBs, Approved FWAs, and Documents Received in Last 60 Days. http://ohrp.cit.nih.gov/search/search.aspx?styp=bsc. Accessed 01 May 2018.
Taljaard M, Brehaut JC, Weijer C, Boruch R, Donner A, Eccles MP, et al. Variability in research ethics review of cluster randomized trials: a scenario-based survey in three countries. Trials. 2014;15:48.
QSR International Pty Ltd. NVivo qualitative data analysis Software. Version 11 edn; 2017.
Marshall C, Rossman G. Designing qualitative research. Newbury Park, CA: Sage; 1989.
Taljaard M, Weijer C, Grimshaw JM, Belle Brown J, Binik A, Boruch R, et al. Ethical and policy issues in cluster randomized trials: rationale and design of a mixed methods research study. Trials. 2009;10:61. https://doi.org/10.1186/1745-6215-10-61.
Kim SY, Miller FG. Ethical complexities in standard of care randomized trials: a case study of morning versus nighttime dosing of blood pressure drugs. Clin Trials. 2015;12:557–63. https://doi.org/10.1177/1740774515607213.
Faden RR, Kass NE, Goodman SN, Pronovost P, Tunis S, Beauchamp TL. An ethics framework for a learning health care system: a departure from traditional research ethics and clinical ethics. Hastings Cent Rep. 2013;Spec No:S16–27. https://doi.org/10.1002/hast.134.
Lantos JD, Wendler D, Septimus E, Wahba S, Madigan R, Bliss G. Considerations in the evaluation and determination of minimal risk in pragmatic clinical trials. Clin Trials. 2015;12:485–93. https://doi.org/10.1177/1740774515597687.
O'Rourke PP, Carrithers J, Patrick-Lake B, Rice TW, Corsmo J, Hart R, et al. Harmonization and streamlining of research oversight for pragmatic clinical trials. Clin Trials. 2015;12:449–56. https://doi.org/10.1177/1740774515597685.
McKinney RE Jr, Beskow LM, Ford DE, Lantos JD, McCall J, Patrick-Lake B, et al. Use of altered informed consent in pragmatic clinical research. Clin Trials. 2015;12:494–502. https://doi.org/10.1177/1740774515597688.
Faden RR, Beauchamp TL, Kass NE. Informed consent, comparative effectiveness, and learning health care. N Engl J Med. 2014;370:766–8.
Selby JV, Krumholz HM. Ethical oversight: serving the best interests of patients. Hastings Cent Rep. 2013;43:S34–6. https://doi.org/10.1002/hast.138.
Taljaard M, Weijer C, Grimshaw JM, Eccles MP, Ottawa Ethics of Cluster Randomised Trials Consensus Group. The Ottawa Statement on the ethical design and conduct of cluster randomised trials: précis for researchers and research ethics committees. BMJ. 2013;346:f2838. https://doi.org/10.1136/bmj.f2838.
Guide to Knowledge Translation Planning at CIHR: Integrated and End-of-Grant Approaches. http://www.cihr-irsc.gc.ca/e/45321.html. Accessed 01 May 2018.
Funding
This work is supported by the Canadian Institutes of Health Research through the Project Grant competition (competitive, peer-reviewed), award number PJT-153045. Jeremy Grimshaw holds a Canada Research Chair in Health Knowledge Transfer and Uptake. Charles Weijer holds a Canada Research Chair in Bioethics. Sarah Edwards is funded by the UCL/UCLH Biomedical Research Centre (BRC). Vipul Jairath hold a personal Endowed Chair at Western University (John and Susan McDonald Endowed Chair). Joanne McKenzie is supported by an NHMRC Career Development Fellowship (1143429). The University of Aberdeen’s Health Services Research Unit is core-funded by the Chief Scientist Office of the Scottish Government Health and Social Care Directorates. Ian D Graham is a CIHR Foundation Grant recipient (FDN# 143237).
Author information
Authors and Affiliations
Contributions
MT, CW, JMG, and DAF conceived the project idea and co-led the development of the study protocol and funding application with substantial contributions from all authors. MT wrote the initial draft of the manuscript. MT, CW, JMG, AA, JCB, MC, KC, SE, SE, CBF, BG, CEG, IDG, KH, SH, AH, VJ, TK, AJL, SM, JM, LM, JM, SN, AP, MZ, and DAF contributed critical revisions and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This manuscript does not report on or involve the use of any animal or human data or tissue. Research ethics approval for specific future studies within this project will be sought from Clinical Trials Ontario or the Ottawa Health Science Network Research Ethics Board and/or any applicable local research ethics committees in relevant jurisdictions.
Consent for publication
All authors have approved the manuscript for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Taljaard, M., Weijer, C., Grimshaw, J.M. et al. Developing a framework for the ethical design and conduct of pragmatic trials in healthcare: a mixed methods research protocol. Trials 19, 525 (2018). https://doi.org/10.1186/s13063-018-2895-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2895-x